
A Novel Hydrogel-Based 3D In Vitro Tumor Panel of 30 PDX Models Incorporates Tumor, Stromal and Immune Cell Compartments of the TME for the Screening of Oncology and Immuno-Therapies

 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Therapeutic Compounds
2.2. Cultivation of PDX Lines and Fibroblasts In Vitro
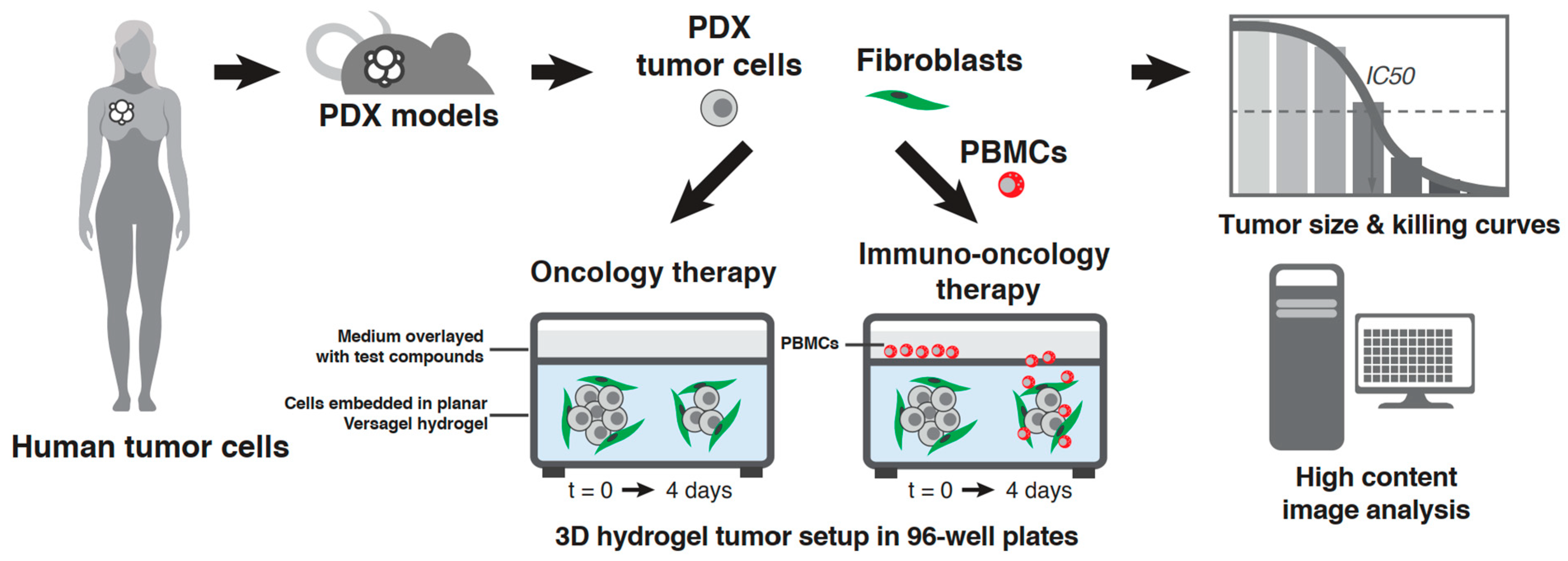
2.3. PDX In Vitro Model Setup in the VersaGel Hydrogel Platform
2.4. Drug Dosing and PBMC Addition
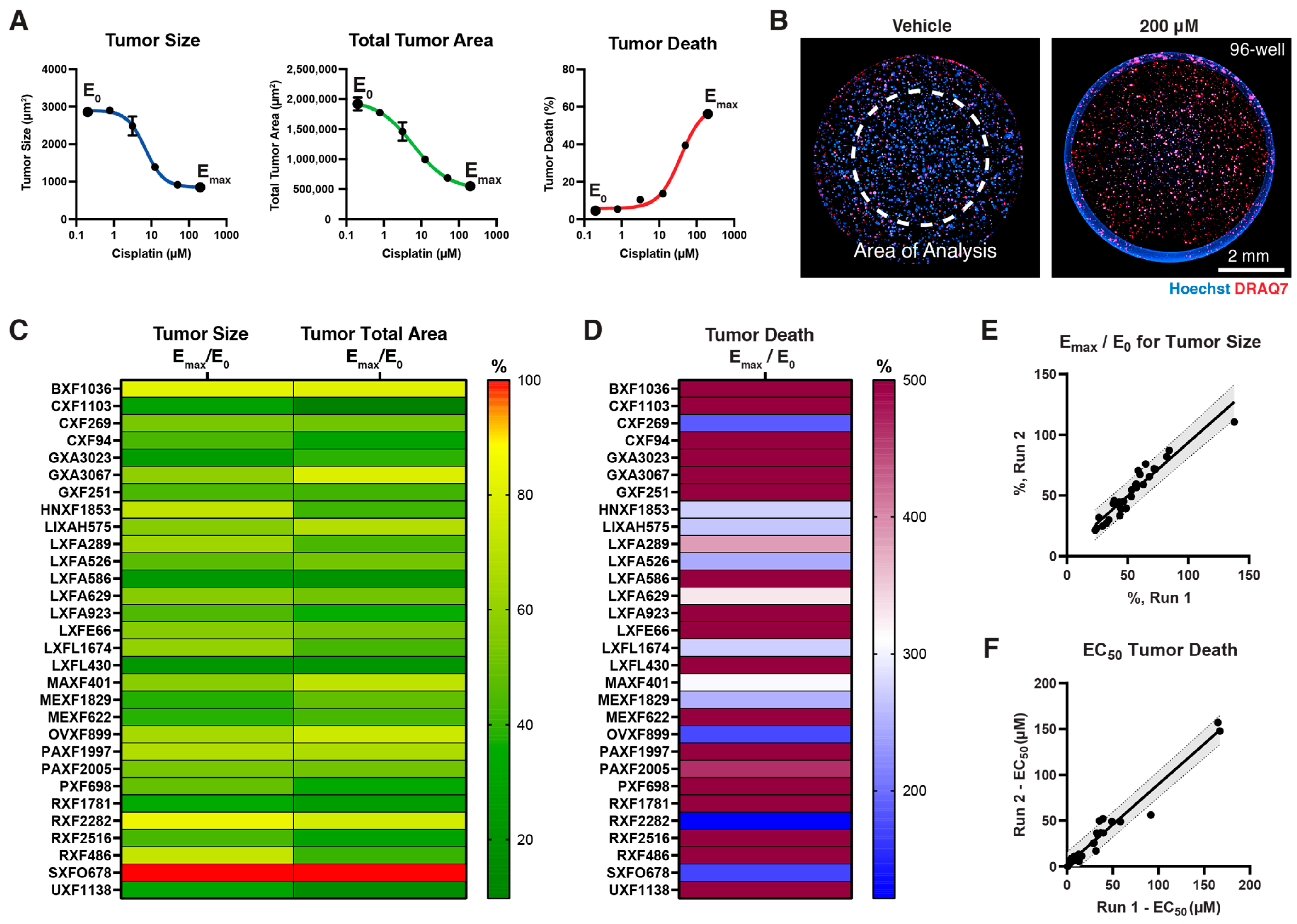
2.5. High-Content Image Analysis
2.6. Calculation of Drug Response Curves
2.7. Multiplex Cytokine Analysis
2.8. Immunofluorescence Staining
3. Results
3.1. Setup in Cypre 3D Platform and Selection of 30 PDX Panel
3.2. Model Development
3.3. Screening Small Molecule Chemotherapeutics on Cypre 30 PDX Panel
3.4. Screening Immuno-Oncology Antibodies on Cypre 30 PDX Panel
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Turley, S.J.; Cremasco, V.; Astarita, J.L. Immunological Hallmarks of Stromal Cells in the Tumour Microenvironment. Nat. Rev. Immunol. 2015, 15, 669–682. [Google Scholar] [CrossRef]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The Tumor Microenvironment at a Glance. J. Cell Sci. 2012, 125, 5591–5596. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.M.; Simon, M.C. The Tumor Microenvironment. Curr. Biol. 2020, 30, R921–R925. [Google Scholar] [CrossRef] [PubMed]
- Kather, J.N.; Suarez-Carmona, M.; Charoentong, P.; Weis, C.-A.; Hirsch, D.; Bankhead, P.; Horning, M.; Ferber, D.; Kel, I.; Herpel, E.; et al. Topography of Cancer-Associated Immune Cells in Human Solid Tumors. eLife 2018, 7, e36967. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. The Tumor Microenvironment and Its Role in Promoting Tumor Growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E.; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFβ Attenuates Tumour Response to PD-L1 Blockade by Contributing to Exclusion of T Cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Nicolas-Boluda, A.; Vaquero, J.; Vimeux, L.; Guilbert, T.; Barrin, S.; Kantari-Mimoun, C.; Ponzo, M.; Renault, G.; Deptula, P.; Pogoda, K.; et al. Tumor Stiffening Reversion through Collagen Crosslinking Inhibition Improves T Cell Migration and Anti-PD-1 Treatment. eLife 2021, 10, e58688. [Google Scholar] [CrossRef]
- Thurber, G.M.; Schmidt, M.M.; Wittrup, K.D. Antibody Tumor Penetration: Transport Opposed by Systemic and Antigen-Mediated Clearance. Adv. Drug Deliv. Rev. 2008, 60, 1421–1434. [Google Scholar] [CrossRef]
- Cancer Cell Line Encyclopedia Consortium; Genomics of Drug Sensitivity in Cancer Consortium. Pharmacogenomic Agreement between Two Cancer Cell Line Data Sets. Nature 2015, 528, 84–87. [Google Scholar] [CrossRef]
- Danku, A.E.; Dulf, E.-H.; Braicu, C.; Jurj, A.; Berindan-Neagoe, I. Organ-on-a-Chip: A Survey of Technical Results and Problems. Front. Bioeng. Biotechnol. 2022, 10, 840674. [Google Scholar] [CrossRef]
- Ingber, D.E. Human Organs-on-Chips for Disease Modelling, Drug Development and Personalized Medicine. Nat. Rev. Genet. 2022, 23, 467–491. [Google Scholar] [CrossRef]
- Yakavets, I.; Francois, A.; Benoit, A.; Merlin, J.-L.; Bezdetnaya, L.; Vogin, G. Advanced Co-Culture 3D Breast Cancer Model for Investigation of Fibrosis Induced by External Stimuli: Optimization Study. Sci. Rep. 2020, 10, 21273. [Google Scholar] [CrossRef] [PubMed]
- Edwards, S.J.; Carannante, V.; Kuhnigk, K.; Ring, H.; Tararuk, T.; Hallböök, F.; Blom, H.; Önfelt, B.; Brismar, H. High-Resolution Imaging of Tumor Spheroids and Organoids Enabled by Expansion Microscopy. Front. Mol. Biosci. 2020, 7, 208. [Google Scholar] [CrossRef] [PubMed]
- Vukicevic, S.; Kleinman, H.K.; Luyten, F.P.; Roberts, A.B.; Roche, N.S.; Reddi, A.H. Identification of Multiple Active Growth Factors in Basement Membrane Matrigel Suggests Caution in Interpretation of Cellular Activity Related to Extracellular Matrix Components. Exp. Cell Res. 1992, 202, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.K.; Umino, T.; Liu, X.D.; Wang, H.J.; Romberger, D.J.; Spurzem, J.R.; Rennard, S.I. Contraction of Fibroblast-Containing Collagen Gels: Initial Collagen Concentration Regulates the Degree of Contraction and Cell Survival. Vitr. Cell. Dev. Biol. Anim. 2001, 37, 10–16. [Google Scholar] [CrossRef]
- Roth, T.; Burger, A.M.; Dengler, W.; Willmann, H.; Fiebig, H.H. Human Tumor Cell Lines Demonstrating the Characteristics of Patient Tumors as Useful Models for Anticancer Drug Screening. Contrib. Oncol. 1999, 54, 145–156. [Google Scholar] [CrossRef]
- Hribar, K.C.; Wheeler, C.J.; Bazarov, A.; Varshneya, K.; Yamada, R.; Buckley, P.; Patil, C.G. A Simple Three-Dimensional Hydrogel Platform Enables Ex Vivo Cell Culture of Patient and PDX Tumors for Assaying Their Response to Clinically Relevant Therapies. Mol. Cancer Ther. 2019, 18, 718–725. [Google Scholar] [CrossRef]
- N-Parameter Logistic Regression, R Package Nplr Version 0.1-7; GitHub, Inc.: San Francisco, CA, USA, 2016.
- Rajkumar, P.; Mathew, B.S.; Das, S.; Isaiah, R.; John, S.; Prabha, R.; Fleming, D.H. Cisplatin Concentrations in Long and Short Duration Infusion: Implications for the Optimal Time of Radiation Delivery. J. Clin. Diagn. Res. 2016, 10, XC01. [Google Scholar] [CrossRef]
- Richter, M.; Piwocka, O.; Musielak, M.; Piotrowski, I.; Suchorska, W.M.; Trzeciak, T. From Donor to the Lab: A Fascinating Journey of Primary Cell Lines. Front. Cell Dev. Biol. 2021, 9, 711381. [Google Scholar] [CrossRef]
- Bhowmick, N.A.; Neilson, E.G.; Moses, H.L. Stromal Fibroblasts in Cancer Initiation and Progression. Nature 2004, 432, 332–337. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A Framework for Advancing Our Understanding of Cancer-Associated Fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [PubMed]
- Olumi, A.; Grossfeld, G.; Hayward, S.; Carroll, P.; Cunha, G.; Hein, P.; Tlsty, T. Carcinoma-Associated Fibroblasts Stimulate Tumor Progression of Initiated Human Epithelium. Breast Cancer Res. 2000, 2, S.19. [Google Scholar] [CrossRef]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal Fibroblasts Present in Invasive Human Breast Carcinomas Promote Tumor Growth and Angiogenesis through Elevated SDF-1/CXCL12 Secretion. Cell 2005, 121, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Higa, A.; Hiyama, G.; Tamura, H.; Hoshi, H.; Dobashi, Y.; Katahira, K.; Ishihara, H.; Takagi, K.; Goda, K.; et al. Construction of in Vitro Patient-Derived Tumor Models to Evaluate Anticancer Agents and Cancer Immunotherapy. Oncol. Lett. 2021, 21, 406. [Google Scholar] [CrossRef] [PubMed]
- Rudisch, A.; Dewhurst, M.R.; Horga, L.G.; Kramer, N.; Harrer, N.; Dong, M.; Van Der Kuip, H.; Wernitznig, A.; Bernthaler, A.; Dolznig, H.; et al. High EMT Signature Score of Invasive Non-Small Cell Lung Cancer (NSCLC) Cells Correlates with NFκB Driven Colony-Stimulating Factor 2 (CSF2/GM-CSF) Secretion by Neighboring Stromal Fibroblasts. PLoS ONE 2015, 10, e0124283. [Google Scholar] [CrossRef] [PubMed]
- Morillon, Y.M.; Sabzevari, A.; Schlom, J.; Greiner, J.W. The Development of Next-Generation PBMC Humanized Mice for Preclinical Investigation of Cancer Immunotherapeutic Agents. Anticancer Res. 2020, 40, 5329. [Google Scholar] [CrossRef] [PubMed]
- Pearson, T.; Greiner, D.L.; Shultz, L.D. Creation of “Humanized” Mice to Study Human Immunity. In Current Protocols in Immunology; Coligan, J.E., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2008. [Google Scholar] [CrossRef]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. 2016, 39, 98. [Google Scholar] [CrossRef]
- Simon, S.; Labarriere, N. PD-1 Expression on Tumor-Specific T Cells: Friend or Foe for Immunotherapy? Oncoimmunology 2018, 7, e1364828. [Google Scholar] [CrossRef]
- Dijkstra, K.K.; Cattaneo, C.M.; Weeber, F.; Chalabi, M.; van de Haar, J.; Fanchi, L.F.; Slagter, M.; van der Velden, D.L.; Kaing, S.; Kelderman, S.; et al. Generation of Tumor-Reactive T Cells by Co-Culture of Peripheral Blood Lymphocytes and Tumor Organoids. Cell 2018, 174, 1586–1598.e12. [Google Scholar] [CrossRef]
- Jenkins, R.W.; Aref, A.R.; Lizotte, P.H.; Ivanova, E.; Stinson, S.; Zhou, C.W.; Bowden, M.; Deng, J.; Liu, H.; Miao, D.; et al. Ex Vivo Profiling of PD-1 Blockade Using Organotypic Tumor Spheroids. Cancer Discov. 2018, 8, 196–215. [Google Scholar] [CrossRef]
- Aref, A.R.; Campisi, M.; Ivanova, E.; Portell, A.; Larios, D.; Piel, B.P.; Mathur, N.; Zhou, C.; Coakley, R.V.; Bartels, A.; et al. 3D Microfluidic Ex Vivo Culture of Organotypic Tumor Spheroids to Model Immune Checkpoint Blockade. Lab Chip 2018, 18, 3129. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer Type | Tumor Designation | Model # | Histology | Primary/Metastasis/Recurrent |
|---|---|---|---|---|
| Bladder * | BXF | 1036 | urothelial carcinoma | Metastasis |
| Colon | CXF | 94 | adenocarcinoma | Primary |
| Colon | CXF | 269 | adenocarcinoma | Primary |
| Colon | CXF | 1103 | adenocarcinoma | Metastasis |
| Gastric | GXA | 3023 | adenocarcinoma Lauren intestinal type | Primary |
| Gastric | GXA | 3067 | adenocarcinoma | Primary |
| Gastric * | GXF | 251 | adenocarcinoma | Primary |
| Head neck | HNXF | 1853 | squamous cell carcinoma | Recurrent |
| Liver * | LIXFC | 2050 | cholangiocarcinoma | Primary |
| Liver | LIXFC | 575 | cholangiocarcinoma | Primary |
| NSCLC (adeno) | LXFA | 289 | adenocarcinoma | Metastasis |
| NSCLC (adeno) | LXFA | 526 | adenocarcinoma | Metastasis |
| NSCLC (adeno) | LXFA | 586 | adenocarcinoma | Primary |
| NSCLC (adeno) | LXFA | 629 | adenocarcinoma | Primary |
| NSCLC (adeno) | LXFA | 923 | adenocarcinoma | Metastasis |
| NSCLC (epidermoid) | LXFE | 66 | squamous cell carcinoma | Primary |
| NSCLC (large cell) | LXFL | 1674 | large cell carcinoma | Primary |
| NSCLC (large cell) | LXFL | 430 | large cell carcinoma | Primary |
| Breast (Triple Negative) | MAXFTN | 401 | adenocarcinoma | Metastasis |
| Melanoma | MEXF | 1829 | melanoma | Metastasis |
| Melanoma | MEXF | 622 | melanoma | Metastasis |
| Ovarian | OVXF | 899 | serous adenocarcinoma | Primary |
| Pancreatic | PAXF | 1997 | adenocarcinoma | Primary |
| Pancreatic | PAXF | 2005 | adenocarcinoma | Primary |
| Mesothelioma | PXF | 698 | pleural mesothelioma | Primary |
| Renal | RXF | 1781 | clear cell carcinoma | Recurrent |
| Renal | RXF | 2282 | clear cell carcinoma | not known |
| Renal | RXF | 2516 | clear cell carcinoma | not known |
| Renal | RXF | 486 | hypernephroma | Primary |
| Osteosarcoma | SXFO | 678 | osteosarcoma | Primary |
| Soft tissue sarcoma | SXFS | 1301 | rhabdomyosarcoma | Metastasis |
| Uterus | UXF | 1138 | sarcoma | Metastasis |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xue, B.; Schüler, J.; Harrod, C.M.; Lashuk, K.; Bomya, Z.; Hribar, K.C. A Novel Hydrogel-Based 3D In Vitro Tumor Panel of 30 PDX Models Incorporates Tumor, Stromal and Immune Cell Compartments of the TME for the Screening of Oncology and Immuno-Therapies. Cells 2023, 12, 1145. https://doi.org/10.3390/cells12081145
Xue B, Schüler J, Harrod CM, Lashuk K, Bomya Z, Hribar KC. A Novel Hydrogel-Based 3D In Vitro Tumor Panel of 30 PDX Models Incorporates Tumor, Stromal and Immune Cell Compartments of the TME for the Screening of Oncology and Immuno-Therapies. Cells. 2023; 12(8):1145. https://doi.org/10.3390/cells12081145
Chicago/Turabian StyleXue, Bin, Julia Schüler, Christopher M. Harrod, Kanstantsin Lashuk, Zoji Bomya, and Kolin C. Hribar. 2023. "A Novel Hydrogel-Based 3D In Vitro Tumor Panel of 30 PDX Models Incorporates Tumor, Stromal and Immune Cell Compartments of the TME for the Screening of Oncology and Immuno-Therapies" Cells 12, no. 8: 1145. https://doi.org/10.3390/cells12081145
APA StyleXue, B., Schüler, J., Harrod, C. M., Lashuk, K., Bomya, Z., & Hribar, K. C. (2023). A Novel Hydrogel-Based 3D In Vitro Tumor Panel of 30 PDX Models Incorporates Tumor, Stromal and Immune Cell Compartments of the TME for the Screening of Oncology and Immuno-Therapies. Cells, 12(8), 1145. https://doi.org/10.3390/cells12081145