

A New Wave of Targeting ‘Undruggable’ Wnt Signaling for Cancer Therapy: Challenges and Opportunities


Abstract
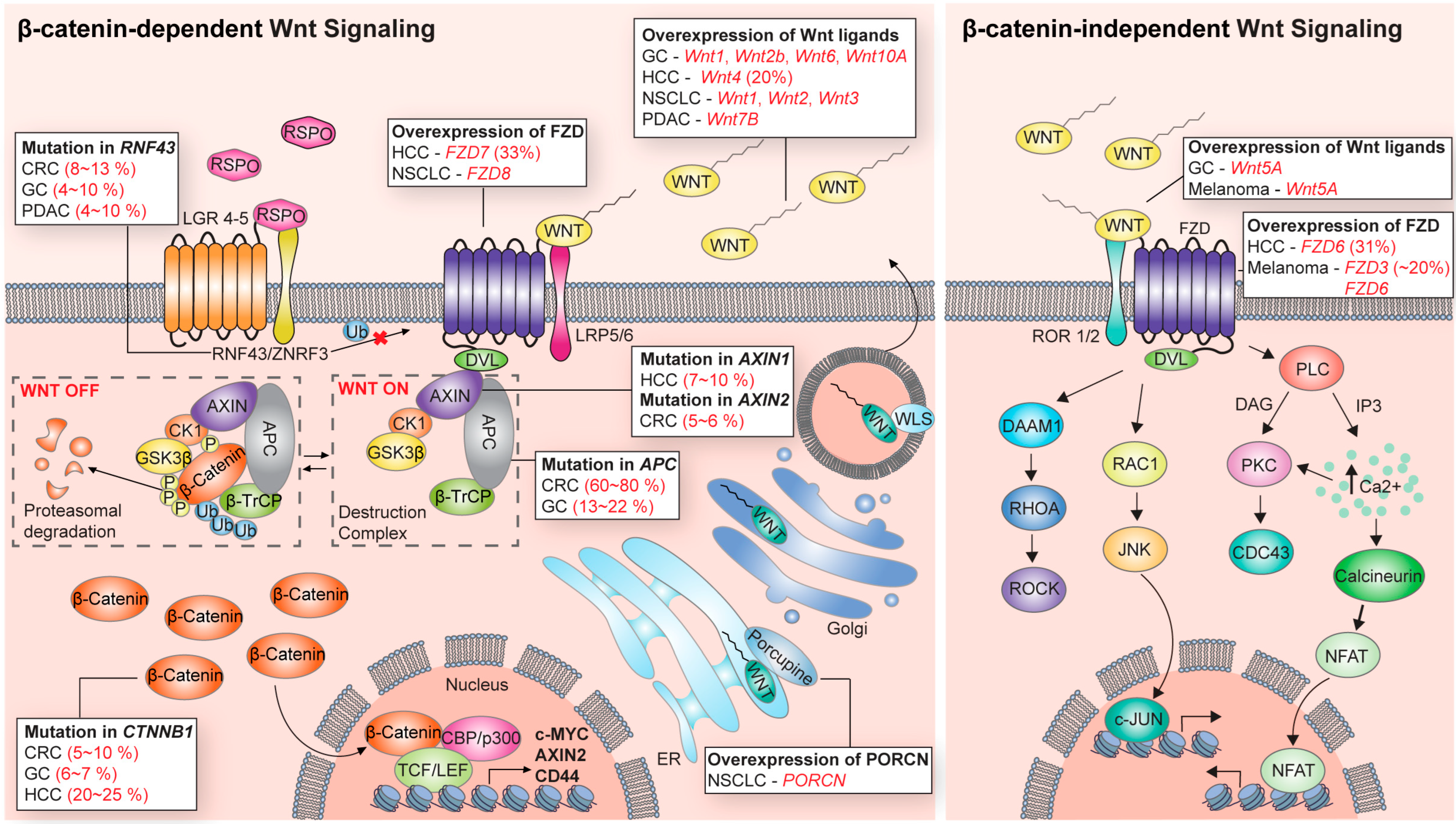
1. Wnt Signaling Pathway
1.1. Wnt Ligands and Receptors
1.2. β-Catenin-Dependent Signaling
1.3. β-Catenin-Independent Signaling
2. Alterations of Wnt Signaling in Cancers
2.1. Driver Alterations of Wnt Signaling in Cancers
2.2. Cancer-Promoting Alterations of Wnt Signaling in Cancers
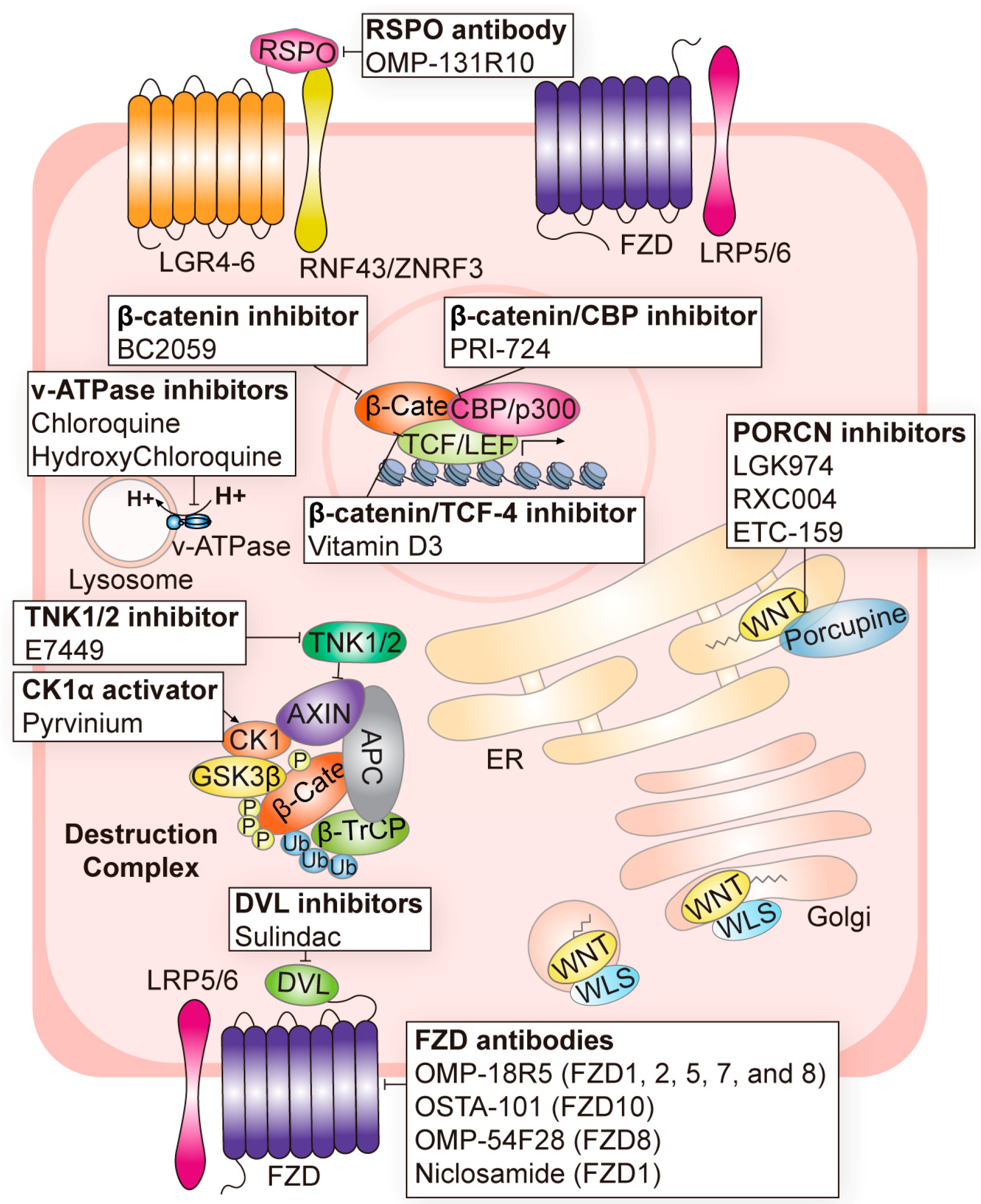
3. Targeting Strategies for Wnt Signaling in Cancer
3.1. Targeting Wnt Ligands
3.2. Targeting Wnt Receptors
3.3. Targeting the β-Catenin Destruction Complex
3.4. Targeting β-Catenin and Its Transcription Partners
3.5. Current Caveats of Targeting Wnt Signaling
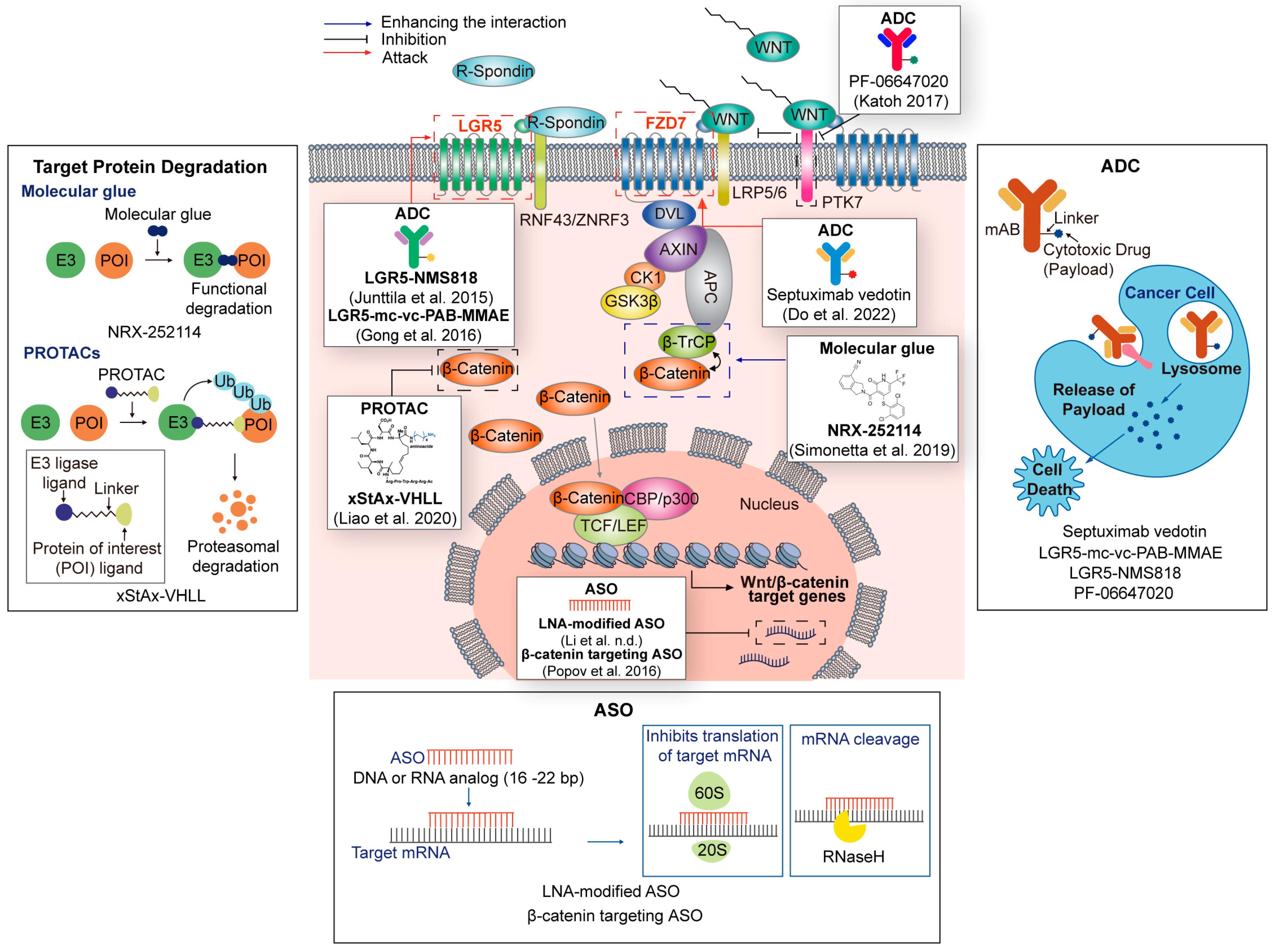
4. New Targeting Strategies for Wnt Signaling in Cancer
4.1. PROTAC/Molecular Glue-Based Wnt Signaling Targeting
4.1.1. PROTAC
4.1.2. Molecular Glue
4.1.3. Other Protein Degradation Technologies
4.2. Antibody–Drug Conjugate (ADC)-Based Wnt Signaling Targeting
4.3. Oligonucleotide-Based Wnt Signaling Targeting
4.3.1. MicroRNAs and siRNAs
4.3.2. Antisense Oligonucleotides
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nüsslein-Volhard, C.; Wieschaus, E. Mutations affecting segment number and polarity in Drosophila. Nature 1980, 287, 795–801. [Google Scholar] [CrossRef]
- Nusse, R.; Varmus, H.E. Many tumors induced by the mouse mammary tumor virus contain a provirus integrated in the same region of the host genome. Cell 1982, 31, 99–109. [Google Scholar] [CrossRef]
- Yu, F.; Yu, C.; Li, F.; Zuo, Y.; Wang, Y.; Yao, L.; Wu, C.; Wang, C.; Ye, L. Wnt/β-catenin signaling in cancers and targeted therapies. Signal Transduct. Target. Ther. 2021, 6, 307. [Google Scholar] [CrossRef]
- Jackstadt, R.; Hodder, M.C.; Sansom, O.J. WNT and β-Catenin in Cancer: Genes and Therapy. Annu. Rev. Cancer Biol. 2020, 4, 177–196. [Google Scholar] [CrossRef]
- Xu, X.; Zhang, M.; Xu, F.; Jiang, S. Wnt signaling in breast cancer: Biological mechanisms, challenges and opportunities. Mol. Cancer 2020, 19, 165. [Google Scholar] [CrossRef]
- Clevers, H.; Loh, K.M.; Nusse, R. Stem cell signaling. An integral program for tissue renewal and regeneration: Wnt signaling and stem cell control. Science 2014, 346, 1248012. [Google Scholar] [CrossRef]
- Galli, L.M.; Zebarjadi, N.; Li, L.; Lingappa, V.R.; Burrus, L.W. Divergent effects of Porcupine and Wntless on WNT1 trafficking, secretion, and signaling. Exp. Cell Res. 2016, 347, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Niehrs, C. The complex world of WNT receptor signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 767–779. [Google Scholar] [CrossRef]
- Schulte, G.; Bryja, V. The Frizzled family of unconventional G-protein-coupled receptors. Trends Pharmacol. Sci. 2007, 28, 518–525. [Google Scholar] [CrossRef]
- Nusse, R.; Clevers, H. Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef] [PubMed]
- Koo, B.K.; Spit, M.; Jordens, I.; Low, T.Y.; Stange, D.E.; van de Wetering, M.; van Es, J.H.; Mohammed, S.; Heck, A.J.; Maurice, M.M.; et al. Tumour suppressor RNF43 is a stem-cell E3 ligase that induces endocytosis of Wnt receptors. Nature 2012, 488, 665–669. [Google Scholar] [CrossRef]
- Hao, H.X.; Xie, Y.; Zhang, Y.; Charlat, O.; Oster, E.; Avello, M.; Lei, H.; Mickanin, C.; Liu, D.; Ruffner, H.; et al. ZNRF3 promotes Wnt receptor turnover in an R-spondin-sensitive manner. Nature 2012, 485, 195–200. [Google Scholar] [CrossRef]
- Zebisch, M.; Xu, Y.; Krastev, C.; MacDonald, B.T.; Chen, M.; Gilbert, R.J.; He, X.; Jones, E.Y. Structural and molecular basis of ZNRF3/RNF43 transmembrane ubiquitin ligase inhibition by the Wnt agonist R-spondin. Nat. Commun. 2013, 4, 2787. [Google Scholar] [CrossRef] [PubMed]
- Junge, H.J.; Yang, S.; Burton, J.B.; Paes, K.; Shu, X.; French, D.M.; Costa, M.; Rice, D.S.; Ye, W. TSPAN12 regulates retinal vascular development by promoting Norrin- but not Wnt-induced FZD4/beta-catenin signaling. Cell 2009, 139, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Wang, Y.; Dabdoub, A.; Smallwood, P.M.; Williams, J.; Woods, C.; Kelley, M.W.; Jiang, L.; Tasman, W.; Zhang, K.; et al. Vascular development in the retina and inner ear: Control by Norrin and Frizzled-4, a high-affinity ligand-receptor pair. Cell 2004, 116, 883–895. [Google Scholar] [CrossRef]
- Morin, P.J. beta-catenin signaling and cancer. Bioessays 1999, 21, 1021–1030. [Google Scholar] [CrossRef]
- Yu, W.; Yang, L.; Li, T.; Zhang, Y. Cadherin Signaling in Cancer: Its Functions and Role as a Therapeutic Target. Front. Oncol. 2019, 9, 989. [Google Scholar] [CrossRef] [PubMed]
- Orsulic, S.; Huber, O.; Aberle, H.; Arnold, S.; Kemler, R. E-cadherin binding prevents beta-catenin nuclear localization and beta-catenin/LEF-1-mediated transactivation. J. Cell Sci. 1999, 112, 1237–1245. [Google Scholar] [CrossRef]
- Kim, M.J.; Huang, Y.; Park, J.I. Targeting Wnt Signaling for Gastrointestinal Cancer Therapy: Present and Evolving Views. Cancers 2020, 12, 3638. [Google Scholar] [CrossRef]
- Takemaru, K.I.; Moon, R.T. The transcriptional coactivator CBP interacts with beta-catenin to activate gene expression. J. Cell Biol. 2000, 149, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Hecht, A.; Vleminckx, K.; Stemmler, M.P.; van Roy, F.; Kemler, R. The p300/CBP acetyltransferases function as transcriptional coactivators of beta-catenin in vertebrates. EMBO J. 2000, 19, 1839–1850. [Google Scholar] [CrossRef]
- Schlessinger, K.; Hall, A.; Tolwinski, N. Wnt signaling pathways meet Rho GTPases. Genes Dev. 2009, 23, 265–277. [Google Scholar] [CrossRef] [PubMed]
- De, A. Wnt/Ca2+ signaling pathway: A brief overview. Acta Biochim. Biophys. Sin. (Shanghai) 2011, 43, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.R.; Hocking, A.M.; Brown, J.D.; Moon, R.T. Mechanism and function of signal transduction by the Wnt/beta-catenin and Wnt/Ca2+ pathways. Oncogene 1999, 18, 7860–7872. [Google Scholar] [CrossRef] [PubMed]
- Network, C.G.A. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Giles, R.H.; van Es, J.H.; Clevers, H. Caught up in a Wnt storm: Wnt signaling in cancer. Biochim. Biophys. Acta 2003, 1653, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Bugter, J.M.; Fenderico, N.; Maurice, M.M. Mutations and mechanisms of WNT pathway tumour suppressors in cancer. Nat. Rev. Cancer 2021, 21, 5–21. [Google Scholar] [CrossRef]
- Li, S.; Lavrijsen, M.; Bakker, A.; Magierowski, M.; Magierowska, K.; Liu, P.; Wang, W.; Peppelenbosch, M.P.; Smits, R. Commonly observed RNF43 mutations retain functionality in attenuating Wnt/β-catenin signaling and unlikely confer Wnt-dependency onto colorectal cancers. Oncogene 2020, 39, 3458–3472. [Google Scholar] [CrossRef]
- Wang, K.; Yuen, S.T.; Xu, J.; Lee, S.P.; Yan, H.H.; Shi, S.T.; Siu, H.C.; Deng, S.; Chu, K.M.; Law, S.; et al. Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nat. Genet. 2014, 46, 573–582. [Google Scholar] [CrossRef]
- Ratti, M.; Lampis, A.; Hahne, J.C.; Passalacqua, R.; Valeri, N. Microsatellite instability in gastric cancer: Molecular bases, clinical perspectives, and new treatment approaches. Cell. Mol. Life Sci. 2018, 75, 4151–4162. [Google Scholar] [CrossRef]
- Wang, W.; Xu, L.; Liu, P.; Jairam, K.; Yin, Y.; Chen, K.; Sprengers, D.; Peppelenbosch, M.P.; Pan, Q.; Smits, R. Blocking Wnt Secretion Reduces Growth of Hepatocellular Carcinoma Cell Lines Mostly Independent of β-Catenin Signaling. Neoplasia 2016, 18, 711–723. [Google Scholar] [CrossRef]
- Mao, J.; Fan, S.; Ma, W.; Fan, P.; Wang, B.; Zhang, J.; Wang, H.; Tang, B.; Zhang, Q.; Yu, X.; et al. Roles of Wnt/β-catenin signaling in the gastric cancer stem cells proliferation and salinomycin treatment. Cell Death Dis. 2014, 5, e1039. [Google Scholar] [CrossRef] [PubMed]
- Yuan, G.; Regel, I.; Lian, F.; Friedrich, T.; Hitkova, I.; Hofheinz, R.D.; Ströbel, P.; Langer, R.; Keller, G.; Röcken, C.; et al. WNT6 is a novel target gene of caveolin-1 promoting chemoresistance to epirubicin in human gastric cancer cells. Oncogene 2013, 32, 375–387. [Google Scholar] [CrossRef]
- Oshima, H.; Matsunaga, A.; Fujimura, T.; Tsukamoto, T.; Taketo, M.M.; Oshima, M. Carcinogenesis in mouse stomach by simultaneous activation of the Wnt signaling and prostaglandin E2 pathway. Gastroenterology 2006, 131, 1086–1095. [Google Scholar] [CrossRef]
- Zuo, W.; Yang, H.; Li, N.; Ouyang, Y.; Xu, X.; Hong, J. Helicobacter pylori infection activates Wnt/β-catenin pathway to promote the occurrence of gastritis by upregulating ASCL1 and AQP5. Cell Death Discov. 2022, 8, 257. [Google Scholar] [CrossRef]
- Wu, C.; Zhuang, Y.; Jiang, S.; Liu, S.; Zhou, J.; Wu, J.; Teng, Y.; Xia, B.; Wang, R.; Zou, X. Interaction between Wnt/β-catenin pathway and microRNAs regulates epithelial-mesenchymal transition in gastric cancer (Review). Int. J. Oncol. 2016, 48, 2236–2246. [Google Scholar] [CrossRef] [PubMed]
- Lachenmayer, A.; Alsinet, C.; Savic, R.; Cabellos, L.; Toffanin, S.; Hoshida, Y.; Villanueva, A.; Minguez, B.; Newell, P.; Tsai, H.W.; et al. Wnt-pathway activation in two molecular classes of hepatocellular carcinoma and experimental modulation by sorafenib. Clin. Cancer Res. 2012, 18, 4997–5007. [Google Scholar] [CrossRef]
- Taniguchi, K.; Roberts, L.R.; Aderca, I.N.; Dong, X.; Qian, C.; Murphy, L.M.; Nagorney, D.M.; Burgart, L.J.; Roche, P.C.; Smith, D.I.; et al. Mutational spectrum of beta-catenin, AXIN1, and AXIN2 in hepatocellular carcinomas and hepatoblastomas. Oncogene 2002, 21, 4863–4871. [Google Scholar] [CrossRef]
- Bengochea, A.; de Souza, M.M.; Lefrançois, L.; Le Roux, E.; Galy, O.; Chemin, I.; Kim, M.; Wands, J.R.; Trepo, C.; Hainaut, P.; et al. Common dysregulation of Wnt/Frizzled receptor elements in human hepatocellular carcinoma. Br. J. Cancer 2008, 99, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Colnot, S.; Decaens, T.; Niwa-Kawakita, M.; Godard, C.; Hamard, G.; Kahn, A.; Giovannini, M.; Perret, C. Liver-targeted disruption of Apc in mice activates beta-catenin signaling and leads to hepatocellular carcinomas. Proc. Natl. Acad. Sci. USA 2004, 101, 17216–17221. [Google Scholar] [CrossRef]
- Satoh, S.; Daigo, Y.; Furukawa, Y.; Kato, T.; Miwa, N.; Nishiwaki, T.; Kawasoe, T.; Ishiguro, H.; Fujita, M.; Tokino, T.; et al. AXIN1 mutations in hepatocellular carcinomas, and growth suppression in cancer cells by virus-mediated transfer of AXIN1. Nat. Genet. 2000, 24, 245–250. [Google Scholar] [CrossRef]
- Jiang, X.; Hao, H.X.; Growney, J.D.; Woolfenden, S.; Bottiglio, C.; Ng, N.; Lu, B.; Hsieh, M.H.; Bagdasarian, L.; Meyer, R.; et al. Inactivating mutations of RNF43 confer Wnt dependency in pancreatic ductal adenocarcinoma. Proc. Natl. Acad. Sci. USA 2013, 110, 12649–12654. [Google Scholar] [CrossRef] [PubMed]
- Tammela, T.; Sanchez-Rivera, F.J.; Cetinbas, N.M.; Wu, K.; Joshi, N.S.; Helenius, K.; Park, Y.; Azimi, R.; Kerper, N.R.; Wesselhoeft, R.A.; et al. A Wnt-producing niche drives proliferative potential and progression in lung adenocarcinoma. Nature 2017, 545, 355–359. [Google Scholar] [CrossRef] [PubMed]
- Suda, T.; Arai, F. Wnt signaling in the niche. Cell 2008, 132, 729–730. [Google Scholar] [CrossRef]
- Ohgaki, H.; Kros, J.M.; Okamoto, Y.; Gaspert, A.; Huang, H.; Kurrer, M.O. APC mutations are infrequent but present in human lung cancer. Cancer Lett. 2004, 207, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Thomas de Montpréville, V.; Lacroix, L.; Rouleau, E.; Mamodaly, M.; Leclerc, J.; Tutuianu, L.; Planchard, D.; Boulate, D.; Mercier, O.; Besse, B.; et al. Non-small cell lung carcinomas with CTNNB1 (beta-catenin) mutations: A clinicopathological study of 26 cases. Ann. Diagn. Pathol. 2020, 46, 151522. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Wang, H.; Zhu, D. Wnt/β-catenin signaling pathway in lung cancer. Med. Drug Discov. 2022, 13, 100113. [Google Scholar] [CrossRef]
- Wagner, A.H.; Devarakonda, S.; Skidmore, Z.L.; Krysiak, K.; Ramu, A.; Trani, L.; Kunisaki, J.; Masood, A.; Waqar, S.N.; Spies, N.C.; et al. Recurrent WNT pathway alterations are frequent in relapsed small cell lung cancer. Nat. Commun. 2018, 9, 3787. [Google Scholar] [CrossRef]
- Stewart, D.J. Wnt signaling pathway in non-small cell lung cancer. J. Natl. Cancer Inst. 2014, 106, djt356. [Google Scholar] [CrossRef]
- Zhang, Y.; Morris, J.P.; Yan, W.; Schofield, H.K.; Gurney, A.; Simeone, D.M.; Millar, S.E.; Hoey, T.; Hebrok, M.; Pasca di Magliano, M. Canonical wnt signaling is required for pancreatic carcinogenesis. Cancer Res. 2013, 73, 4909–4922. [Google Scholar] [CrossRef]
- Steinhart, Z.; Pavlovic, Z.; Chandrashekhar, M.; Hart, T.; Wang, X.; Zhang, X.; Robitaille, M.; Brown, K.R.; Jaksani, S.; Overmeer, R.; et al. Genome-wide CRISPR screens reveal a Wnt-FZD5 signaling circuit as a druggable vulnerability of RNF43-mutant pancreatic tumors. Nat. Med. 2017, 23, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.M.; Yeung, V.P.; Cattaruzza, F.; Hussein, R.; Yen, W.C.; Murriel, C.; Evans, J.W.; O’Young, G.; Brunner, A.L.; Wang, M.; et al. RSPO3 antagonism inhibits growth and tumorigenicity in colorectal tumors harboring common Wnt pathway mutations. Sci. Rep. 2017, 7, 15270. [Google Scholar] [CrossRef] [PubMed]
- Säfholm, A.; Tuomela, J.; Rosenkvist, J.; Dejmek, J.; Härkönen, P.; Andersson, T. The Wnt-5a-derived hexapeptide Foxy-5 inhibits breast cancer metastasis in vivo by targeting cell motility. Clin. Cancer Res. 2008, 14, 6556–6563. [Google Scholar] [CrossRef]
- Rodon, J.; Argilés, G.; Connolly, R.M.; Vaishampayan, U.; de Jonge, M.; Garralda, E.; Giannakis, M.; Smith, D.C.; Dobson, J.R.; McLaughlin, M.E.; et al. Phase 1 study of single-agent WNT974, a first-in-class Porcupine inhibitor, in patients with advanced solid tumours. Br. J. Cancer 2021, 125, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Phillips, C.; Bhamra, I.; Eagle, C.; Flanagan, E.; Armer, R.; Jones, C.D.; Bingham, M.; Calcraft, P.; Edmenson Cook, A.; Thompson, B. The Wnt Pathway Inhibitor RXC004 Blocks Tumor Growth and Reverses Immune Evasion in Wnt Ligand–dependent Cancer Models. Cancer Res. Commun. 2022, 2, 914–928. [Google Scholar] [CrossRef]
- Kelly, J.; Woodcock, S.; Bhamra, I.; Jones, C.; Armer, R.; Robertson, J.; Phillips, C. Pre-clinical efficacy of the Wnt pathway inhibitor RXC004 in combination with anti-cancer therapies. Cancer Res. 2022, 82, 2566. [Google Scholar] [CrossRef]
- Bhamra, I.; Adams, N.; Armer, R.; Bingham, M.; McKeever, H.; Phillips, C.; Thompson, B.; Woodcock, S. Novel porcupine (PORCN) inhibitor RXC004: Evaluation in models of RNF43 loss of function cancers. J. Clin. Oncol. 2017, 35, e14094. [Google Scholar] [CrossRef]
- Ng, M.; Tan, D.S.; Subbiah, V.; Weekes, C.D.; Teneggi, V.; Diermayr, V.; Ethirajulu, K.; Yeo, P.; Chen, D.; Blanchard, S. First-in-human phase 1 study of ETC-159 an oral PORCN inhbitor in patients with advanced solid tumours. J. Clin. Oncol. 2017, 35, 2584. [Google Scholar] [CrossRef]
- Madan, B.; Ke, Z.; Harmston, N.; Ho, S.Y.; Frois, A.O.; Alam, J.; Jeyaraj, D.A.; Pendharkar, V.; Ghosh, K.; Virshup, I.H.; et al. Wnt addiction of genetically defined cancers reversed by PORCN inhibition. Oncogene 2016, 35, 2197–2207. [Google Scholar] [CrossRef]
- Jimeno, A.; Gordon, M.; Chugh, R.; Messersmith, W.; Mendelson, D.; Dupont, J.; Stagg, R.; Kapoun, A.M.; Xu, L.; Uttamsingh, S.; et al. A First-in-Human Phase I Study of the Anticancer Stem Cell Agent Ipafricept (OMP-54F28), a Decoy Receptor for Wnt Ligands, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2017, 23, 7490–7497. [Google Scholar] [CrossRef]
- Smith, D.C.; Gordon, M.; Messersmith, W.; Chugh, R.; Mendelson, D.; Dupont, J.; Stagg, R.; Kapoun, A.M.; Xu, L.; Brachmann, R.K. Abstract B79: A first-in-human Phase 1 study of anti-cancer stem cell (CSC) agent OMP-54F28 (FZD8-Fc) targeting the WNT pathway in patients with advanced solid tumors. Mol. Cancer Ther. 2013, 12, B79. [Google Scholar] [CrossRef]
- Yeung, P.; Beviglia, L.; Cancilla, B.; Dee-Hoskins, C.; Evans, J.W.; Fischer, M.M.; Yen, W.-C.; Gurney, A.; Lewicki, J.; Hoey, T. Wnt pathway antagonist OMP-54F28 (FZD8-Fc) inhibits tumor growth and reduces tumor-initiating cell frequency in patient-derived hepatocellular carcinoma and ovarian cancer xenograft models. Cancer Res. 2014, 74, 1907. [Google Scholar] [CrossRef]
- Chen, W.; Mook, R.A.; Premont, R.T.; Wang, J. Niclosamide: Beyond an antihelminthic drug. Cell. Signal. 2018, 41, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Diamond, J.R.; Becerra, C.; Richards, D.; Mita, A.; Osborne, C.; O’Shaughnessy, J.; Zhang, C.; Henner, R.; Kapoun, A.M.; Xu, L.; et al. Phase Ib clinical trial of the anti-frizzled antibody vantictumab (OMP-18R5) plus paclitaxel in patients with locally advanced or metastatic HER2-negative breast cancer. Breast Cancer Res. Treat. 2020, 184, 53–62. [Google Scholar] [CrossRef]
- Gurney, A.; Axelrod, F.; Bond, C.J.; Cain, J.; Chartier, C.; Donigan, L.; Fischer, M.; Chaudhari, A.; Ji, M.; Kapoun, A.M.; et al. Wnt pathway inhibition via the targeting of Frizzled receptors results in decreased growth and tumorigenicity of human tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 11717–11722. [Google Scholar] [CrossRef] [PubMed]
- Giraudet, A.L.; Cassier, P.A.; Iwao-Fukukawa, C.; Garin, G.; Badel, J.N.; Kryza, D.; Chabaud, S.; Gilles-Afchain, L.; Clapisson, G.; Desuzinges, C.; et al. A first-in-human study investigating biodistribution, safety and recommended dose of a new radiolabeled MAb targeting FZD10 in metastatic synovial sarcoma patients. BMC Cancer 2018, 18, 646. [Google Scholar] [CrossRef]
- Inglis, D.J.; Licari, J.; Georgiou, K.R.; Wittwer, N.L.; Hamilton, R.W.; Beaumont, D.M.; Scherer, M.A.; Lavranos, T.C. Characterization of BNC101 a human specific monoclonal antibody targeting the GPCR LGR5: First-in-human evidence of target engagement. Cancer Res. 2018, 78, 3910. [Google Scholar] [CrossRef]
- Edenfield, W.J.; Richards, D.A.; Vukelja, S.J.; Weiss, G.J.; Sirard, C.A.; Landau, S.B.; Ramanathan, R.K. A phase 1 study evaluating the safety and efficacy of DKN-01, an investigational monoclonal antibody (Mab) in patients (pts) with advanced non-small cell lung cancer. J. Clin. Oncol. 2014, 32, 8068. [Google Scholar] [CrossRef]
- Eads, J.R.; Goyal, L.; Stein, S.; El-Khoueiry, A.B.; Manji, G.A.; Abrams, T.A.; Landau, S.B.; Sirard, C.A. Phase I study of DKN-01, an anti-DKK1 antibody, in combination with gemcitabine (G) and cisplatin (C) in patients (pts) with advanced biliary cancer. J. Clin. Oncol. 2016, 34, e15603. [Google Scholar] [CrossRef]
- Goyal, L.; Sirard, C.; Schrag, M.; Kagey, M.H.; Eads, J.R.; Stein, S.; El-Khoueiry, A.B.; Manji, G.A.; Abrams, T.A.; Khorana, A.A.; et al. Phase I and Biomarker Study of the Wnt Pathway Modulator DKN-01 in Combination with Gemcitabine/Cisplatin in Advanced Biliary Tract Cancer. Clin. Cancer Res. 2020, 26, 6158–6167. [Google Scholar] [CrossRef]
- Arend, R.C.; Castro, C.M.; Matulonis, U.A.; Hamilton, E.; Gunderson, C.C.; Lybarger, K.S.S.; Goodman, H.M.; Duska, L.R.; Mahdi, H.; ElNaggar, A.C. Dkn-01 treated patients with recurrent epithelial endometrial (EEC) or ovarian (EOC) cancers which harbor Wnt activating mutations have longer progression-free survival and improved clinical benefit. Gynecol. Oncol. 2020, 159, 5–6. [Google Scholar] [CrossRef]
- Klempner, S.J.; Bendell, J.C.; Villaflor, V.M.; Tenner, L.L.; Stein, S.; Naik, G.S.; Sirard, C.A.; Kagey, M.; Chaney, M.F.; Strickler, J.H. DKN-01 in combination with pembrolizumab in patients with advanced gastroesophageal adenocarcinoma (GEA): Tumoral DKK1 expression as a predictor of response and survival. J. Clin. Oncol. 2020, 38, 357. [Google Scholar] [CrossRef]
- Lee, H.J.; Wang, N.X.; Shi, D.L.; Zheng, J.J. Sulindac inhibits canonical Wnt signaling by blocking the PDZ domain of the protein Dishevelled. Angew. Chem. Int. Ed. Engl. 2009, 48, 6448–6452. [Google Scholar] [CrossRef] [PubMed]
- Thorne, C.A.; Hanson, A.J.; Schneider, J.; Tahinci, E.; Orton, D.; Cselenyi, C.S.; Jernigan, K.K.; Meyers, K.C.; Hang, B.I.; Waterson, A.G.; et al. Small-molecule inhibition of Wnt signaling through activation of casein kinase 1α. Nat. Chem. Biol. 2010, 6, 829–836. [Google Scholar] [CrossRef]
- McGonigle, S.; Chen, Z.; Wu, J.; Chang, P.; Kolber-Simonds, D.; Ackermann, K.; Twine, N.C.; Shie, J.L.; Miu, J.T.; Huang, K.C.; et al. E7449: A dual inhibitor of PARP1/2 and tankyrase1/2 inhibits growth of DNA repair deficient tumors and antagonizes Wnt signaling. Oncotarget 2015, 6, 41307–41323. [Google Scholar] [CrossRef]
- Plummer, R.; Dua, D.; Cresti, N.; Drew, Y.; Stephens, P.; Foegh, M.; Knudsen, S.; Sachdev, P.; Mistry, B.M.; Dixit, V.; et al. First-in-human study of the PARP/tankyrase inhibitor E7449 in patients with advanced solid tumours and evaluation of a novel drug-response predictor. Br. J. Cancer 2020, 123, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Cranmer, L.D.; Abdul Razak, A.R.; Ratan, R.; Choy, E.; George, S.; Liebner, D.A.; Stenehjem, D.D.; Gounder, M.M. Results of a phase I dose escalation and expansion study of tegavivint (BC2059), a first-in-class TBL1 inhibitor for patients with progressive, unresectable desmoid tumor. J. Clin. Oncol. 2022, 40, 11523. [Google Scholar] [CrossRef]
- Fiskus, W.; Sharma, S.; Saha, S.; Shah, B.; Devaraj, S.G.; Sun, B.; Horrigan, S.; Leveque, C.; Zu, Y.; Iyer, S.; et al. Pre-clinical efficacy of combined therapy with novel β-catenin antagonist BC2059 and histone deacetylase inhibitor against AML cells. Leukemia 2015, 29, 1267–1278. [Google Scholar] [CrossRef]
- Savvidou, I.; Khong, T.; Cuddihy, A.; McLean, C.; Horrigan, S.; Spencer, A. β-catenin inhibitor BC2059 is efficacious as monotherapy or in combination with proteasome inhibitor bortezomib in multiple myeloma. Mol. Cancer Ther. 2017, 16, 1765–1778. [Google Scholar] [CrossRef]
- Osawa, Y.; Oboki, K.; Imamura, J.; Kojika, E.; Hayashi, Y.; Hishima, T.; Saibara, T.; Shibasaki, F.; Kohara, M.; Kimura, K. Inhibition of Cyclic Adenosine Monophosphate (cAMP)-response Element-binding Protein (CREB)-binding Protein (CBP)/β-Catenin Reduces Liver Fibrosis in Mice. EBioMedicine 2015, 2, 1751–1758. [Google Scholar] [CrossRef]
- McWilliams, R.R.; Ko, A.H.; Chiorean, E.G.; Kwak, E.L.; Lenz, H.-J.; Nadler, P.I.; Wood, D.L.; Fujimori, M.; Morita, K.; Inada, T. A phase Ib dose-escalation study of PRI-724, a CBP/beta-catenin modulator, plus gemcitabine (GEM) in patients with advanced pancreatic adenocarcinoma (APC) as second-line therapy after FOLFIRINOX or FOLFOX. J. Clin. Oncol. 2015, 33, e15270. [Google Scholar] [CrossRef]
- Scott, A.; Call, J.; Chandana, S.; Borazanci, E.; Falchook, G.; Bordoni, R.; Richey, S.; Starodub, A.; Chung, V.; Lakhani, N. 451O Preliminary evidence of clinical activity from phase I and Ib trials of the CLK/DYRK inhibitor cirtuvivint (CIRT) in subjects with advanced solid tumors. Ann. Oncol. 2022, 33, S742–S743. [Google Scholar] [CrossRef]
- Molenaar, R.J.; Coelen, R.J.S.; Khurshed, M.; Roos, E.; Caan, M.W.A.; van Linde, M.E.; Kouwenhoven, M.; Bramer, J.A.M.; Bovée, J.V.M.G.; Mathôt, R.A.; et al. Study protocol of a phase IB/II clinical trial of metformin and chloroquine in patients with. BMJ Open 2017, 7, e014961. [Google Scholar] [CrossRef]
- Arora, S.P.; Tenner, L.; Sarantopoulos, J.; Morris, J.; Liu, Q.; Mendez, J.A.; Curiel, T.; Michalek, J.; Mahalingam, D. Modulation of autophagy: A Phase II study of vorinostat plus hydroxychloroquine versus regorafenib in chemotherapy-refractory metastatic colorectal cancer (mCRC). Br. J. Cancer 2022, 127, 1153–1161. [Google Scholar] [CrossRef] [PubMed]
- Boone, B.A.; Bahary, N.; Zureikat, A.H.; Moser, A.J.; Normolle, D.P.; Wu, W.C.; Singhi, A.D.; Bao, P.; Bartlett, D.L.; Liotta, L.A.; et al. Safety and Biologic Response of Pre-operative Autophagy Inhibition in Combination with Gemcitabine in Patients with Pancreatic Adenocarcinoma. Ann. Surg. Oncol. 2015, 22, 4402–4410. [Google Scholar] [CrossRef]
- Loaiza-Bonilla, A.; O’Hara, M.H.; Redlinger, M.; Damjanov, N.; Teitelbaum, U.R.; Vasilevskaya, I.; Rosen, M.A.; Heitjan, D.F.; Amaravadi, R.K.; O’Dwyer, P.J. Phase II trial of autophagy inhibition using hydroxychloroquine (HCQ) with FOLFOX/bevacizumab in the first-line treatment of advanced colorectal cancer. J. Clin. Oncol. 2015, 33, 3614. [Google Scholar] [CrossRef]
- Mahalingam, D.; Mita, M.; Sarantopoulos, J.; Wood, L.; Amaravadi, R.K.; Davis, L.E.; Mita, A.C.; Curiel, T.J.; Espitia, C.M.; Nawrocki, S.T.; et al. Combined autophagy and HDAC inhibition: A phase I safety, tolerability, pharmacokinetic, and pharmacodynamic analysis of hydroxychloroquine in combination with the HDAC inhibitor vorinostat in patients with advanced solid tumors. Autophagy 2014, 10, 1403–1414. [Google Scholar] [CrossRef] [PubMed]
- Wolpin, B.M.; Rubinson, D.A.; Wang, X.; Chan, J.A.; Cleary, J.M.; Enzinger, P.C.; Fuchs, C.S.; McCleary, N.J.; Meyerhardt, J.A.; Ng, K.; et al. Phase II and pharmacodynamic study of autophagy inhibition using hydroxychloroquine in patients with metastatic pancreatic adenocarcinoma. Oncologist 2014, 19, 637–638. [Google Scholar] [CrossRef]
- Chen, B.; Dodge, M.E.; Tang, W.; Lu, J.; Ma, Z.; Fan, C.W.; Wei, S.; Hao, W.; Kilgore, J.; Williams, N.S.; et al. Small molecule-mediated disruption of Wnt-dependent signaling in tissue regeneration and cancer. Nat. Chem. Biol. 2009, 5, 100–107. [Google Scholar] [CrossRef]
- Wang, X.; Moon, J.; Dodge, M.E.; Pan, X.; Zhang, L.; Hanson, J.M.; Tuladhar, R.; Ma, Z.; Shi, H.; Williams, N.S.; et al. The development of highly potent inhibitors for porcupine. J. Med. Chem. 2013, 56, 2700–2704. [Google Scholar] [CrossRef]
- Proffitt, K.D.; Madan, B.; Ke, Z.; Pendharkar, V.; Ding, L.; Lee, M.A.; Hannoush, R.N.; Virshup, D.M. Pharmacological inhibition of the Wnt acyltransferase PORCN prevents growth of WNT-driven mammary cancer. Cancer Res. 2013, 73, 502–507. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.; Liu, J.; Han, D.; Zhang, G.; Gao, W.; Hsieh, M.H.; Ng, N.; Kasibhatla, S.; Tompkins, C.; Li, J.; et al. Discovery of Pyridinyl Acetamide Derivatives as Potent, Selective, and Orally Bioavailable Porcupine Inhibitors. ACS Med. Chem. Lett. 2016, 7, 676–680. [Google Scholar] [CrossRef] [PubMed]
- Mook, R.A.; Wang, J.; Ren, X.R.; Chen, M.; Spasojevic, I.; Barak, L.S.; Lyerly, H.K.; Chen, W. Structure-activity studies of Wnt/β-catenin inhibition in the Niclosamide chemotype: Identification of derivatives with improved drug exposure. Bioorg. Med. Chem. 2015, 23, 5829–5838. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Mook, R.A.; Ren, X.R.; Zhang, Q.; Jing, G.; Lu, M.; Spasojevic, I.; Lyerly, H.K.; Hsu, D.; Chen, W. Identification of DK419, a potent inhibitor of Wnt/β-catenin signaling and colorectal cancer growth. Bioorg. Med. Chem. 2018, 26, 5435–5442. [Google Scholar] [CrossRef] [PubMed]
- Nile, A.H.; de Sousa, E.; Melo, F.; Mukund, S.; Piskol, R.; Hansen, S.; Zhou, L.; Zhang, Y.; Fu, Y.; Gogol, E.B.; et al. A selective peptide inhibitor of Frizzled 7 receptors disrupts intestinal stem cells. Nat. Chem. Biol. 2018, 14, 582–590. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Lu, W.; Ananthan, S.; Suto, M.J.; Li, Y. Discovery of novel frizzled-7 inhibitors by targeting the receptor’s transmembrane domain. Oncotarget 2017, 8, 91459–91470. [Google Scholar] [CrossRef] [PubMed]
- Nambotin, S.B.; Lefrancois, L.; Sainsily, X.; Berthillon, P.; Kim, M.; Wands, J.R.; Chevallier, M.; Jalinot, P.; Scoazec, J.Y.; Trepo, C.; et al. Pharmacological inhibition of Frizzled-7 displays anti-tumor properties in hepatocellular carcinoma. J. Hepatol. 2011, 54, 288–299. [Google Scholar] [CrossRef]
- Lee, H.J.; Bao, J.; Miller, A.; Zhang, C.; Wu, J.; Baday, Y.C.; Guibao, C.; Li, L.; Wu, D.; Zheng, J.J. Structure-based Discovery of Novel Small Molecule Wnt Signaling Inhibitors by Targeting the Cysteine-rich Domain of Frizzled. J. Biol. Chem. 2015, 290, 30596–30606. [Google Scholar] [CrossRef]
- Choi, J.; Ma, S.; Kim, H.Y.; Yun, J.H.; Heo, J.N.; Lee, W.; Choi, K.Y.; No, K.T. Identification of small-molecule compounds targeting the dishevelled PDZ domain by virtual screening and binding studies. Bioorg. Med. Chem. 2016, 24, 3259–3266. [Google Scholar] [CrossRef]
- Grandy, D.; Shan, J.; Zhang, X.; Rao, S.; Akunuru, S.; Li, H.; Zhang, Y.; Alpatov, I.; Zhang, X.A.; Lang, R.A.; et al. Discovery and characterization of a small molecule inhibitor of the PDZ domain of dishevelled. J. Biol. Chem. 2009, 284, 16256–16263. [Google Scholar] [CrossRef]
- Shan, J.; Zhang, X.; Bao, J.; Cassell, R.; Zheng, J.J. Synthesis of potent dishevelled PDZ domain inhibitors guided by virtual screening and NMR studies. Chem. Biol. Drug Des. 2012, 79, 376–383. [Google Scholar] [CrossRef]
- Fujii, N.; You, L.; Xu, Z.; Uematsu, K.; Shan, J.; He, B.; Mikami, I.; Edmondson, L.R.; Neale, G.; Zheng, J.; et al. An antagonist of dishevelled protein-protein interaction suppresses beta-catenin-dependent tumor cell growth. Cancer Res. 2007, 67, 573–579. [Google Scholar] [CrossRef]
- Kim, H.Y.; Choi, S.; Yoon, J.H.; Lim, H.J.; Lee, H.; Choi, J.; Ro, E.J.; Heo, J.N.; Lee, W.; No, K.T.; et al. Small molecule inhibitors of the Dishevelled-CXXC5 interaction are new drug candidates for bone anabolic osteoporosis therapy. EMBO Mol. Med. 2016, 8, 375–387. [Google Scholar] [CrossRef]
- Shan, J.; Zheng, J.J. Optimizing Dvl PDZ domain inhibitor by exploring chemical space. J. Comput. Aided Mol. Des. 2009, 23, 37–47. [Google Scholar] [CrossRef]
- Zhang, Y.; Appleton, B.A.; Wiesmann, C.; Lau, T.; Costa, M.; Hannoush, R.N.; Sidhu, S.S. Inhibition of Wnt signaling by Dishevelled PDZ peptides. Nat. Chem. Biol. 2009, 5, 217–219. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Orton, D.; Neitzel, L.R.; Astudillo, L.; Shen, C.; Long, J.; Chen, X.; Kirkbride, K.C.; Doundoulakis, T.; Guerra, M.L.; et al. Differential abundance of CK1α provides selectivity for pharmacological CK1α activators to target WNT-dependent tumors. Sci. Signal. 2017, 10, 485. [Google Scholar] [CrossRef] [PubMed]
- Ewan, K.; Pajak, B.; Stubbs, M.; Todd, H.; Barbeau, O.; Quevedo, C.; Botfield, H.; Young, R.; Ruddle, R.; Samuel, L.; et al. A useful approach to identify novel small-molecule inhibitors of Wnt-dependent transcription. Cancer Res. 2010, 70, 5963–5973. [Google Scholar] [CrossRef] [PubMed]
- Ríos, J.A.; Godoy, J.A.; Inestrosa, N.C. Wnt3a ligand facilitates autophagy in hippocampal neurons by modulating a novel GSK-3β-AMPK axis. Cell Commun. Signal. 2018, 16, 15. [Google Scholar] [CrossRef]
- Scarborough, H.A.; Helfrich, B.A.; Casás-Selves, M.; Schuller, A.G.; Grosskurth, S.E.; Kim, J.; Tan, A.C.; Chan, D.C.; Zhang, Z.; Zaberezhnyy, V.; et al. AZ1366: An Inhibitor of Tankyrase and the Canonical Wnt Pathway that Limits the Persistence of Non-Small Cell Lung Cancer Cells Following EGFR Inhibition. Clin. Cancer Res. 2017, 23, 1531–1541. [Google Scholar] [CrossRef]
- Lau, T.; Chan, E.; Callow, M.; Waaler, J.; Boggs, J.; Blake, R.A.; Magnuson, S.; Sambrone, A.; Schutten, M.; Firestein, R.; et al. A novel tankyrase small-molecule inhibitor suppresses APC mutation-driven colorectal tumor growth. Cancer Res. 2013, 73, 3132–3144. [Google Scholar] [CrossRef]
- Okada-Iwasaki, R.; Takahashi, Y.; Watanabe, Y.; Ishida, H.; Saito, J.; Nakai, R.; Asai, A. The Discovery and Characterization of K-756, a Novel Wnt/β-Catenin Pathway Inhibitor Targeting Tankyrase. Mol. Cancer Ther. 2016, 15, 1525–1534. [Google Scholar] [CrossRef]
- Shultz, M.D.; Cheung, A.K.; Kirby, C.A.; Firestone, B.; Fan, J.; Chen, C.H.; Chen, Z.; Chin, D.N.; Dipietro, L.; Fazal, A.; et al. Identification of NVP-TNKS656: The use of structure-efficiency relationships to generate a highly potent, selective, and orally active tankyrase inhibitor. J. Med. Chem. 2013, 56, 6495–6511. [Google Scholar] [CrossRef]
- Narwal, M.; Koivunen, J.; Haikarainen, T.; Obaji, E.; Legala, O.E.; Venkannagari, H.; Joensuu, P.; Pihlajaniemi, T.; Lehtiö, L. Discovery of tankyrase inhibiting flavones with increased potency and isoenzyme selectivity. J. Med. Chem. 2013, 56, 7880–7889. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, A.; Yashiroda, Y.; Muramatsu, Y.; Yoshida, H.; Chikada, T.; Tsumura, T.; Okue, M.; Shirai, F.; Fukami, T.; Yoshida, M.; et al. RK-287107, a potent and specific tankyrase inhibitor, blocks colorectal cancer cell growth in a preclinical model. Cancer Sci. 2018, 109, 4003–4014. [Google Scholar] [CrossRef] [PubMed]
- James, R.G.; Davidson, K.C.; Bosch, K.A.; Biechele, T.L.; Robin, N.C.; Taylor, R.J.; Major, M.B.; Camp, N.D.; Fowler, K.; Martins, T.J.; et al. WIKI4, a novel inhibitor of tankyrase and Wnt/ß-catenin signaling. PLoS ONE 2012, 7, e50457. [Google Scholar] [CrossRef] [PubMed]
- Menon, M.; Elliott, R.; Bowers, L.; Balan, N.; Rafiq, R.; Costa-Cabral, S.; Munkonge, F.; Trinidade, I.; Porter, R.; Campbell, A.D.; et al. A novel tankyrase inhibitor, MSC2504877, enhances the effects of clinical CDK4/6 inhibitors. Sci. Rep. 2019, 9, 201. [Google Scholar] [CrossRef]
- Voronkov, A.; Holsworth, D.D.; Waaler, J.; Wilson, S.R.; Ekblad, B.; Perdreau-Dahl, H.; Dinh, H.; Drewes, G.; Hopf, C.; Morth, J.P.; et al. Structural basis and SAR for G007-LK, a lead stage 1,2,4-triazole based specific tankyrase 1/2 inhibitor. J. Med. Chem. 2013, 56, 3012–3023. [Google Scholar] [CrossRef] [PubMed]
- Waaler, J.; Machon, O.; Tumova, L.; Dinh, H.; Korinek, V.; Wilson, S.R.; Paulsen, J.E.; Pedersen, N.M.; Eide, T.J.; Machonova, O.; et al. A novel tankyrase inhibitor decreases canonical Wnt signaling in colon carcinoma cells and reduces tumor growth in conditional APC mutant mice. Cancer Res. 2012, 72, 2822–2832. [Google Scholar] [CrossRef]
- Waaler, J.; Machon, O.; von Kries, J.P.; Wilson, S.R.; Lundenes, E.; Wedlich, D.; Gradl, D.; Paulsen, J.E.; Machonova, O.; Dembinski, J.L.; et al. Novel synthetic antagonists of canonical Wnt signaling inhibit colorectal cancer cell growth. Cancer Res. 2011, 71, 197–205. [Google Scholar] [CrossRef]
- Shetti, D.; Zhang, B.; Fan, C.; Mo, C.; Lee, B.H.; Wei, K. Low Dose of Paclitaxel Combined with XAV939 Attenuates Metastasis, Angiogenesis and Growth in Breast Cancer by Suppressing Wnt Signaling. Cells 2019, 8, 892. [Google Scholar] [CrossRef] [PubMed]
- Cha, P.H.; Cho, Y.H.; Lee, S.K.; Lee, J.; Jeong, W.J.; Moon, B.S.; Yun, J.H.; Yang, J.S.; Choi, S.; Yoon, J.; et al. Small-molecule binding of the axin RGS domain promotes β-catenin and Ras degradation. Nat. Chem. Biol. 2016, 12, 593–600. [Google Scholar] [CrossRef]
- Trosset, J.Y.; Dalvit, C.; Knapp, S.; Fasolini, M.; Veronesi, M.; Mantegani, S.; Gianellini, L.M.; Catana, C.; Sundström, M.; Stouten, P.F.; et al. Inhibition of protein-protein interactions: The discovery of druglike beta-catenin inhibitors by combining virtual and biophysical screening. Proteins 2006, 64, 60–67. [Google Scholar] [CrossRef]
- Yao, H.; Ashihara, E.; Strovel, J.W.; Nakagawa, Y.; Kuroda, J.; Nagao, R.; Tanaka, R.; Yokota, A.; Takeuchi, M.; Hayashi, Y.; et al. AV-65, a novel Wnt/β-catenin signal inhibitor, successfully suppresses progression of multiple myeloma in a mouse model. Blood Cancer J. 2011, 1, e43. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Ao, A.; Zhou, L.; Murphy, C.K.; Frist, A.Y.; Keel, J.J.; Thorne, C.A.; Kim, K.; Lee, E.; Hong, C.C. Selective small molecule targeting β-catenin function discovered by in vivo chemical genetic screen. Cell Rep. 2013, 4, 898–904. [Google Scholar] [CrossRef] [PubMed]
- Bordonaro, M.; Lazarova, D.L. Determination of the Role of CBP- and p300-Mediated Wnt Signaling on Colonic Cells. JMIR Res. Protoc. 2016, 5, e66. [Google Scholar] [CrossRef]
- Takada, K.; Zhu, D.; Bird, G.H.; Sukhdeo, K.; Zhao, J.J.; Mani, M.; Lemieux, M.; Carrasco, D.E.; Ryan, J.; Horst, D.; et al. Targeted disruption of the BCL9/β-catenin complex inhibits oncogenic Wnt signaling. Sci. Transl. Med. 2012, 4, 148ra117. [Google Scholar] [CrossRef]
- De la Roche, M.; Rutherford, T.J.; Gupta, D.; Veprintsev, D.B.; Saxty, B.; Freund, S.M.; Bienz, M. An intrinsically labile α-helix abutting the BCL9-binding site of β-catenin is required for its inhibition by carnosic acid. Nat. Commun. 2012, 3, 680. [Google Scholar] [PubMed]
- Zhang, H.; Liu, C.; Zhu, D.; Zhang, Q.; Li, J. Medicinal Chemistry Strategies for the Development of Inhibitors Disrupting β-Catenin’s Interactions with Its Nuclear Partners. J. Med. Chem. 2023, 66, 1–31. [Google Scholar] [CrossRef] [PubMed]
- Lavergne, E.; Hendaoui, I.; Coulouarn, C.; Ribault, C.; Leseur, J.; Eliat, P.A.; Mebarki, S.; Corlu, A.; Clément, B.; Musso, O. Blocking Wnt signaling by SFRP-like molecules inhibits in vivo cell proliferation and tumor growth in cells carrying active β-catenin. Oncogene 2011, 30, 423–433. [Google Scholar] [CrossRef]
- Bovolenta, P.; Esteve, P.; Ruiz, J.M.; Cisneros, E.; Lopez-Rios, J. Beyond Wnt inhibition: New functions of secreted Frizzled-related proteins in development and disease. J. Cell Sci. 2008, 121, 737–746. [Google Scholar] [CrossRef]
- Glinka, A.; Wu, W.; Delius, H.; Monaghan, A.P.; Blumenstock, C.; Niehrs, C. Dickkopf-1 is a member of a new family of secreted proteins and functions in head induction. Nature 1998, 391, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Killick, R.; Ribe, E.M.; Al-Shawi, R.; Malik, B.; Hooper, C.; Fernandes, C.; Dobson, R.; Nolan, P.M.; Lourdusamy, A.; Furney, S.; et al. Clusterin regulates β-amyloid toxicity via Dickkopf-1-driven induction of the wnt-PCP-JNK pathway. Mol. Psychiatry 2014, 19, 88–98. [Google Scholar] [CrossRef]
- Krause, U.; Ryan, D.M.; Clough, B.H.; Gregory, C.A. An unexpected role for a Wnt-inhibitor: Dickkopf-1 triggers a novel cancer survival mechanism through modulation of aldehyde-dehydrogenase-1 activity. Cell Death Dis. 2014, 5, e1093. [Google Scholar] [CrossRef] [PubMed]
- Endo, Y.; Beauchamp, E.; Woods, D.; Taylor, W.G.; Toretsky, J.A.; Uren, A.; Rubin, J.S. Wnt-3a and Dickkopf-1 stimulate neurite outgrowth in Ewing tumor cells via a Frizzled3- and c-Jun N-terminal kinase-dependent mechanism. Mol. Cell. Biol. 2008, 28, 2368–2379. [Google Scholar] [CrossRef]
- Caneparo, L.; Huang, Y.L.; Staudt, N.; Tada, M.; Ahrendt, R.; Kazanskaya, O.; Niehrs, C.; Houart, C. Dickkopf-1 regulates gastrulation movements by coordinated modulation of Wnt/beta catenin and Wnt/PCP activities, through interaction with the Dally-like homolog Knypek. Genes Dev. 2007, 21, 465–480. [Google Scholar] [CrossRef]
- Zhu, G.; Song, J.; Chen, W.; Yuan, D.; Wang, W.; Chen, X.; Liu, H.; Su, H.; Zhu, J. Expression and Role of Dickkopf-1 (Dkk1) in Tumors: From the Cells to the Patients. Cancer Manag. Res. 2021, 13, 659–675. [Google Scholar] [CrossRef] [PubMed]
- Hirata, H.; Hinoda, Y.; Nakajima, K.; Kawamoto, K.; Kikuno, N.; Ueno, K.; Yamamura, S.; Zaman, M.S.; Khatri, G.; Chen, Y.; et al. Wnt antagonist DKK1 acts as a tumor suppressor gene that induces apoptosis and inhibits proliferation in human renal cell carcinoma. Int. J. Cancer 2011, 128, 1793–1803. [Google Scholar] [CrossRef]
- Mikheev, A.M.; Mikheeva, S.A.; Maxwell, J.P.; Rivo, J.V.; Rostomily, R.; Swisshelm, K.; Zarbl, H. Dickkopf-1 mediated tumor suppression in human breast carcinoma cells. Breast Cancer Res. Treat. 2008, 112, 263–273. [Google Scholar] [CrossRef]
- Cowling, V.H.; D’Cruz, C.M.; Chodosh, L.A.; Cole, M.D. c-Myc transforms human mammary epithelial cells through repression of the Wnt inhibitors DKK1 and SFRP1. Mol. Cell. Biol. 2007, 27, 5135–5146. [Google Scholar] [CrossRef]
- Kuphal, S.; Lodermeyer, S.; Bataille, F.; Schuierer, M.; Hoang, B.H.; Bosserhoff, A.K. Expression of Dickkopf genes is strongly reduced in malignant melanoma. Oncogene 2006, 25, 5027–5036. [Google Scholar] [CrossRef]
- Barker, N.; Clevers, H. Mining the Wnt pathway for cancer therapeutics. Nat. Rev. Drug Discov. 2006, 5, 997–1014. [Google Scholar] [PubMed]
- Kimelman, D.; Xu, W. beta-catenin destruction complex: Insights and questions from a structural perspective. Oncogene 2006, 25, 7482–7491. [Google Scholar] [CrossRef]
- Turner, J.A.; Johnson, P.E. Pyrvinium pamoate in the treatment of pinworm infection (enterobiasis) in the home. J. Pediatr. 1962, 60, 243–251. [Google Scholar] [CrossRef]
- Li, B.; Flaveny, C.A.; Giambelli, C.; Fei, D.L.; Han, L.; Hang, B.I.; Bai, F.; Pei, X.H.; Nose, V.; Burlingame, O.; et al. Repurposing the FDA-approved pinworm drug pyrvinium as a novel chemotherapeutic agent for intestinal polyposis. PLoS ONE 2014, 9, e101969. [Google Scholar] [CrossRef]
- Marchetti, M.; Resnick, L.; Gamliel, E.; Kesaraju, S.; Weissbach, H.; Binninger, D. Sulindac enhances the killing of cancer cells exposed to oxidative stress. PLoS ONE 2009, 4, e5804. [Google Scholar] [CrossRef]
- Riffell, J.L.; Lord, C.J.; Ashworth, A. Tankyrase-targeted therapeutics: Expanding opportunities in the PARP family. Nat. Rev. Drug Discov. 2012, 11, 923–936. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.M.; Mishina, Y.M.; Liu, S.; Cheung, A.; Stegmeier, F.; Michaud, G.A.; Charlat, O.; Wiellette, E.; Zhang, Y.; Wiessner, S.; et al. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 2009, 461, 614–620. [Google Scholar] [CrossRef]
- Cruciat, C.M.; Ohkawara, B.; Acebron, S.P.; Karaulanov, E.; Reinhard, C.; Ingelfinger, D.; Boutros, M.; Niehrs, C. Requirement of prorenin receptor and vacuolar H+-ATPase-mediated acidification for Wnt signaling. Science 2010, 327, 459–463. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.S.; Jun, S.; Kim, M.J.; Lee, S.H.; Suh, H.N.; Lien, E.M.; Jung, H.Y.; Lee, S.; Zhang, J.; Yang, J.I.; et al. TMEM9 promotes intestinal tumorigenesis through vacuolar-ATPase-activated Wnt/β-catenin signalling. Nat. Cell Biol. 2018, 20, 1421–1433. [Google Scholar] [CrossRef] [PubMed]
- Pálmer, H.G.; González-Sancho, J.M.; Espada, J.; Berciano, M.T.; Puig, I.; Baulida, J.; Quintanilla, M.; Cano, A.; de Herreros, A.G.; Lafarga, M.; et al. Vitamin D3 promotes the differentiation of colon carcinoma cells by the induction of E-cadherin and the inhibition of beta-catenin signaling. J. Cell Biol. 2001, 154, 369–387. [Google Scholar] [CrossRef]
- Kahn, M. Can we safely target the WNT pathway? Nat. Rev. Drug Discov. 2014, 13, 513–532. [Google Scholar] [CrossRef] [PubMed]
- Raman, S.; Beilschmidt, M.; To, M.; Lin, K.; Lui, F.; Jmeian, Y.; Ng, M.; Fernandez, M.; Fu, Y.; Mascall, K.; et al. Structure-guided design fine-tunes pharmacokinetics, tolerability, and antitumor profile of multispecific frizzled antibodies. Proc. Natl. Acad. Sci. USA 2019, 116, 6812–6817. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.; Li, X.; Zhao, L.; Wang, Y.; Wang, X.; Wu, Y.; Zhou, X.; Fu, W.; Liu, L.; Hu, H.G.; et al. A PROTAC peptide induces durable β-catenin degradation and suppresses Wnt-dependent intestinal cancer. Cell Discov. 2020, 6, 35. [Google Scholar] [CrossRef]
- Békés, M.; Langley, D.R.; Crews, C.M. PROTAC targeted protein degraders: The past is prologue. Nat. Rev. Drug Discov. 2022, 21, 181–200. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Gao, H.; Yang, Y.; He, M.; Wu, Y.; Song, Y.; Tong, Y.; Rao, Y. PROTACs: Great opportunities for academia and industry. Signal Transduct. Target Ther. 2019, 4, 64. [Google Scholar] [CrossRef] [PubMed]
- Simonetta, K.R.; Taygerly, J.; Boyle, K.; Basham, S.E.; Padovani, C.; Lou, Y.; Cummins, T.J.; Yung, S.L.; von Soly, S.K.; Kayser, F.; et al. Prospective discovery of small molecule enhancers of an E3 ligase-substrate interaction. Nat. Commun. 2019, 10, 1402. [Google Scholar] [CrossRef]
- Do, M.; Wu, C.C.N.; Sonavane, P.R.; Juarez, E.F.; Adams, S.R.; Ross, J.; Rodriguez, Y.; Baena, A.; Patel, C.; Mesirov, J.P.; et al. A FZD7-specific Antibody-Drug Conjugate Induces Ovarian Tumor Regression in Preclinical Models. Mol. Cancer Ther. 2022, 21, 113–124. [Google Scholar] [CrossRef]
- Katoh, M. Antibody-drug conjugate targeting protein tyrosine kinase 7, a receptor tyrosine kinase-like molecule involved in WNT and vascular endothelial growth factor signaling: Effects on cancer stem cells, tumor microenvironment and whole-body homeostasis. Ann. Transl. Med. 2017, 5, 462. [Google Scholar] [CrossRef]
- Junttila, M.R.; Mao, W.; Wang, X.; Wang, B.E.; Pham, T.; Flygare, J.; Yu, S.F.; Yee, S.; Goldenberg, D.; Fields, C.; et al. Targeting LGR5+ cells with an antibody-drug conjugate for the treatment of colon cancer. Sci. Transl. Med. 2015, 7, 314ra186. [Google Scholar] [CrossRef]
- Li, M.; Ding, X.; Zhang, Y.; Li, X.; Zhou, H.; Yang, L.; Li, Y.; Yang, P.; Zhang, X.; Hu, J.; et al. Antisense oligonucleotides targeting lncRNA AC104041.1 induces antitumor activity through Wnt2B/β-catenin pathway in head and neck squamous cell carcinomas. Cell Death Dis. 2020, 11, 672. [Google Scholar] [CrossRef] [PubMed]
- Popov, V.B.; Jornayvaz, F.R.; Akgul, E.O.; Kanda, S.; Jurczak, M.J.; Zhang, D.; Abudukadier, A.; Majumdar, S.K.; Guigni, B.; Petersen, K.F.; et al. Second-generation antisense oligonucleotides against β-catenin protect mice against diet-induced hepatic steatosis and hepatic and peripheral insulin resistance. FASEB J. 2016, 30, 1207–1217. [Google Scholar] [CrossRef]
- Khan, S.; Zhang, X.; Lv, D.; Zhang, Q.; He, Y.; Zhang, P.; Liu, X.; Thummuri, D.; Yuan, Y.; Wiegand, J.S.; et al. A selective BCL-X. Nat. Med. 2019, 25, 1938–1947. [Google Scholar] [CrossRef]
- Ji, C.H.; Kim, H.Y.; Lee, M.J.; Heo, A.J.; Park, D.Y.; Lim, S.; Shin, S.; Ganipisetti, S.; Yang, W.S.; Jung, C.A.; et al. The AUTOTAC chemical biology platform for targeted protein degradation via the autophagy-lysosome system. Nat. Commun. 2022, 13, 904. [Google Scholar] [CrossRef] [PubMed]
- Banik, S.M.; Pedram, K.; Wisnovsky, S.; Ahn, G.; Riley, N.M.; Bertozzi, C.R. Lysosome-targeting chimaeras for degradation of extracellular proteins. Nature 2020, 584, 291–297. [Google Scholar] [CrossRef]
- Marei, H.; Tsai, W.K.; Kee, Y.S.; Ruiz, K.; He, J.; Cox, C.; Sun, T.; Penikalapati, S.; Dwivedi, P.; Choi, M.; et al. Antibody targeting of E3 ubiquitin ligases for receptor degradation. Nature 2022, 610, 182–189. [Google Scholar] [CrossRef]
- Trail, P.A. Antibody drug conjugates as cancer therapeutics. Antibodies 2013, 2, 113–129. [Google Scholar]
- Barker, N.; Ridgway, R.A.; van Es, J.H.; van de Wetering, M.; Begthel, H.; van den Born, M.; Danenberg, E.; Clarke, A.R.; Sansom, O.J.; Clevers, H. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 2009, 457, 608–611. [Google Scholar] [CrossRef] [PubMed]
- de Lau, W.; Barker, N.; Low, T.Y.; Koo, B.K.; Li, V.S.; Teunissen, H.; Kujala, P.; Haegebarth, A.; Peters, P.J.; van de Wetering, M.; et al. Lgr5 homologues associate with Wnt receptors and mediate R-spondin signalling. Nature 2011, 476, 293–297. [Google Scholar] [CrossRef]
- Schepers, A.G.; Snippert, H.J.; Stange, D.E.; van den Born, M.; van Es, J.H.; van de Wetering, M.; Clevers, H. Lineage tracing reveals Lgr5+ stem cell activity in mouse intestinal adenomas. Science 2012, 337, 730–735. [Google Scholar] [CrossRef] [PubMed]
- Gong, X.; Azhdarinia, A.; Ghosh, S.C.; Xiong, W.; An, Z.; Liu, Q.; Carmon, K.S. LGR5-Targeted Antibody-Drug Conjugate Eradicates Gastrointestinal Tumors and Prevents Recurrence. Mol. Cancer Ther. 2016, 15, 1580–1590. [Google Scholar] [CrossRef]
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in oligonucleotide drug delivery. Nat. Rev. Drug Discov. 2020, 19, 673–694. [Google Scholar] [CrossRef]
- Garzon, R.; Calin, G.A.; Croce, C.M. MicroRNAs in cancer. Annu. Rev. Med. 2009, 60, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Bonci, D.; Coppola, V.; Musumeci, M.; Addario, A.; Giuffrida, R.; Memeo, L.; D’Urso, L.; Pagliuca, A.; Biffoni, M.; Labbaye, C.; et al. The miR-15a-miR-16-1 cluster controls prostate cancer by targeting multiple oncogenic activities. Nat. Med. 2008, 14, 1271–1277. [Google Scholar] [CrossRef]
- van Zandwijk, N.; Pavlakis, N.; Kao, S.C.; Linton, A.; Boyer, M.J.; Clarke, S.; Huynh, Y.; Chrzanowska, A.; Fulham, M.J.; Bailey, D.L.; et al. Safety and activity of microRNA-loaded minicells in patients with recurrent malignant pleural mesothelioma: A first-in-man, phase 1, open-label, dose-escalation study. Lancet Oncol. 2017, 18, 1386–1396. [Google Scholar] [CrossRef]
- Rennoll, S.; Yochum, G. Regulation of MYC gene expression by aberrant Wnt/β-catenin signaling in colorectal cancer. World J. Biol. Chem. 2015, 6, 290–300. [Google Scholar] [CrossRef]
- Tolcher, A.W.; Papadopoulos, K.P.; Patnaik, A.; Rasco, D.W.; Martinez, D.; Wood, D.L.; Fielman, B.; Sharma, M.; Janisch, L.A.; Brown, B.D. Safety and activity of DCR-MYC, a first-in-class Dicer-substrate small interfering RNA (DsiRNA) targeting MYC, in a phase I study in patients with advanced solid tumors. J. Clin. Oncol. 2015, 33, 11006. [Google Scholar] [CrossRef]
- Lam, J.K.; Chow, M.Y.; Zhang, Y.; Leung, S.W. siRNA versus miRNA as therapeutics for gene silencing. Mol. Ther.-Nucleic Acids 2015, 4, e252. [Google Scholar] [CrossRef] [PubMed]
- Dhuri, K.; Bechtold, C.; Quijano, E.; Pham, H.; Gupta, A.; Vikram, A.; Bahal, R. Antisense oligonucleotides: An emerging area in drug discovery and development. J. Clin. Med. 2020, 9, 2004. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; Krieg, A.M. FDA Approves Eteplirsen for Duchenne Muscular Dystrophy: The Next Chapter in the Eteplirsen Saga. Nucleic Acid Ther. 2017, 27, 1–3. [Google Scholar] [CrossRef]
- Heo, Y.A. Golodirsen: First Approval. Drugs 2020, 80, 329–333. [Google Scholar] [CrossRef]
- Aartsma-Rus, A. FDA Approval of Nusinersen for Spinal Muscular Atrophy Makes 2016 the Year of Splice Modulating Oligonucleotides. Nucleic Acid Ther. 2017, 27, 67–69. [Google Scholar] [CrossRef]
- Xiong, H.; Veedu, R.N.; Diermeier, S.D. Recent Advances in Oligonucleotide Therapeutics in Oncology. Int. J. Mol. Sci. 2021, 22, 3295. [Google Scholar] [CrossRef]
- Corey, D.R. Nusinersen, an antisense oligonucleotide drug for spinal muscular atrophy. Nat. Neurosci. 2017, 20, 497–499. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.; Kurzrock, R.; Kim, Y.; Woessner, R.; Younes, A.; Nemunaitis, J.; Fowler, N.; Zhou, T.; Schmidt, J.; Jo, M.; et al. AZD9150, a next-generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer. Sci. Transl. Med. 2015, 7, 314ra185. [Google Scholar] [CrossRef] [PubMed]
- De Velasco, M.A.; Kura, Y.; Sakai, K.; Hatanaka, Y.; Davies, B.R.; Campbell, H.; Klein, S.; Kim, Y.; MacLeod, A.R.; Sugimoto, K.; et al. Targeting castration-resistant prostate cancer with androgen receptor antisense oligonucleotide therapy. JCI Insight 2019, 4, e122688. [Google Scholar] [CrossRef]
- Cohen, P.; Cross, D.; Jänne, P.A. Kinase drug discovery 20 years after imatinib. Nat. Rev. Drug Discov. 2021, 20, 551–569. [Google Scholar] [CrossRef] [PubMed]
- Sliwkowski, M.X.; Lofgren, J.A.; Lewis, G.D.; Hotaling, T.E.; Fendly, B.M.; Fox, J.A. Nonclinical studies addressing the mechanism of action of trastuzumab (Herceptin). Semin. Oncol. 1999, 26, 60–70. [Google Scholar]
- Carter, P.; Presta, L.; Gorman, C.M.; Ridgway, J.; Henner, D.; Wong, W.; Rowland, A.M.; Kotts, C.; Carver, M.E.; Shepard, H.M. Humanization of an anti-p185HER2 antibody for human cancer therapy. Proc. Natl. Acad. Sci. USA 1992, 89, 4285–4289. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Components Name | Target | Cancer | Clinical Phase | Inhibition of Canonical or Non-Canonical Wnt Signaling | Refs. |
|---|---|---|---|---|---|
| OMP-131R10 | RSPO |
| Phase 1 (NCT02482441) | Canonical | [52] |
| Foxy-5 | WNT5A mimic |
| Phase 2 (NCT03883802) Phase 1 (NCT02020291) Phase 1 (NCT02655952) | Non-canonical | [53] |
| LGK974 | PORCN |
| Phase 1/2 (NCT02278133) Phase 2 (NCT02649530) Phase 1 (NCT01351103) | Canonical/Non-canonical | [3,43,54] |
| RXC004 | PORCN |
| Phase 2 (NCT04907539) Phase 1 (NCT03447470) Phase 2 (NCT04907851) | Canonical/Non-canonical | [55,56,57] |
| ETC-159 | PORCN |
| Phase 1 (NCT02521844) | Canonical/Non-canonical | [58,59] |
| OMP-54F28 | FZD8 |
| Phase 1 (NCT02069145) Phase 1 (NCT02092363) Phase 1 (NCT02050178) Phase 1 (NCT01608867) | Canonical | [60,61,62] |
| Niclosamide | FZD1 |
| Phase 1 (NCT02687009) Phase 1 (NCT03123978) Phase 1 (NCT02532114) Phase 2 (NCT02807805) Phase 1 (NCT05188170) FDA-approved antihelminth | Canonical | [63] |
| OMP-18R5 | FZD1/2/5/7/8 |
| Phase 1 (NCT01345201) Phase 1 (NCT01957007) Phase 1 (NCT02005315) Phase 1 (NCT01973309) | Canonical | [64,65] |
| OTSA-101 | FZD10 |
| Phase 1 (NCT01469975) | Canonical | [66] |
| BNC101 | LGR5 |
| Phase 1 (NCT02726334) | Canonical/Non-canonical | [67] |
| DKN-01 | DKK1 |
| Phase 1 (NCT01457417) Phase 1 (NCT01711671) Phase 1 (NCT02013154) Phase 1 (NCT02375880) Phase 2 (NCT03395080) | Canonical/Non-canonical | [68,69,70,71,72] |
| Sulindac | DVL |
| Phase 1 (NCT00245024) Phase 2 (NCT04542135) Phase 2 (NCT01856322) Phase 2 (NCT00062023) Phase 2 (NCT00368927) FDA-approved nonsteroidal anti-inflammatory drug | Canonical | [73] |
| Pyrvinium | CK1 |
| Phase 1 (NCT05055323) FDA-approved antihelminth | Canonical | [74] |
| E7449 | TNK1/2 |
| Phase 2 (NCT03878849) Phase 1 (NCT01618136) | Canonical | [75,76] |
| BC2059 | β-catenin |
| Phase 1 (NCT03459469) Phase 1 (NCT04780568) Phase 1 (NCT04874480) Phase 1/2 (NCT04851119) | Canonical | [77,78,79] |
| PRI-724 | β-catenin/CBP |
| Phase 1 (NCT01302405) Phase 1 (NCT01764477) Phase 1/2 (NCT01606579) | Canonical | [80,81] |
| SM08502 | CLK |
| Phase 1 (NCT05084859) Phase 1 (NCT03355066) | Canonical | [82] |
| Chloroquine | v-ATPase inhibitor |
| Phase 1 (NCT01777477) Phase 1 (NCT02071537) Phase 1/2 (NCT01023477) Phase 1/2 (NCT02496741) Phase 3 (NCT00224978) | Canonical | [83] |
| Hydroxy chloroquine | v-ATPase inhibitor |
| Phase 2 (NCT01006369), etc. (total of 30 trials completed) | Canonical | [84,85,86,87,88] |
| Target | Pre-Clinical Agents | Refs. |
|---|---|---|
| PORCN | IWP1, IWP2, IWP3, IWP4, IWP12, IWP L6, C59, GNF-6231, GNF-1331 | [31,89,90,91,92] |
| FZD1 | DK-520, DK-419 | [93,94] |
| FZD5 | IgG-2919 | [51] |
| FZD7 | Fz7-21, RHPD-P1, SRI37892 | [95,96,97] |
| FZD8 | 1094-0205, 2124-0331, 3235-0367, NSC36784, NSC654259, IgG-2919 | [98] |
| WNT/FZD/LRP complex | Salinomycin | [32] |
| DVL | BMD4702, 3289-8625, J01-017a, FJ9, KY-02061, KY-02327, NSC668036, Peptide Pen-N3 | [99,100,101,102,103,104,105] |
| CK1 | SSTC3, CCT031374 | [106,107] |
| GSK3β mimic | TCS 183 | [108] |
| TNKS | XAV939, AZ1366, G007-LK, MSC2504877, G244-LM, IWR-1, JW74, JW55, K-756, NVP-TNKS656, MN-64, RK-287107, WIKI4 | [109,110,111,112,113,114,115,116,117,118,119,120] |
| β-catenin | KY1220, KYA1797K, MSAB, | [78,121] |
| β-catenin/TCF | PKF115-584, CGP049090, AV-65, PNU-74654 | [122,123] |
| β-catenin/EP300 | Windorphen, IQ-1 | [124,125] |
| β-catenin/BCL9 | PNPB-29, ZW4864, SAH-BCL9, Carnosic acid | [126,127,128] |
| Technologies | Name | Target | Refs. |
|---|---|---|---|
| PROTACs | xStAx-VHL | β-catenin | [153] |
| Molecular glue | NRX-252114 | β-catenin/ β-TrCP | [156] |
| ADC | Septuximab vedotin | FZD7 | [157] |
| PF-06647020 | PTK7 | [158] | |
| LGR5-mc-vc-PAB-MMAE | LGR5 | [159] | |
| LGR5-NMS818 | LGR5 | ||
| ASO | LNA-modified ASO | AC104041.1 (lncRNA) | [160] |
| β-catenin targeting ASO | β-catenin | [161] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, W.-J.; Kim, M.J. A New Wave of Targeting ‘Undruggable’ Wnt Signaling for Cancer Therapy: Challenges and Opportunities. Cells 2023, 12, 1110. https://doi.org/10.3390/cells12081110
Park W-J, Kim MJ. A New Wave of Targeting ‘Undruggable’ Wnt Signaling for Cancer Therapy: Challenges and Opportunities. Cells. 2023; 12(8):1110. https://doi.org/10.3390/cells12081110
Chicago/Turabian StylePark, Woo-Jung, and Moon Jong Kim. 2023. "A New Wave of Targeting ‘Undruggable’ Wnt Signaling for Cancer Therapy: Challenges and Opportunities" Cells 12, no. 8: 1110. https://doi.org/10.3390/cells12081110
APA StylePark, W.-J., & Kim, M. J. (2023). A New Wave of Targeting ‘Undruggable’ Wnt Signaling for Cancer Therapy: Challenges and Opportunities. Cells, 12(8), 1110. https://doi.org/10.3390/cells12081110