
The Functions and Phenotypes of Microglia in Alzheimer’s Disease

Abstract
:
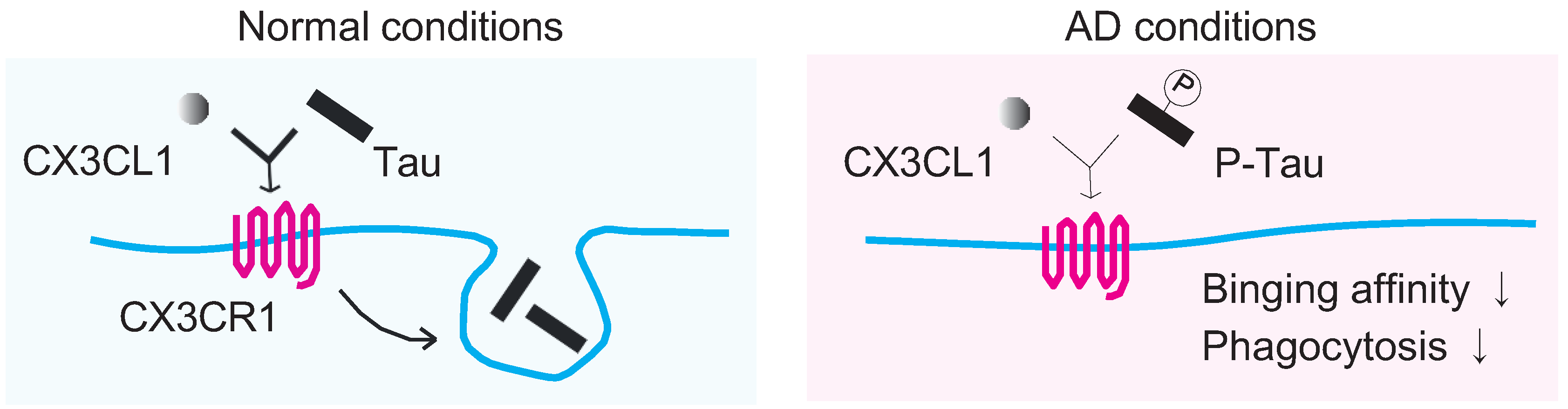
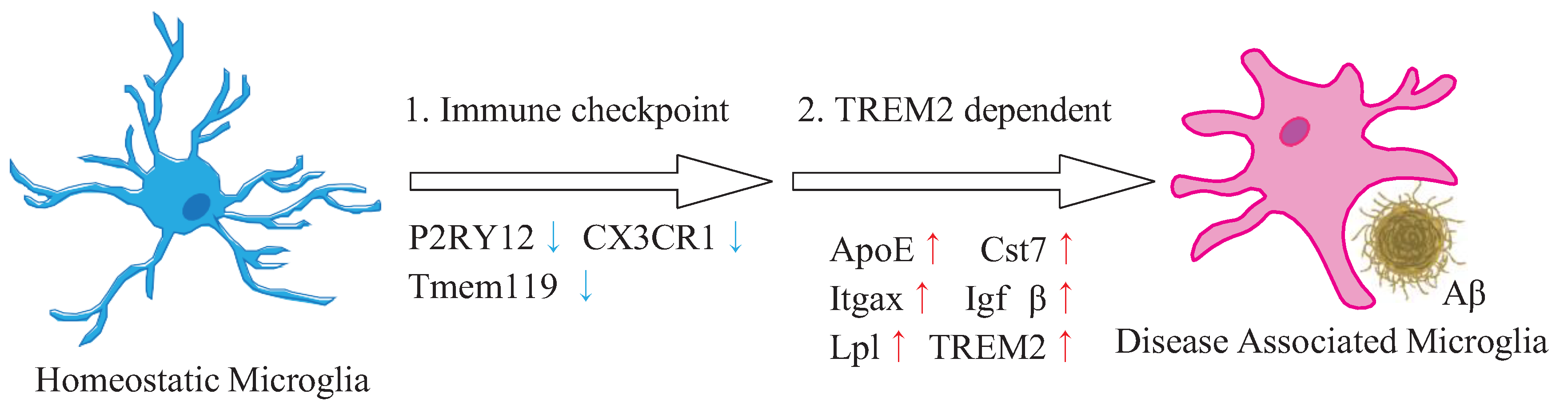
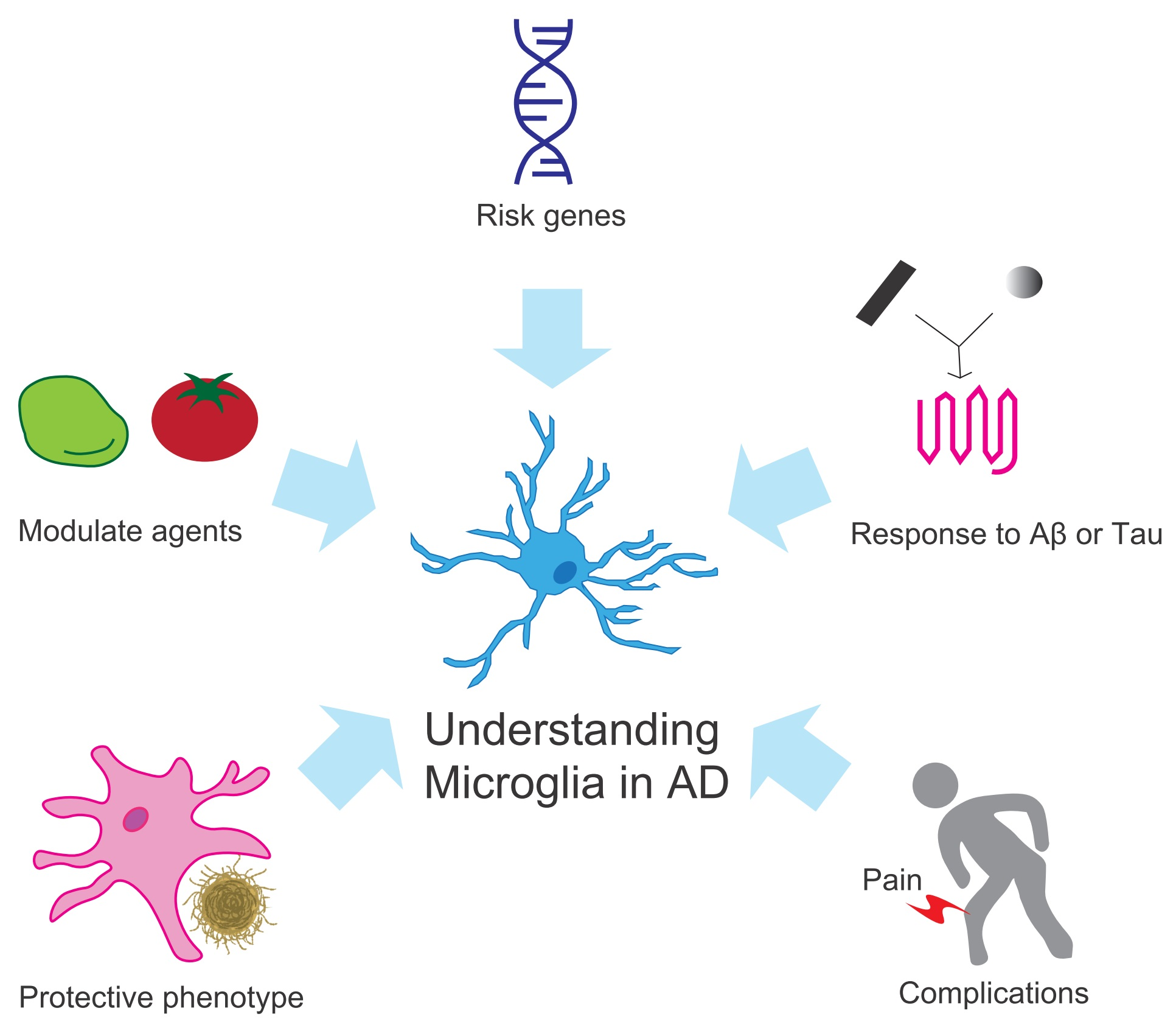{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Microglia
3. Expression of AD Risk Gene in Microglia
4. Microglial Responses to Aβ Plaques
5. Microglia Responses to Tau Tangles
6. Pain Is a Risk Factor for AD: Possible Involvement of Microglial CNS Inflammation
7. Microglia Phenotypes, Especially Protective Microglia, as a New Therapeutic Target
8. Pharmacological or Diet Modulation on Microglial Responses in AD
9. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef]
- Naghavi, M.; on behalf of the Global Burden of Disease Self-Harm Collaborators. Global, regional, and national burden of suicide mortality 1990 to 2016: Systematic analysis for the Global Burden of Disease Study 2016. BMJ 2019, 364, l94. [Google Scholar] [CrossRef] [PubMed]
- Wimo, A.; Guerchet, M.; Ali, G.C.; Wu, Y.T.; Prina, A.M.; Winblad, B.; Jönsson, L.; Liu, Z.; Prince, M. The worldwide costs of dementia 2015 and comparisons with 2010. Alzheimers Dement. 2017, 13, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Elmaleh, D.R.; Farlow, M.R.; Conti, P.S.; Tompkins, R.G.; Kundakovic, L.; Tanzi, R.E. Developing Effective Alzheimer’s Disease Therapies: Clinical Experience and Future Directions. J. Alzheimers Dis. 2019, 71, 715–732. [Google Scholar] [CrossRef] [PubMed]
- Webers, A.; Heneka, M.T.; Gleeson, P.A. The role of innate immune responses and neuroinflammation in amyloid accumulation and progression of Alzheimer’s disease. Immunol. Cell Biol. 2020, 98, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Colonna, M. Microglia in Alzheimer’s disease at single-cell level. Are there common patterns in humans and mice? J. Exp. Med. 2021, 218, e20202717. [Google Scholar] [CrossRef]
- Lewcock, J.W.; Schlepckow, K.; Di Paolo, G.; Tahirovic, S.; Monroe, K.M.; Haass, C. Emerging Microglia Biology Defines Novel Therapeutic Approaches for Alzheimer’s Disease. Neuron 2020, 108, 801–821. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, S.E.; Medeiros, M.; Porfirio, J.; Tavares, W.; Pessôa, L.; Grinberg, L.; Leite, R.E.P.; Ferretti-Rebustini, R.E.L.; Suemoto, C.K.; Filho, W.J.; et al. Similar Microglial Cell Densities across Brain Structures and Mammalian Species: Implications for Brain Tissue Function. J. Neurosci. 2020, 40, 4622–4643. [Google Scholar] [CrossRef]
- Barca, C.; Foray, C.; Hermann, S.; Herrlinger, U.; Remory, I.; Laoui, D.; Schäfers, M.; Grauer, O.M.; Zinnhardt, B.; Jacobs, A.H. The Colony Stimulating Factor-1 Receptor (CSF-1R)-Mediated Regulation of Microglia/Macrophages as a Target for Neurological Disorders (Glioma, Stroke). Front. Immunol. 2021, 12, 787307. [Google Scholar] [CrossRef]
- Lelios, I.; Cansever, D.; Utz, S.G.; Mildenberger, W.; Stifter, S.A.; Greter, M. Emerging roles of IL-34 in health and disease. J. Exp. Med. 2020, 217, e20190290. [Google Scholar] [CrossRef]
- Pawelec, P.; Ziemka-Nalecz, M.; Sypecka, J.; Zalewska, T. The Impact of the CX3CL1/CX3CR1 Axis in Neurological Disorders. Cells 2020, 9, 2277. [Google Scholar] [CrossRef] [PubMed]
- Vainchtein, I.D.; Chin, G.; Cho, F.S.; Kelley, K.W.; Miller, J.G.; Chien, E.C.; Liddelow, S.A.; Nguyen, P.T.; Nakao-Inoue, H.; Dorman, L.C.; et al. Astrocyte-derived interleukin-33 promotes microglial synapse engulfment and neural circuit development. Science 2018, 359, 1269–1273. [Google Scholar] [CrossRef] [PubMed]
- De Waard, D.M.; Bugiani, M. Astrocyte-Oligodendrocyte-Microglia Crosstalk in Astrocytopathies. Front. Cell. Neurosci. 2020, 14, 608073. [Google Scholar] [CrossRef] [PubMed]
- Wendimu, M.Y.; Hooks, S.B. Microglia Phenotypes in Aging and Neurodegenerative Diseases. Cells 2022, 11, 2091. [Google Scholar] [CrossRef]
- Fujikawa, R.; Jinno, S. Identification of hyper-ramified microglia in the CA1 region of the mouse hippocampus potentially associated with stress resilience. Eur. J. Neurosci. 2022, 56, 5137–5153. [Google Scholar] [CrossRef]
- Morrison, H.; Young, K.; Qureshi, M.; Rowe, R.K.; Lifshitz, J. Quantitative microglia analyses reveal diverse morphologic responses in the rat cortex after diffuse brain injury. Sci. Rep. 2017, 7, 13211. [Google Scholar] [CrossRef]
- Plescher, M.; Seifert, G.; Hansen, J.N.; Bedner, P.; Steinhäuser, C.; Halle, A. Plaque-dependent morphological and electrophysiological heterogeneity of microglia in an Alzheimer’s disease mouse model. Glia 2018, 66, 1464–1480. [Google Scholar] [CrossRef]
- Franco-Bocanegra, D.K.; Gourari, Y.; McAuley, C.; Chatelet, D.S.; Johnston, D.A.; Nicoll, J.A.R.; Boche, D. Microglial morphology in Alzheimer’s disease and after Aβ immunotherapy. Sci. Rep. 2021, 11, 15955. [Google Scholar] [CrossRef]
- Galloway, D.A.; Phillips, A.E.M.; Owen, D.R.J.; Moore, C.S. Phagocytosis in the Brain: Homeostasis and Disease. Front. Immunol. 2019, 10, 790. [Google Scholar] [CrossRef]
- Butler, C.A.; Popescu, A.S.; Kitchener, E.J.A.; Allendorf, D.H.; Puigdellívol, M.; Brown, G.C. Microglial phagocytosis of neurons in neurodegeneration, and its regulation. J. Neurochem. 2021, 158, 621–639. [Google Scholar] [CrossRef]
- Miyanishi, K.; Sato, A.; Kihara, N.; Utsunomiya, R.; Tanaka, J. Synaptic elimination by microglia and disturbed higher brain functions. Neurochem. Int. 2021, 142, 104901. [Google Scholar] [CrossRef]
- Yang, Q.Q.; Zhou, J.W. Neuroinflammation in the central nervous system: Symphony of glial cells. Glia 2019, 67, 1017–1035. [Google Scholar] [CrossRef] [PubMed]
- Beaino, W.; Janssen, B.; Vugts, D.J.; de Vries, H.E.; Windhorst, A.D. Towards PET imaging of the dynamic phenotypes of microglia. Clin. Exp. Immunol. 2021, 206, 282–300. [Google Scholar] [CrossRef] [PubMed]
- Al-Khishman, N.U.; Qi, Q.; Roseborough, A.D.; Levit, A.; Allman, B.L.; Anazodo, U.C.; Fox, M.S.; Whitehead, S.N.; Thiessen, J.D. TSPO PET detects acute neuroinflammation but not diffuse chronically activated MHCII microglia in the rat. EJNMMI Res. 2020, 10, 113. [Google Scholar] [CrossRef]
- Roseborough, A.D.; Myers, S.J.; Khazaee, R.; Zhu, Y.; Zhao, L.; Iorio, E.; Elahi, F.M.; Pasternak, S.H.; Whitehead, S.N. Plasma derived extracellular vesicle biomarkers of microglia activation in an experimental stroke model. J. Neuroinflammation 2023, 20, 20. [Google Scholar] [CrossRef] [PubMed]
- McQuade, A.; Blurton-Jones, M. Microglia in Alzheimer’s Disease: Exploring How Genetics and Phenotype Influence Risk. J. Mol. Biol. 2019, 431, 1805–1817. [Google Scholar] [CrossRef]
- Marioni, R.E.; Harris, S.E.; Zhang, Q.; McRae, A.F.; Hagenaars, S.P.; Hill, W.D.; Davies, G.; Ritchie, C.W.; Gale, C.R.; Starr, J.M.; et al. Correction: GWAS on family history of Alzheimer’s disease. Transl. Psychiatry 2019, 9, 161. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef]
- Husain, M.A.; Laurent, B.; Plourde, M. APOE and Alzheimer’s Disease: From Lipid Transport to Physiopathology and Therapeutics. Front. Neurosci. 2021, 15, 630502. [Google Scholar] [CrossRef]
- Corder, E.H.; Saunders, A.M.; Risch, N.J.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Rimmler, J.B.; Locke, P.A.; Conneally, P.M.; Schmader, K.E. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat. Genet. 1994, 7, 180–184. [Google Scholar] [CrossRef]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef]
- Spangenberg, E.; Severson, P.L.; Hohsfield, L.A.; Crapser, J.; Zhang, J.; Burton, E.A.; Zhang, Y.; Spevak, W.; Lin, J.; Phan, N.Y.; et al. Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer’s disease model. Nat. Commun. 2019, 10, 3758. [Google Scholar] [CrossRef]
- Uchihara, T.; Duyckaerts, C.; He, Y.; Kobayashi, K.; Seilhean, D.; Amouyel, P.; Hauw, J.J. ApoE immunoreactivity and microglial cells in Alzheimer’s disease brain. Neurosci. Lett. 1995, 195, 5–8. [Google Scholar] [CrossRef]
- LaDu, M.J.; Falduto, M.T.; Manelli, A.M.; Reardon, C.A.; Getz, G.S.; Frail, D.E. Isoform-specific binding of apolipoprotein E to beta-amyloid. J. Biol. Chem. 1994, 269, 23403–23406. [Google Scholar] [CrossRef] [PubMed]
- Atagi, Y.; Liu, C.C.; Painter, M.M.; Chen, X.F.; Verbeeck, C.; Zheng, H.; Li, X.; Rademakers, R.; Kang, S.S.; Xu, H.; et al. Apolipoprotein E Is a Ligand for Triggering Receptor Expressed on Myeloid Cells 2 (TREM2). J. Biol. Chem. 2015, 290, 26043–26050. [Google Scholar] [CrossRef] [PubMed]
- Kanekiyo, T.; Bu, G. The low-density lipoprotein receptor-related protein 1 and amyloid-β clearance in Alzheimer’s disease. Front. Aging Neurosci. 2014, 6, 93. [Google Scholar] [CrossRef]
- Henningfield, C.M.; Arreola, M.A.; Soni, N.; Spangenberg, E.E.; Green, K.N. Microglia-specific ApoE knock-out does not alter Alzheimer’s disease plaque pathogenesis or gene expression. Glia 2022, 70, 287–302. [Google Scholar] [CrossRef] [PubMed]
- Colonna, M. TREMs in the immune system and beyond. Nat. Rev. Immunol. 2003, 3, 445–453. [Google Scholar] [CrossRef]
- Qin, Q.; Teng, Z.; Liu, C.; Li, Q.; Yin, Y.; Tang, Y. TREM2, microglia, and Alzheimer’s disease. Mech. Ageing Dev. 2021, 195, 111438. [Google Scholar] [CrossRef] [PubMed]
- Schmid, C.D.; Sautkulis, L.N.; Danielson, P.E.; Cooper, J.; Hasel, K.W.; Hilbush, B.S.; Sutcliffe, J.G.; Carson, M.J. Heterogeneous expression of the triggering receptor expressed on myeloid cells-2 on adult murine microglia. J. Neurochem. 2002, 83, 1309–1320. [Google Scholar] [CrossRef]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.; Younkin, S.; et al. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, T.; Stefansson, H.; Steinberg, S.; Jonsdottir, I.; Jonsson, P.V.; Snaedal, J.; Bjornsson, S.; Huttenlocher, J.; Levey, A.I.; Lah, J.J.; et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Yeh, F.L.; Wang, Y.; Tom, I.; Gonzalez, L.C.; Sheng, M. TREM2 Binds to Apolipoproteins, Including APOE and CLU/APOJ, and Thereby Facilitates Uptake of Amyloid-Beta by Microglia. Neuron 2016, 91, 328–340. [Google Scholar] [CrossRef] [PubMed]
- Lue, L.F.; Schmitz, C.T.; Serrano, G.; Sue, L.I.; Beach, T.G.; Walker, D.G. TREM2 Protein Expression Changes Correlate with Alzheimer’s Disease Neurodegenerative Pathologies in Post-Mortem Temporal Cortices. Brain Pathol. 2015, 25, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Tan, L.; Zhu, X.C.; Zhang, Q.Q.; Cao, L.; Tan, M.S.; Gu, L.Z.; Wang, H.F.; Ding, Z.Z.; Zhang, Y.D.; et al. Upregulation of TREM2 ameliorates neuropathology and rescues spatial cognitive impairment in a transgenic mouse model of Alzheimer’s disease. Neuropsychopharmacology 2014, 39, 2949–2962. [Google Scholar] [CrossRef] [PubMed]
- Celarain, N.; de Gordoa, J.S.-R.; Zelaya, M.V.; Roldán, M.; Larumbe, R.; Pulido, L.; Echavarri, C.; Mendioroz, M. TREM2 upregulation correlates with 5-hydroxymethycytosine enrichment in Alzheimer’s disease hippocampus. Clin. Epigenetics 2016, 8, 37. [Google Scholar] [CrossRef]
- Wang, S.; Sudan, R.; Peng, V.; Zhou, Y.; Du, S.; Yuede, C.M.; Lei, T.; Hou, J.; Cai, Z.; Cella, M.; et al. TREM2 drives microglia response to amyloid-β via SYK-dependent and -independent pathways. Cell 2022, 185, 4153–4169.e19. [Google Scholar] [CrossRef]
- Wißfeld, J.; Nozaki, I.; Mathews, M.; Raschka, T.; Ebeling, C.; Hornung, V.; Brüstle, O.; Neumann, H. Deletion of Alzheimer’s disease-associated CD33 results in an inflammatory human microglia phenotype. Glia 2021, 69, 1393–1412. [Google Scholar] [CrossRef]
- Clark, E.A.; Giltiay, N.V. CD22: A Regulator of Innate and Adaptive B Cell Responses and Autoimmunity. Front. Immunol. 2018, 9, 2235. [Google Scholar] [CrossRef]
- Yao, H.; Coppola, K.; Schweig, J.E.; Crawford, F.; Mullan, M.; Paris, D. Distinct Signaling Pathways Regulate TREM2 Phagocytic and NFκB Antagonistic Activities. Front. Cell. Neurosci. 2019, 13, 457. [Google Scholar] [CrossRef]
- Ennerfelt, H.; Frost, E.L.; Shapiro, D.A.; Holliday, C.; Zengeler, K.E.; Voithofer, G.; Bolte, A.C.; Lammert, C.R.; Kulas, J.A.; Ulland, T.K.; et al. SYK coordinates neuroprotective microglial responses in neurodegenerative disease. Cell 2022, 185, 4135–4152.e22. [Google Scholar] [CrossRef] [PubMed]
- Suárez-Calvet, M.; Kleinberger, G.; Araque Caballero, M.; Brendel, M.; Rominger, A.; Alcolea, D.; Fortea, J.; Lleó, A.; Blesa, R.; Gispert, J.D.; et al. sTREM2 cerebrospinal fluid levels are a potential biomarker for microglia activity in early-stage Alzheimer’s disease and associate with neuronal injury markers. EMBO Mol. Med. 2016, 8, 466–476. [Google Scholar] [CrossRef] [PubMed]
- Galatro, T.F.; Holtman, I.R.; Lerario, A.M.; Vainchtein, I.D.; Brouwer, N.; Sola, P.R.; Veras, M.M.; Pereira, T.F.; Leite, R.E.P.; Möller, T.; et al. Transcriptomic analysis of purified human cortical microglia reveals age-associated changes. Nat. Neurosci. 2017, 20, 1162–1171. [Google Scholar] [CrossRef] [PubMed]
- Crocker, P.R.; McMillan, S.J.; Richards, H.E. CD33-related siglecs as potential modulators of inflammatory responses. Ann. N. Y. Acad. Sci. 2012, 1253, 102–111. [Google Scholar] [CrossRef]
- Hollingworth, P.; Harold, D.; Sims, R.; Gerrish, A.; Lambert, J.C.; Carrasquillo, M.M.; Abraham, R.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat. Genet. 2011, 43, 429–435. [Google Scholar] [CrossRef]
- Griciuc, A.; Serrano-Pozo, A.; Parrado, A.R.; Lesinski, A.N.; Asselin, C.N.; Mullin, K.; Hooli, B.; Choi, S.H.; Hyman, B.T.; Tanzi, R.E. Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron 2013, 78, 631–643. [Google Scholar] [CrossRef]
- Hung, A.S.; Liang, Y.; Chow, T.C.; Tang, H.C.; Wu, S.L.; Wai, M.S.; Yew, D.T. Mutated tau, amyloid and neuroinflammation in Alzheimer disease-A brief review. Prog. Histochem. Cytochem. 2016, 51, 1–8. [Google Scholar] [CrossRef]
- Ashrafian, H.; Zadeh, E.H.; Khan, R.H. Review on Alzheimer’s disease: Inhibition of amyloid beta and tau tangle formation. Int. J. Biol. Macromol. 2021, 167, 382–394. [Google Scholar] [CrossRef]
- Chen, G.F.; Xu, T.H.; Yan, Y.; Zhou, Y.R.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid beta: Structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef]
- Dunn, B.; Stein, P.; Cavazzoni, P. Approval of Aducanumab for Alzheimer Disease-The FDA’s Perspective. JAMA Intern. Med. 2021, 181, 1276–1278. [Google Scholar] [CrossRef]
- Tampi, R.R.; Forester, B.P.; Agronin, M. Aducanumab: Evidence from clinical trial data and controversies. Drugs Context 2021, 10, 2021-7-3. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.F.; Fu, A.K.Y.; Ip, N.Y. Cytokine signaling convergence regulates the microglial state transition in Alzheimer’s disease. Cell. Mol. Life Sci. 2021, 78, 4703–4712. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.Y.; Tan, M.S.; Yu, J.T.; Tan, L. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann. Transl. Med. 2015, 3, 136. [Google Scholar]
- Zhong, L.; Wang, Z.; Wang, D.; Martens, Y.A.; Wu, L.; Xu, Y.; Wang, K.; Li, J.; Huang, R.; Can, D.; et al. Amyloid-beta modulates microglial responses by binding to the triggering receptor expressed on myeloid cells 2 (TREM2). Mol. Neurodegener. 2018, 13, 15. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, S.; Atluri, V.; Kaushik, A.; Yndart, A.; Nair, M. Alzheimer’s disease: Pathogenesis, diagnostics, and therapeutics. Int. J. Nanomed. 2019, 14, 5541–5554. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Nam, H.; Kim, L.E.; Jeon, Y.; Min, H.; Ha, S.; Lee, Y.; Kim, S.Y.; Lee, S.J.; Kim, E.K.; et al. TLR4 (toll-like receptor 4) activation suppresses autophagy through inhibition of FOXO3 and impairs phagocytic capacity of microglia. Autophagy 2019, 15, 753–770. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.T.; Wang, K.; Hu, G.; Wang, X.; Miao, Z.; Azevedo, J.A.; Suh, E.; Van Deerlin, V.M.; Choi, D.; Roeder, K.; et al. APOE and TREM2 regulate amyloid-responsive microglia in Alzheimer’s disease. Acta Neuropathol. 2020, 140, 477–493. [Google Scholar] [CrossRef]
- Chiozzi, P.; Sarti, A.C.; Sanz, J.M.; Giuliani, A.L.; Adinolfi, E.; Vultaggio-Poma, V.; Falzoni, S.; Di Virgilio, F. Amyloid β-dependent mitochondrial toxicity in mouse microglia requires P2X7 receptor expression and is prevented by nimodipine. Sci. Rep. 2019, 9, 6475. [Google Scholar] [CrossRef]
- Li, Q.; Wu, Y.; Chen, J.; Xuan, A.; Wang, X. Microglia and immunotherapy in Alzheimer’s disease. Acta Neurol. Scand. 2022, 145, 273–278. [Google Scholar] [CrossRef]
- Yang, S.H. Cellular and Molecular Mediators of Neuroinflammation in Alzheimer Disease. Int. Neurourol. J. 2019, 23, S54–S62. [Google Scholar] [CrossRef]
- Combs, C.K.; Karlo, J.C.; Kao, S.C.; Landreth, G.E. beta-Amyloid stimulation of microglia and monocytes results in TNFalpha-dependent expression of inducible nitric oxide synthase and neuronal apoptosis. J. Neurosci. 2001, 21, 1179–1188. [Google Scholar] [CrossRef] [PubMed]
- Blasko, I.; Veerhuis, R.; Stampfer-Kountchev, M.; Saurwein-Teissl, M.; Eikelenboom, P.; Grubeck-Loebenstein, B. Costimulatory effects of interferon-gamma and interleukin-1beta or tumor necrosis factor alpha on the synthesis of Abeta1-40 and Abeta1-42 by human astrocytes. Neurobiol. Dis. 2000, 7, 682–689. [Google Scholar] [CrossRef]
- Liao, Y.F.; Wang, B.J.; Cheng, H.T.; Kuo, L.H.; Wolfe, M.S. Tumor necrosis factor-alpha, interleukin-1beta, and interferon-gamma stimulate gamma-secretase-mediated cleavage of amyloid precursor protein through a JNK-dependent MAPK pathway. J. Biol. Chem. 2004, 279, 49523–49532. [Google Scholar] [CrossRef] [PubMed]
- Fillit, H.; Ding, W.H.; Buee, L.; Kalman, J.; Altstiel, L.; Lawlor, B.; Wolf-Klein, G. Elevated circulating tumor necrosis factor levels in Alzheimer’s disease. Neurosci. Lett. 1991, 129, 318–320. [Google Scholar] [CrossRef] [PubMed]
- Tarkowski, E.; Andreasen, N.; Tarkowski, A.; Blennow, K. Intrathecal inflammation precedes development of Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 2003, 74, 1200–1205. [Google Scholar] [CrossRef]
- Zhou, M.; Xu, R.; Kaelber, D.C.; Gurney, M.E. Tumor Necrosis Factor (TNF) blocking agents are associated with lower risk for Alzheimer’s disease in patients with rheumatoid arthritis and psoriasis. PLoS ONE 2020, 15, e0229819. [Google Scholar] [CrossRef]
- Nilsson, L.N.; Bales, K.R.; DiCarlo, G.; Gordon, M.N.; Morgan, D.; Paul, S.M.; Potter, H. Alpha-1-antichymotrypsin promotes beta-sheet amyloid plaque deposition in a transgenic mouse model of Alzheimer’s disease. J. Neurosci. 2001, 21, 1444–1451. [Google Scholar] [CrossRef]
- Willis, E.F.; MacDonald, K.P.A.; Nguyen, Q.H.; Garrido, A.L.; Gillespie, E.R.; Harley, S.B.R.; Bartlett, P.F.; Schroder, W.A.; Yates, A.G.; Anthony, D.C.; et al. Repopulating Microglia Promote Brain Repair in an IL-6-Dependent Manner. Cell 2020, 180, 833–846.e16. [Google Scholar] [CrossRef]
- Kann, O.; Almouhanna, F.; Chausse, B. Interferon γ: A master cytokine in microglia-mediated neural network dysfunction and neurodegeneration. Trends Neurosci. 2022, 45, 913–927. [Google Scholar] [CrossRef]
- Spiteri, A.G.; Wishart, C.L.; Pamphlett, R.; Locatelli, G.; King, N.J.C. Microglia and monocytes in inflammatory CNS disease: Integrating phenotype and function. Acta Neuropathol. 2022, 143, 179–224. [Google Scholar] [CrossRef]
- Mildner, A.; Schmidt, H.; Nitsche, M.; Merkler, D.; Hanisch, U.K.; Mack, M.; Heikenwalder, M.; Brück, W.; Priller, J.; Prinz, M. Microglia in the adult brain arise from Ly-6ChiCCR2+ monocytes only under defined host conditions. Nat. Neurosci. 2007, 10, 1544–1553. [Google Scholar] [CrossRef]
- Mildner, A.; Schlevogt, B.; Kierdorf, K.; Böttcher, C.; Erny, D.; Kummer, M.P.; Quinn, M.; Brück, W.; Bechmann, I.; Heneka, M.T.; et al. Distinct and non-redundant roles of microglia and myeloid subsets in mouse models of Alzheimer’s disease. J. Neurosci. 2011, 31, 11159–11171. [Google Scholar] [CrossRef] [PubMed]
- Michaud, J.P.; Bellavance, M.A.; Préfontaine, P.; Rivest, S. Real-time in vivo imaging reveals the ability of monocytes to clear vascular amyloid beta. Cell Rep. 2013, 5, 646–653. [Google Scholar] [CrossRef] [PubMed]
- Prokop, S.; Miller, K.R.; Drost, N.; Handrick, S.; Mathur, V.; Luo, J.; Wegner, A.; Wyss-Coray, T.; Heppner, F.L. Impact of peripheral myeloid cells on amyloid-β pathology in Alzheimer’s disease-like mice. J. Exp. Med. 2015, 212, 1811–1818. [Google Scholar] [CrossRef] [PubMed]
- Perea, J.R.; Bolós, M.; Avila, J. Microglia in Alzheimer’s Disease in the Context of Tau Pathology. Biomolecules 2020, 10, 1439. [Google Scholar] [CrossRef]
- Tapia-Rojas, C.; Cabezas-Opazo, F.; Deaton, C.A.; Vergara, E.H.; Johnson, G.V.W.; Quintanilla, R.A. It’s all about tau. Prog. Neurobiol. 2019, 175, 54–76. [Google Scholar] [CrossRef]
- Gong, C.X.; Iqbal, K. Hyperphosphorylation of microtubule-associated protein tau: A promising therapeutic target for Alzheimer disease. Curr. Med. Chem. 2008, 15, 2321–2328. [Google Scholar] [CrossRef]
- Biernat, J.; Gustke, N.; Drewes, G.; Mandelkow, E.M.; Mandelkow, E. Phosphorylation of Ser262 strongly reduces binding of tau to microtubules: Distinction between PHF-like immunoreactivity and microtubule binding. Neuron 1993, 11, 153–163. [Google Scholar] [CrossRef]
- Wu, M.; Zhang, M.; Yin, X.; Chen, K.; Hu, Z.; Zhou, Q.; Cao, X.; Chen, Z.; Liu, D. The role of pathological tau in synaptic dysfunction in Alzheimer’s diseases. Transl. Neurodegener. 2021, 10, 45. [Google Scholar] [CrossRef]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.M.; Iwata, N.; Saido, T.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 2007, 53, 337–351. [Google Scholar] [CrossRef]
- Maphis, N.; Xu, G.; Kokiko-Cochran, O.N.; Jiang, S.; Cardona, A.; Ransohoff, R.M.; Lamb, B.T.; Bhaskar, K. Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain 2015, 138, 1738–1755. [Google Scholar] [CrossRef]
- Das, R.; Balmik, A.A.; Chinnathambi, S. Phagocytosis of full-length Tau oligomers by Actin-remodeling of activated microglia. J. Neuroinflammation 2020, 17, 10. [Google Scholar] [CrossRef] [PubMed]
- Perea, J.R.; Llorens-Martín, M.; Ávila, J.; Bolós, M. The Role of Microglia in the Spread of Tau: Relevance for Tauopathies. Front. Cell. Neurosci. 2018, 12, 172. [Google Scholar] [CrossRef]
- Nishiyori, A.; Minami, M.; Ohtani, Y.; Takami, S.; Yamamoto, J.; Kawaguchi, N.; Kume, T.; Akaike, A.; Satoh, M. Localization of fractalkine and CX3CR1 mRNAs in rat brain: Does fractalkine play a role in signaling from neuron to microglia? FEBS Lett. 1998, 429, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Bolós, M.; Perea, J.R.; Terreros-Roncal, J.; Pallas-Bazarra, N.; Jurado-Arjona, J.; Ávila, J.; Llorens-Martín, M. Absence of microglial CX3CR1 impairs the synaptic integration of adult-born hippocampal granule neurons. Brain Behav. Immun. 2018, 68, 76–89. [Google Scholar] [CrossRef] [PubMed]
- Bolós, M.; Llorens-Martín, M.; Perea, J.R.; Jurado-Arjona, J.; Rábano, A.; Hernández, F.; Avila, J. Absence of CX3CR1 impairs the internalization of Tau by microglia. Mol. Neurodegener. 2017, 12, 59. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.F.; Hu, H.; Tan, L.; Yu, J.T. Microglia Biomarkers in Alzheimer’s Disease. Mol. Neurobiol. 2021, 58, 3388–3404. [Google Scholar] [CrossRef]
- Kulczyńska-Przybik, A.; Słowik, A.; Mroczko, P.; Borawski, B.; Groblewska, M.; Borawska, R.; Mroczko, B. Cerebrospinal Fluid and Blood CX3CL1 as a Potential Biomarker in Early Diagnosis and Prognosis of Dementia. Curr. Alzheimer Res. 2020, 17, 709–721. [Google Scholar] [CrossRef]
- Kim, T.S.; Lim, H.K.; Lee, J.Y.; Kim, D.J.; Park, S.; Lee, C.; Lee, C.U. Changes in the levels of plasma soluble fractalkine in patients with mild cognitive impairment and Alzheimer’s disease. Neurosci. Lett. 2008, 436, 196–200. [Google Scholar] [CrossRef]
- Inoue, K.; Tsuda, M. Microglia in neuropathic pain: Cellular and molecular mechanisms and therapeutic potential. Nat. Rev. Neurosci. 2018, 19, 138–152. [Google Scholar] [CrossRef]
- Van Kooten, J.; Binnekade, T.T.; van der Wouden, J.C.; Stek, M.L.; Scherder, E.J.; Husebø, B.S.; Smalbrugge, M.; Hertogh, C.M. A Review of Pain Prevalence in Alzheimer’s, Vascular, Frontotemporal and Lewy Body Dementias. Dement. Geriatr. Cogn. Disord. 2016, 41, 220–232. [Google Scholar] [CrossRef] [PubMed]
- Whitlock, E.L.; Diaz-Ramirez, L.G.; Glymour, M.M.; Boscardin, W.J.; Covinsky, K.E.; Smith, A.K. Association between Persistent Pain and Memory Decline and Dementia in a Longitudinal Cohort of Elders. JAMA Intern. Med. 2017, 177, 1146–1153. [Google Scholar] [CrossRef] [PubMed]
- Ezzati, A.; Wang, C.; Katz, M.J.; Derby, C.A.; Zammit, A.R.; Zimmerman, M.E.; Pavlovic, J.M.; Sliwinski, M.J.; Lipton, R.B. The Temporal Relationship between Pain Intensity and Pain Interference and Incident Dementia. Curr. Alzheimer Res. 2019, 16, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Kubota, Y.; Tabuchi, T.; Shirai, K.; Iso, H.; Kondo, N.; Kondo, K. A prospective study of knee pain, low back pain, and risk of dementia: The JAGES project. Sci. Rep. 2019, 9, 10690. [Google Scholar] [CrossRef] [PubMed]
- Morton, R.E.; St John, P.D.; Tyas, S.L. Migraine and the risk of all-cause dementia, Alzheimer’s disease, and vascular dementia: A prospective cohort study in community-dwelling older adults. Int. J. Geriatr. Psychiatry 2019, 34, 1667–1676. [Google Scholar] [CrossRef] [PubMed]
- Tzeng, N.S.; Chung, C.H.; Liu, F.C.; Chiu, Y.H.; Chang, H.A.; Yeh, C.B.; Huang, S.Y.; Lu, R.B.; Yeh, H.W.; Kao, Y.C.; et al. Fibromyalgia and Risk of Dementia-A Nationwide, Population-Based, Cohort Study. Am. J. Med. Sci. 2018, 355, 153–161. [Google Scholar] [CrossRef]
- Chen, C.H.; Lin, C.L.; Kao, C.H. Irritable Bowel Syndrome Is Associated with an Increased Risk of Dementia: A Nationwide Population-Based Study. PLoS ONE 2016, 11, e0144589. [Google Scholar] [CrossRef]
- Jensen-Dahm, C.; Werner, M.U.; Jensen, T.S.; Ballegaard, M.; Andersen, B.B.; Høgh, P.; Waldemar, G. Discrepancy between stimulus response and tolerance of pain in Alzheimer disease. Neurology 2015, 84, 1575–1581. [Google Scholar] [CrossRef]
- Cravello, L.; Di Santo, S.; Varrassi, G.; Benincasa, D.; Marchettini, P.; de Tommaso, M.; Shofany, J.; Assogna, F.; Perotta, D.; Palmer, K.; et al. Chronic Pain in the Elderly with Cognitive Decline: A Narrative Review. Pain Ther. 2019, 8, 53–65. [Google Scholar] [CrossRef]
- Achterberg, W.; Lautenbacher, S.; Husebo, B.; Erdal, A.; Herr, K. Pain in dementia. Pain Rep. 2020, 5, e803. [Google Scholar] [CrossRef]
- Albrecht, D.S.; Forsberg, A.; Sandström, A.; Bergan, C.; Kadetoff, D.; Protsenko, E.; Lampa, J.; Lee, Y.C.; Höglund, C.O.; Catana, C.; et al. Brain glial activation in fibromyalgia—A multi-site positron emission tomography investigation. Brain Behav. Immun. 2019, 75, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, M. Microglia in the spinal cord and neuropathic pain. J. Diabetes Investig. 2016, 7, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Barcelon, E.E.; Cho, W.H.; Jun, S.B.; Lee, S.J. Brain Microglial Activation in Chronic Pain-Associated Affective Disorder. Front. Neurosci. 2019, 13, 213. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.M.; Mehrabani, S.; Liu, S.; Taylor, A.J.; Cahill, C.M. Topography of microglial activation in sensory- and affect-related brain regions in chronic pain. J. Neurosci. Res. 2017, 95, 1330–1335. [Google Scholar] [CrossRef] [PubMed]
- Gui, W.S.; Wei, X.; Mai, C.L.; Murugan, M.; Wu, L.J.; Xin, W.J.; Zhou, L.J.; Liu, X.G. Interleukin-1β overproduction is a common cause for neuropathic pain, memory deficit, and depression following peripheral nerve injury in rodents. Mol. Pain 2016, 12, 1744806916646784. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Fisher, D.W.; Yu, T.; Dong, H. The link between chronic pain and Alzheimer’s disease. J. Neuroinflammation 2019, 16, 204. [Google Scholar] [CrossRef]
- Gupta, D.P.; Lee, Y.S.; Choe, Y.; Kim, K.T.; Song, G.J.; Hwang, S.C. Knee osteoarthritis accelerates amyloid beta deposition and neurodegeneration in a mouse model of Alzheimer’s disease. Mol. Brain 2023, 16, 1. [Google Scholar] [CrossRef] [PubMed]
- Aston-Jones, G.; Cohen, J.D. An integrative theory of locus coeruleus-norepinephrine function: Adaptive gain and optimal performance. Annu. Rev. Neurosci. 2005, 28, 403–450. [Google Scholar] [CrossRef]
- Llorca-Torralba, M.; Borges, G.; Neto, F.; Mico, J.A.; Berrocoso, E. Noradrenergic Locus Coeruleus pathways in pain modulation. Neuroscience 2016, 338, 93–113. [Google Scholar] [CrossRef]
- Alba-Delgado, C.; Llorca-Torralba, M.; Horrillo, I.; Ortega, J.E.; Mico, J.A.; Sánchez-Blázquez, P.; Meana, J.J.; Berrocoso, E. Chronic pain leads to concomitant noradrenergic impairment and mood disorders. Biol. Psychiatry 2013, 73, 54–62. [Google Scholar] [CrossRef]
- Cordeiro Matos, S.; Zamfir, M.; Longo, G.; Ribeiro-da-Silva, A.; Séguéla, P. Noradrenergic fiber sprouting and altered transduction in neuropathic prefrontal cortex. Brain Struct. Funct. 2018, 223, 1149–1164. [Google Scholar] [CrossRef]
- Sugama, S.; Takenouchi, T.; Hashimoto, M.; Ohata, H.; Takenaka, Y.; Kakinuma, Y. Stress-induced microglial activation occurs through β-adrenergic receptor: Noradrenaline as a key neurotransmitter in microglial activation. J. Neuroinflammation 2019, 16, 266. [Google Scholar] [CrossRef]
- Mori, K.; Ozaki, E.; Zhang, B.; Yang, L.; Yokoyama, A.; Takeda, I.; Maeda, N.; Sakanaka, M.; Tanaka, J. Effects of norepinephrine on rat cultured microglial cells that express alpha1, alpha2, beta1 and beta2 adrenergic receptors. Neuropharmacology 2002, 43, 1026–1034. [Google Scholar] [CrossRef]
- Wohleb, E.S.; Hanke, M.L.; Corona, A.W.; Powell, N.D.; Stiner, L.M.; Bailey, M.T.; Nelson, R.J.; Godbout, J.P.; Sheridan, J.F. β-Adrenergic receptor antagonism prevents anxiety-like behavior and microglial reactivity induced by repeated social defeat. J. Neurosci. 2011, 31, 6277–6288. [Google Scholar] [CrossRef]
- Schlachetzki, J.C.; Fiebich, B.L.; Haake, E.; de Oliveira, A.C.; Candelario-Jalil, E.; Heneka, M.T.; Hüll, M. Norepinephrine enhances the LPS-induced expression of COX-2 and secretion of PGE2 in primary rat microglia. J. Neuroinflammation 2010, 7, 2. [Google Scholar] [CrossRef]
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Sierra, A.; Stevens, B.; Tremblay, M.E.; Aguzzi, A.; Ajami, B.; Amit, I.; Audinat, E.; Bechmann, I.; Bennett, M.; et al. Microglia states and nomenclature: A field at its crossroads. Neuron 2022, 110, 3458–3483. [Google Scholar] [CrossRef]
- Masuda, T.; Sankowski, R.; Staszewski, O.; Prinz, M. Microglia Heterogeneity in the Single-Cell Era. Cell Rep. 2020, 30, 1271–1281. [Google Scholar] [CrossRef]
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019, 570, 332–337. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef]
- Deczkowska, A.; Keren-Shaul, H.; Weiner, A.; Colonna, M.; Schwartz, M.; Amit, I. Disease-Associated Microglia: A Universal Immune Sensor of Neurodegeneration. Cell 2018, 173, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581.e9. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, K.; Friedman, B.A.; Etxeberria, A.; Huntley, M.A.; van der Brug, M.P.; Foreman, O.; Paw, J.S.; Modrusan, Z.; Beach, T.G.; Serrano, G.E.; et al. Alzheimer’s Patient Microglia Exhibit Enhanced Aging and Unique Transcriptional Activation. Cell Rep. 2020, 31, 107843. [Google Scholar] [CrossRef]
- Benmamar-Badel, A.; Owens, T.; Wlodarczyk, A. Protective Microglial Subset in Development, Aging, and Disease: Lessons from Transcriptomic Studies. Front. Immunol. 2020, 11, 430. [Google Scholar] [CrossRef] [PubMed]
- Bulloch, K.; Miller, M.M.; Gal-Toth, J.; Milner, T.A.; Gottfried-Blackmore, A.; Waters, E.M.; Kaunzner, U.W.; Liu, K.; Lindquist, R.; Nussenzweig, M.C.; et al. CD11c/EYFP transgene illuminates a discrete network of dendritic cells within the embryonic, neonatal, adult, and injured mouse brain. J. Comp. Neurol. 2008, 508, 687–710. [Google Scholar] [CrossRef]
- Immig, K.; Gericke, M.; Menzel, F.; Merz, F.; Krueger, M.; Schiefenhövel, F.; Lösche, A.; Jäger, K.; Hanisch, U.K.; Biber, K.; et al. CD11c-positive cells from brain, spleen, lung, and liver exhibit site-specific immune phenotypes and plastically adapt to new environments. Glia 2015, 63, 611–625. [Google Scholar] [CrossRef]
- Prodinger, C.; Bunse, J.; Krüger, M.; Schiefenhövel, F.; Brandt, C.; Laman, J.D.; Greter, M.; Immig, K.; Heppner, F.; Becher, B.; et al. CD11c-expressing cells reside in the juxtavascular parenchyma and extend processes into the glia limitans of the mouse nervous system. Acta Neuropathol. 2011, 121, 445–458. [Google Scholar] [CrossRef]
- Wlodarczyk, A.; Løbner, M.; Cédile, O.; Owens, T. Comparison of microglia and infiltrating CD11c⁺ cells as antigen presenting cells for T cell proliferation and cytokine response. J. Neuroinflammation 2014, 11, 57. [Google Scholar] [CrossRef]
- Kohno, K.; Shirasaka, R.; Yoshihara, K.; Mikuriya, S.; Tanaka, K.; Takanami, K.; Inoue, K.; Sakamoto, H.; Ohkawa, Y.; Masuda, T.; et al. A spinal microglia population involved in remitting and relapsing neuropathic pain. Science 2022, 376, 86–90. [Google Scholar] [CrossRef]
- Kamphuis, W.; Kooijman, L.; Schetters, S.; Orre, M.; Hol, E.M. Transcriptional profiling of CD11c-positive microglia accumulating around amyloid plaques in a mouse model for Alzheimer’s disease. Biochim. Biophys. Acta 2016, 1862, 1847–1860. [Google Scholar] [CrossRef]
- Butovsky, O.; Koronyo-Hamaoui, M.; Kunis, G.; Ophir, E.; Landa, G.; Cohen, H.; Schwartz, M. Glatiramer acetate fights against Alzheimer’s disease by inducing dendritic-like microglia expressing insulin-like growth factor 1. Proc. Natl. Acad. Sci. USA 2006, 103, 11784–11789. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Raj, D.; Saiepour, N.; Van Dam, D.; Brouwer, N.; Holtman, I.R.; Eggen, B.J.L.; Möller, T.; Tamm, J.A.; Abdourahman, A.; et al. Immune hyperreactivity of Aβ plaque-associated microglia in Alzheimer’s disease. Neurobiol. Aging 2017, 55, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Wlodarczyk, A.; Benmamar-Badel, A.; Cédile, O.; Jensen, K.N.; Kramer, I.; Elsborg, N.B.; Owens, T. CSF1R Stimulation Promotes Increased Neuroprotection by CD11c+ Microglia in EAE. Front. Cell. Neurosci. 2018, 12, 523. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, T.; Doi, Y.; Mizoguchi, H.; Jin, S.; Noda, M.; Sonobe, Y.; Takeuchi, H.; Suzumura, A. Interleukin-34 selectively enhances the neuroprotective effects of microglia to attenuate oligomeric amyloid-β neurotoxicity. Am. J. Pathol. 2011, 179, 2016–2027. [Google Scholar] [CrossRef]
- Peña-Altamira, E.; Petralla, S.; Massenzio, F.; Virgili, M.; Bolognesi, M.L.; Monti, B. Nutritional and Pharmacological Strategies to Regulate Microglial Polarization in Cognitive Aging and Alzheimer’s Disease. Front. Aging Neurosci. 2017, 9, 175. [Google Scholar] [CrossRef]
- Jope, R.S.; Yuskaitis, C.J.; Beurel, E. Glycogen synthase kinase-3 (GSK3): Inflammation, diseases, and therapeutics. Neurochem. Res. 2007, 32, 577–595. [Google Scholar] [CrossRef]
- Green, H.F.; Nolan, Y.M. GSK-3 mediates the release of IL-1β, TNF-α and IL-10 from cortical glia. Neurochem. Int. 2012, 61, 666–671. [Google Scholar] [CrossRef]
- Licht-Murava, A.; Paz, R.; Vaks, L.; Avrahami, L.; Plotkin, B.; Eisenstein, M.; Eldar-Finkelman, H. A unique type of GSK-3 inhibitor brings new opportunities to the clinic. Sci. Signal. 2016, 9, ra110. [Google Scholar] [CrossRef]
- Lauretti, E.; Dincer, O.; Praticò, D. Glycogen synthase kinase-3 signaling in Alzheimer’s disease. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118664. [Google Scholar] [CrossRef]
- Hu, W.; Jiang, C.; Kim, M.; Xiao, Y.; Richter, H.J.; Guan, D.; Zhu, K.; Krusen, B.M.; Roberts, A.N.; Miller, J.; et al. Isoform-specific functions of PPARγ in gene regulation and metabolism. Genes Dev. 2022, 36, 300–312. [Google Scholar] [CrossRef]
- Ji, H.; Wang, H.; Zhang, F.; Li, X.; Xiang, L.; Aiguo, S. PPARγ agonist pioglitazone inhibits microglia inflammation by blocking p38 mitogen-activated protein kinase signaling pathways. Inflamm. Res. 2010, 59, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Burns, D.K.; Alexander, R.C.; Welsh-Bohmer, K.A.; Culp, M.; Chiang, C.; O’Neil, J.; Evans, R.M.; Harrigan, P.; Plassman, B.L.; Burke, J.R.; et al. Safety and efficacy of pioglitazone for the delay of cognitive impairment in people at risk of Alzheimer’s disease (TOMMORROW): A prognostic biomarker study and a phase 3, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2021, 20, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, S.; Gabriel, H.; Strittmatter, W.; Didsbury, J. An Exploratory Phase IIa Study of the PPAR delta/gamma Agonist T3D-959 Assessing Metabolic and Cognitive Function in Subjects with Mild to Moderate Alzheimer’s Disease. J. Alzheimers Dis. 2020, 73, 1085–1103. [Google Scholar] [CrossRef]
- Jeong, J.W.; Lee, H.H.; Han, M.H.; Kim, G.Y.; Kim, W.J.; Choi, Y.H. Anti-inflammatory effects of genistein via suppression of the toll-like receptor 4-mediated signaling pathway in lipopolysaccharide-stimulated BV2 microglia. Chem. Biol. Interact. 2014, 212, 30–39. [Google Scholar] [CrossRef]
- Fujikawa, R.; Yamada, J.; Iinuma, K.M.; Jinno, S. Phytoestrogen genistein modulates neuron-microglia signaling in a mouse model of chronic social defeat stress. Neuropharmacology 2022, 206, 108941. [Google Scholar] [CrossRef] [PubMed]
- Viña, J.; Escudero, J.; Baquero, M.; Cebrián, M.; Carbonell-Asíns, J.A.; Muñoz, J.E.; Satorres, E.; Meléndez, J.C.; Ferrer-Rebolleda, J.; Cózar-Santiago, M.D.P.; et al. Genistein effect on cognition in prodromal Alzheimer’s disease patients. The GENIAL clinical trial. Alzheimers Res. Ther. 2022, 14, 164. [Google Scholar] [CrossRef]
- Lin, H.Y.; Huang, B.R.; Yeh, W.L.; Lee, C.H.; Huang, S.S.; Lai, C.H.; Lin, H.; Lu, D.Y. Antineuroinflammatory effects of lycopene via activation of adenosine monophosphate-activated protein kinase-α1/heme oxygenase-1 pathways. Neurobiol. Aging 2014, 35, 191–202. [Google Scholar] [CrossRef]
- Wang, J.; Li, L.; Wang, Z.; Cui, Y.; Tan, X.; Yuan, T.; Liu, Q.; Liu, Z.; Liu, X. Supplementation of lycopene attenuates lipopolysaccharide-induced amyloidogenesis and cognitive impairments via mediating neuroinflammation and oxidative stress. J. Nutr. Biochem. 2018, 56, 16–25. [Google Scholar] [CrossRef]
- Crowe-White, K.M.; Phillips, T.A.; Ellis, A.C. Lycopene and cognitive function. J. Nutr. Sci. 2019, 8, e20. [Google Scholar] [CrossRef]
- Neuner, S.M.; Telpoukhovskaia, M.; Menon, V.; O’Connell, K.M.S.; Hohman, T.J.; Kaczorowski, C.C. Translational approaches to understanding resilience to Alzheimer’s disease. Trends Neurosci. 2022, 45, 369–383. [Google Scholar] [CrossRef]
- Ewers, M.; Franzmeier, N.; Suárez-Calvet, M.; Morenas-Rodriguez, E.; Caballero, M.A.A.; Kleinberger, G.; Piccio, L.; Cruchaga, C.; Deming, Y.; Dichgans, M.; et al. Increased soluble TREM2 in cerebrospinal fluid is associated with reduced cognitive and clinical decline in Alzheimer’s disease. Sci. Transl. Med. 2019, 11, eaav6221. [Google Scholar] [CrossRef]
- Yokoyama, M.; Kobayashi, H.; Tatsumi, L.; Tomita, T. Mouse Models of Alzheimer’s Disease. Front. Mol. Neurosci. 2022, 15, 912995. [Google Scholar] [CrossRef] [PubMed]
- Benitez, D.P.; Jiang, S.; Wood, J.; Wang, R.; Hall, C.M.; Peerboom, C.; Wong, N.; Stringer, K.M.; Vitanova, K.S.; Smith, V.C.; et al. Knock-in models related to Alzheimer’s disease: Synaptic transmission, plaques and the role of microglia. Mol. Neurodegener. 2021, 16, 47. [Google Scholar] [CrossRef] [PubMed]
- Frautschy, S.A.; Yang, F.; Irrizarry, M.; Hyman, B.; Saido, T.C.; Hsiao, K.; Cole, G.M. Microglial response to amyloid plaques in APPsw transgenic mice. Am. J. Pathol. 1998, 152, 307–317. [Google Scholar]
- Wright, A.L.; Zinn, R.; Hohensinn, B.; Konen, L.M.; Beynon, S.B.; Tan, R.P.; Clark, I.A.; Abdipranoto, A.; Vissel, B. Neuroinflammation and neuronal loss precede Aβ plaque deposition in the hAPP-J20 mouse model of Alzheimer’s disease. PLoS ONE 2013, 8, e59586. [Google Scholar] [CrossRef] [PubMed]
- Baron, R.; Babcock, A.A.; Nemirovsky, A.; Finsen, B.; Monsonego, A. Accelerated microglial pathology is associated with Aβ plaques in mouse models of Alzheimer’s disease. Aging Cell 2014, 13, 584–595. [Google Scholar] [CrossRef]
- Crapser, J.D.; Spangenberg, E.E.; Barahona, R.A.; Arreola, M.A.; Hohsfield, L.A.; Green, K.N. Microglia facilitate loss of perineuronal nets in the Alzheimer’s disease brain. EBioMedicine 2020, 58, 102919. [Google Scholar] [CrossRef]
- Fagan, S.G.; Bechet, S.; Dev, K.K. Fingolimod Rescues Memory and Improves Pathological Hallmarks in the 3xTg-AD Model of Alzheimer’s Disease. Mol. Neurobiol. 2022, 59, 1882–1895. [Google Scholar] [CrossRef]
- Edler, M.K.; Mhatre-Winters, I.; Richardson, J.R. Microglia in Aging and Alzheimer’s Disease: A Comparative Species Review. Cells 2021, 10, 1138. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fujikawa, R.; Tsuda, M. The Functions and Phenotypes of Microglia in Alzheimer’s Disease. Cells 2023, 12, 1207. https://doi.org/10.3390/cells12081207
Fujikawa R, Tsuda M. The Functions and Phenotypes of Microglia in Alzheimer’s Disease. Cells. 2023; 12(8):1207. https://doi.org/10.3390/cells12081207
Chicago/Turabian StyleFujikawa, Risako, and Makoto Tsuda. 2023. "The Functions and Phenotypes of Microglia in Alzheimer’s Disease" Cells 12, no. 8: 1207. https://doi.org/10.3390/cells12081207