



Natural History of Glaucoma Progression in the DBA/2J Model: Early Contribution of Müller Cell Gliosis

 ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Assessment of IOP
2.3. Immunofluorescence
2.4. Isolectin B4 Staining of Retinal Vessels
2.5. Analysis of BRB Leakage Using Evans Blue Dye
2.6. Western Blot
2.7. Statistical Analysis
3. Results
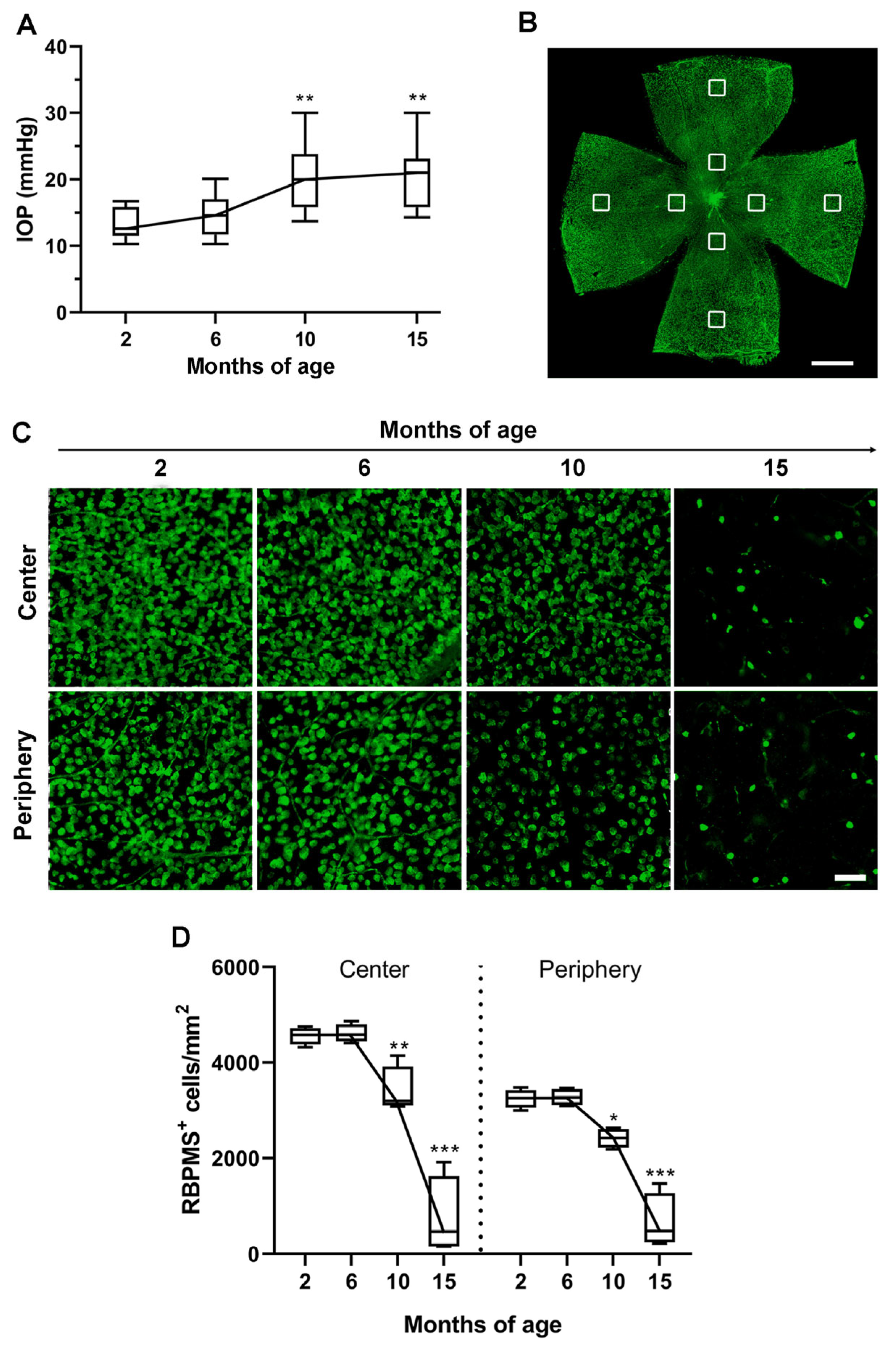
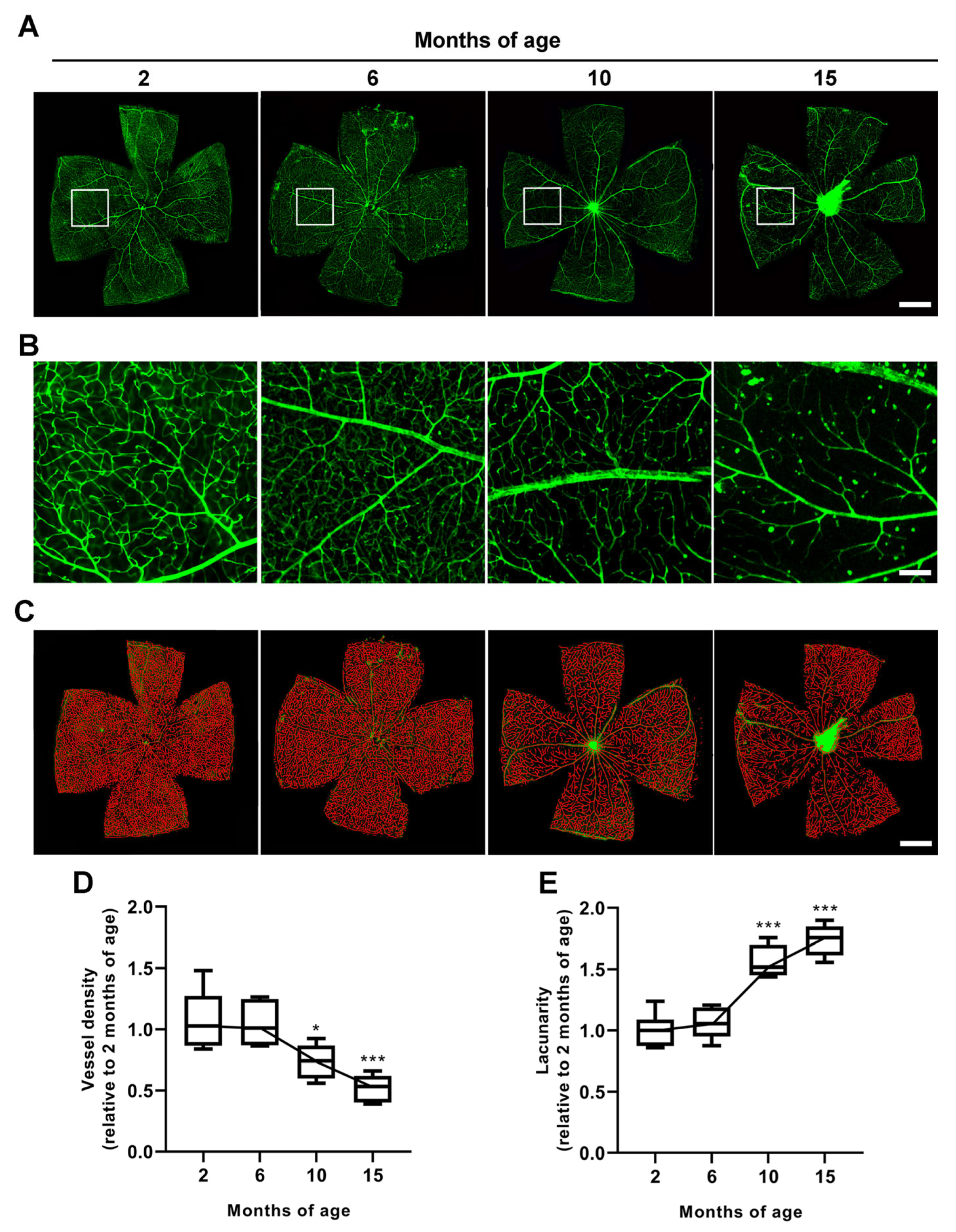
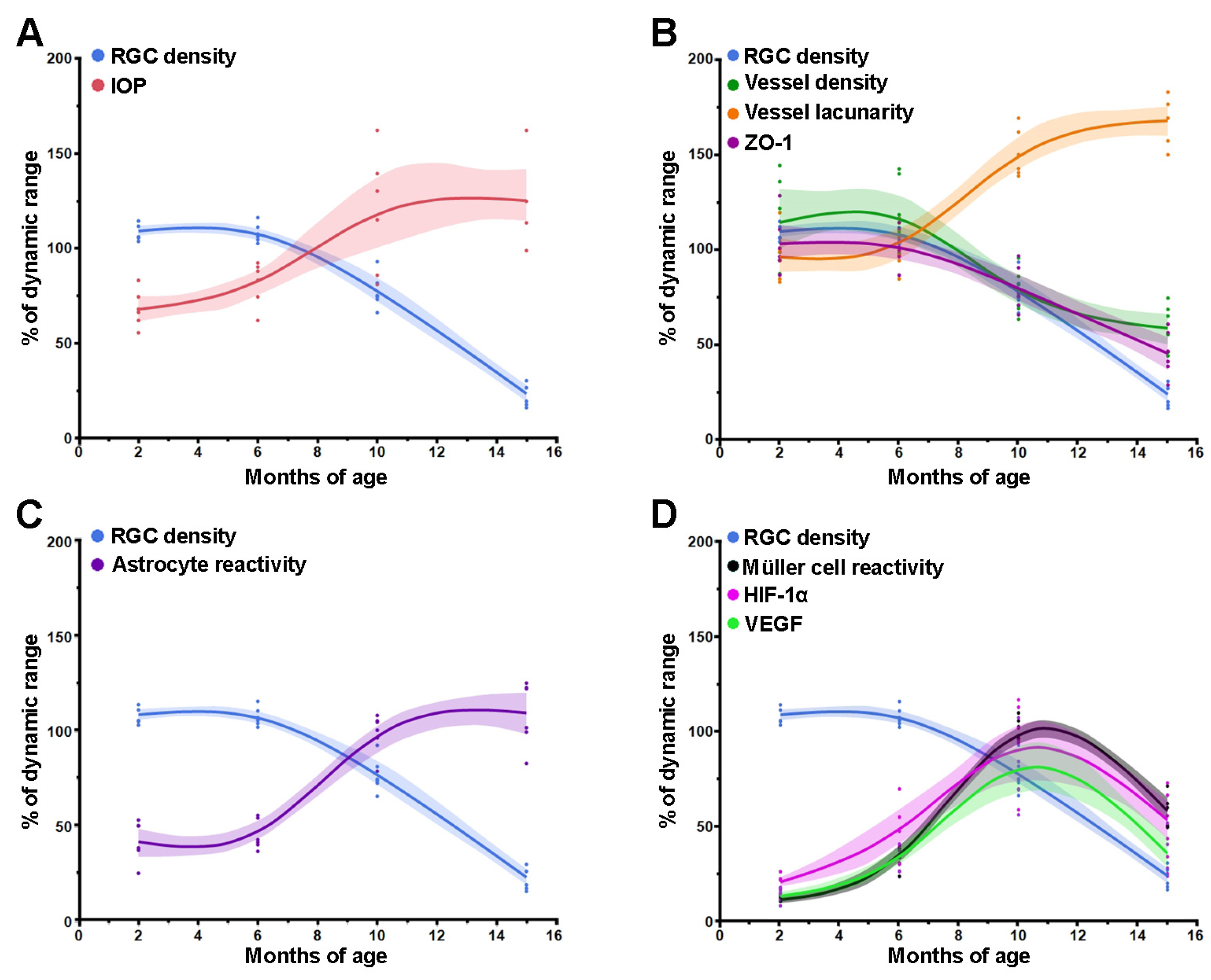
3.1. Age-Dependent Profiling of IOP Increase, RGC Loss and Vascular Regression
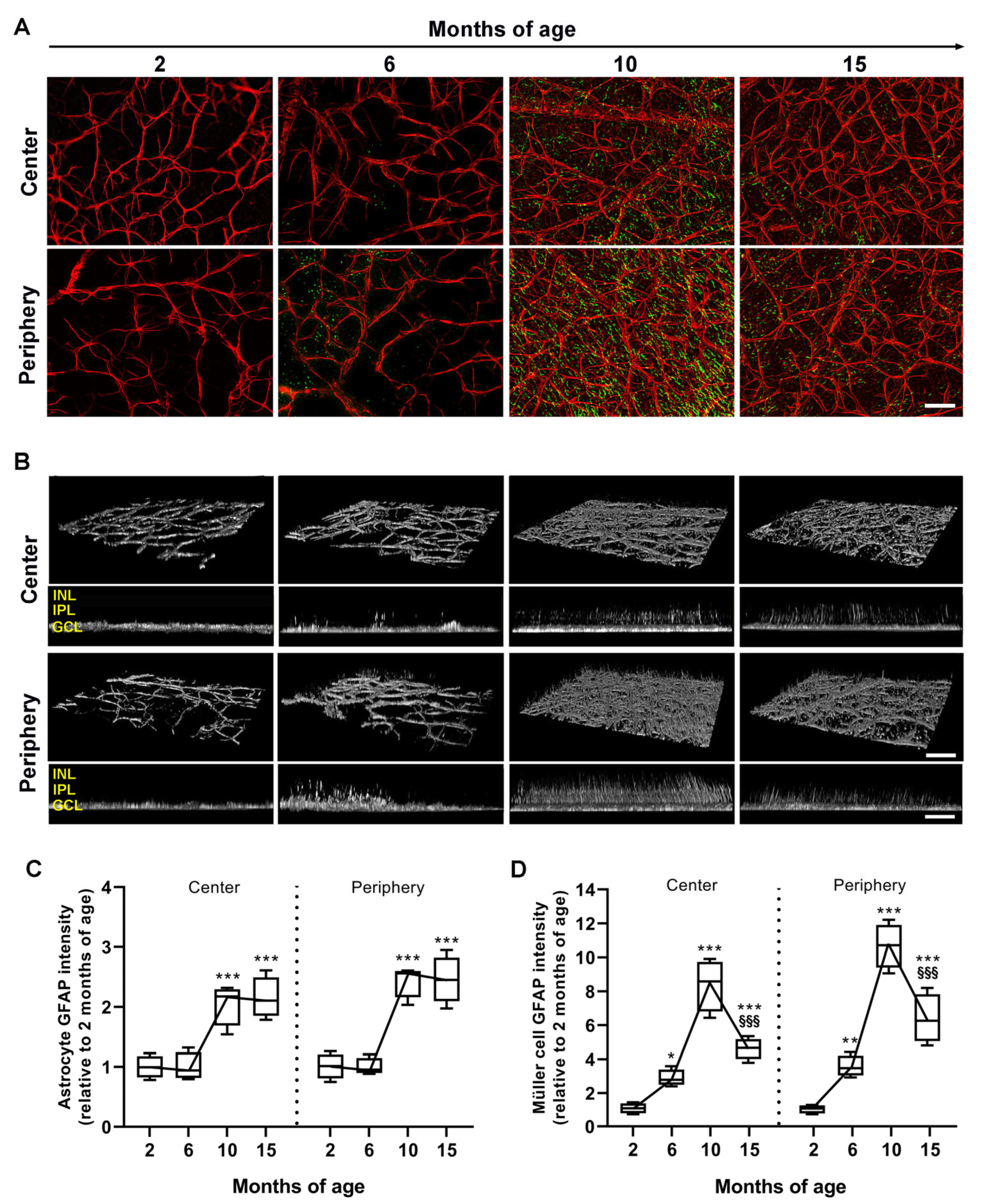
3.2. Age-Dependent Profiling of Astrocyte and Müller Cell Reactivity
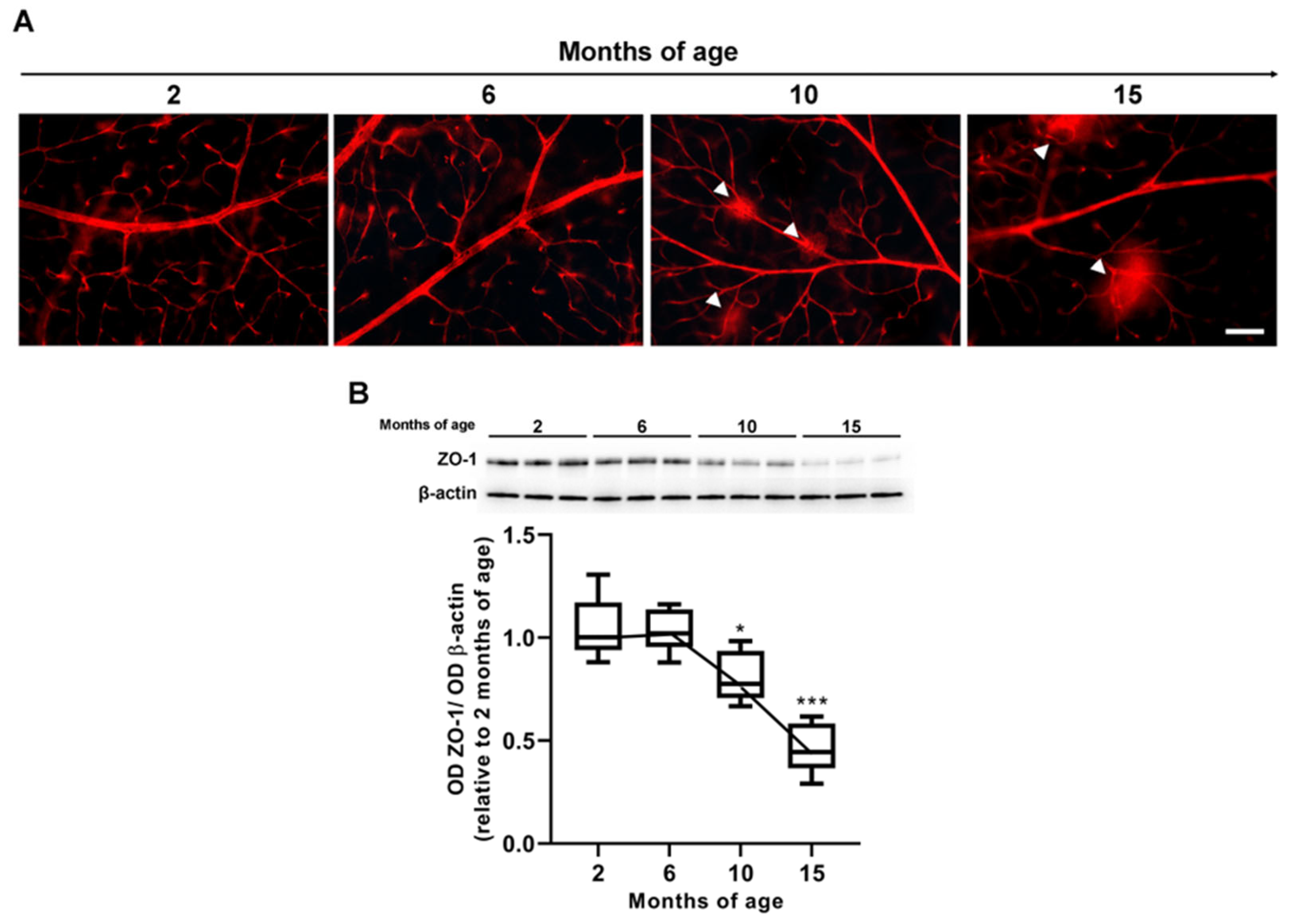
3.3. Age-Dependent Changes in VEGF Levels and Inner BRB Integrity
3.4. Comparison between Time Courses of RGC, IOP and Retinal Factors
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jammal, A.A.; Berchuck, S.I.; Thompson, A.C.; Costa, V.P.; Medeiros, F.A. The Effect of Age on Increasing Susceptibility to Retinal Nerve Fiber Layer Loss in Glaucoma. Investig. Ophthalmol. Vis. Sci. 2020, 61, 8. [Google Scholar] [CrossRef] [PubMed]
- Coleman, A.L.; Miglior, S. Risk factors for glaucoma onset and progression. Surv. Ophthalmol. 2008, 53, S3–S10. [Google Scholar] [CrossRef]
- Casson, R.J. Medical therapy for glaucoma: A review. Clin. Exp. Ophthalmol. 2022, 50, 198–212. [Google Scholar] [CrossRef] [PubMed]
- Chang, B.; Smith, R.S.; Hawes, N.L.; Anderson, M.G.; Zabaleta, A.; Savinova, O.; Roderick, T.H.; Heckenlively, J.R.; Davisson, M.T.; John, S.W. Interacting loci cause severe iris atrophy and glaucoma in DBA/2J mice. Nat. Genet. 1999, 21, 405–409. [Google Scholar] [CrossRef] [PubMed]
- John, S.W.; Smith, R.S.; Savinova, O.V.; Hawes, N.L.; Chang, B.; Turnbull, D.; Davisson, M.; Roderick, T.H.; Heckenlively, J.R. Essential iris atrophy, pigment dispersion, and glaucoma in DBA/2J mice. Investig. Ophthalmol. Vis. Sci. 1998, 39, 951–962, Erratum in Investig. Ophthalmol. Vis. Sci. 1998, 39, 1641. [Google Scholar]
- John, S.W.; Hagaman, J.R.; MacTaggart, T.E.; Peng, L.; Smithes, O. Intraocular pressure in inbred mouse strains. Investig. Ophthalmol. Vis. Sci. 1997, 38, 249–253. [Google Scholar]
- Schlamp, C.L.; Li, Y.; Dietz, J.A.; Janssen, K.T.; Nickells, R.W. Progressive ganglion cell loss and optic nerve degeneration in DBA/2J mice is variable and asymmetric. BMC Neurosci. 2006, 7, 66. [Google Scholar] [CrossRef]
- Libby, R.T.; Anderson, M.G.; Pang, I.H.; Robinson, Z.H.; Savinova, O.V.; Cosma, I.M.; Snow, A.; Wilson, L.A.; Smith, R.S.; Clark, A.F.; et al. Inherited glaucoma in DBA/2J mice: Pertinent disease features for studying the neurodegeneration. Vis. Neurosci. 2005, 22, 637–648. [Google Scholar] [CrossRef]
- Saleh, M.; Nagaraju, M.; Porciatti, V. Longitudinal evaluation of retinal ganglion cell function and IOP in the DBA/2J mouse model of glaucoma. Investig. Ophthalmol. Vis. Sci. 2007, 48, 4564–4572. [Google Scholar] [CrossRef]
- Banitt, M.R.; Ventura, L.M.; Feuer, W.J.; Savatovsky, E.; Luna, G.; Shif, O.; Bosse, B.; Porciatti, V. Progressive loss of retinal ganglion cell function precedes structural loss by several years in glaucoma suspects. Investig. Ophthalmol. Vis. Sci. 2013, 54, 2346–2352. [Google Scholar] [CrossRef]
- Chou, T.H.; Musada, G.R.; Romano, G.L.; Bolton, E.; Porciatti, V. Anesthetic Preconditioning as Endogenous Neuroprotection in Glaucoma. Int. J. Mol. Sci. 2018, 19, 237. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.H.; Park, K.K.; Luo, X.; Porciatti, V. Retrograde signaling in the optic nerve is necessary for electrical responsiveness of retinal ganglion cells. Investig. Ophthalmol. Vis. Sci. 2013, 54, 1236–1243. [Google Scholar] [CrossRef]
- Williams, P.A.; Harder, J.M.; Foxworth, N.E.; Cochran, K.E.; Philip, V.M.; Porciatti, V.; Smithies, O.; John, S.W. Vitamin B3 modulates mitochondrial vulnerability and prevents glaucoma in aged mice. Science. 2017, 355, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Wareham, L.K.; Calkins, D.J. The Neurovascular Unit in Glaucomatous Neurodegeneration. Front. Cell Dev. Biol. 2020, 8, 452. [Google Scholar] [CrossRef] [PubMed]
- Trivli, A.; Koliarakis, I.; Terzidou, C.; Goulielmos, G.N.; Siganos, C.S.; Spandidos, D.A.; Dalianis, G.; Detorakis, E.T. Normal-tension glaucoma: Pathogenesis and genetics. Exp. Ther. Med. 2019, 17, 563–574. [Google Scholar] [CrossRef]
- Ahmad, S.S. Controversies in the vascular theory of glaucomatous optic nerve degeneration. Taiwan. J. Ophthalmol. 2016, 6, 182–186. [Google Scholar] [CrossRef]
- Wang, X.; Jiang, C.; Ko, T.; Kong, X.; Yu, X.; Min, W.; Shi, G.; Sun, X. Correlation between optic disc perfusion and glaucomatous severity in patients with open-angle glaucoma: An optical coherence tomography angiography study. Graefes Arch. Clin. Exp. Ophthalmol. 2015, 253, 1557–1564. [Google Scholar] [CrossRef]
- Pitale, P.M.; Shen, G.; Sigireddi, R.R.; Polo-Prieto, M.; Park, Y.H.; Gibson, S.E.; Westenskow, P.D.; Channa, R.; Frankfort, B.J. Selective vulnerability of the intermediate retinal capillary plexus precedes retinal ganglion cell loss in ocular hypertension. Front. Cell. Neurosci. 2022, 16, 1073786. [Google Scholar] [CrossRef]
- Tribble, J.R.; Otmani, A.; Kokkali, E.; Lardner, E.; Morgan, J.E.; Williams, P.A. Retinal Ganglion Cell Degeneration in a Rat Magnetic Bead Model of Ocular Hypertensive Glaucoma. Transl. Vis. Sci. Technol. 2021, 10, 21. [Google Scholar] [CrossRef]
- Tao, X.; Sigireddi, R.R.; Westenskow, P.D.; Channa, R.; Frankfort, B.J. Single transient intraocular pressure elevations cause prolonged retinal ganglion cell dysfunction and retinal capillary abnormalities in mice. Exp. Eye Res. 2020, 201, 108296. [Google Scholar] [CrossRef]
- Moreno, M.; Ríos, M.C.; Alba, C.; Díaz, F.; Villena, A.; Figueroa-Ortiz, L.C.; García-Campos, J. Morphological and morphometric changes in rat optic nerve microvessels in a glaucoma experimental model. Arch. Soc. Esp. Oftalmol. 2014, 89, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Inman, D.M.; Horner, P.J. Reactive nonproliferative gliosis predominates in a chronic mouse model of glaucoma. Glia 2007, 55, 942–953. [Google Scholar] [CrossRef] [PubMed]
- Wilson, G.N.; Inman, D.M.; Dengler Crish, C.M.; Smith, M.A.; Crish, S.D. Early pro-inflammatory cytokine elevations in the DBA/2J mouse model of glaucoma. J. Neuroinflamm. 2015, 12, 176. [Google Scholar] [CrossRef] [PubMed]
- Quaranta, L.; Bruttini, C.; Micheletti, E.; Konstas, A.G.P.; Michelessi, M.; Oddone, F.; Katsanos, A.; Sbardella, D.; De Angelis, G.; Riva, I. Glaucoma and neuroinflammation: An overview. Surv. Ophthalmol. 2021, 66, 693–713. [Google Scholar] [CrossRef] [PubMed]
- De Hoz, R.; Rojas, B.; Ramírez, A.I.; Salazar, J.J.; Gallego, B.I.; Triviño, A.; Ramírez, J.M. Retinal Macroglial Responses in Health and Disease. Biomed. Res. Int. 2016, 2016, 2954721. [Google Scholar] [CrossRef]
- Fernández-Albarral, J.A.; Salazar, J.J.; de Hoz, R.; Marco, E.M.; Martín-Sánchez, B.; Flores-Salguero, E.; Salobrar-García, E.; López-Cuenca, I.; Barrios-Sabador, V.; Avilés-Trigueros, M.; et al. Retinal Molecular Changes Are Associated with Neuroinflammation and Loss of RGCs in an Experimental Model of Glaucoma. Int. J. Mol. Sci. 2021, 22, 2066. [Google Scholar] [CrossRef]
- Zhou, J.; Chen, F.; Yan, A.; Xia, X. Role of mammalian target of rapamycin in regulating HIF-1α and vascular endothelial growth factor signals in glaucoma. Arch. Physiol. Biochem. 2021, 127, 44–50. [Google Scholar] [CrossRef]
- Zudaire, E.; Gambardella, L.; Kurcz, C.; Vermeren, S. A computational tool for quantitative analysis of vascular networks. PLoS ONE 2011, 6, e27385. [Google Scholar] [CrossRef]
- Buckingham, B.P.; Inman, D.M.; Lambert, W.; Oglesby, E.; Calkins, D.J.; Steele, M.R.; Vetter, M.L.; Marsh-Armstrong, N.; Horner, P.J. Progressive ganglion cell degeneration precedes neuronal loss in a mouse model of glaucoma. J. Neurosci. 2008, 28, 2735–2744. [Google Scholar] [CrossRef]
- Danias, J.; Lee, K.C.; Zamora, M.F.; Chen, B.; Shen, F.; Filippopoulos, T.; Su, Y.; Goldblum, D.; Podos, S.M.; Mittag, T. Quantitative analysis of retinal ganglion cell (RGC) loss in aging DBA/2NNia glaucomatous mice: Comparison with RGC loss in aging C57/BL6 mice. Investig. Ophthalmol. Vis. Sci. 2003, 44, 5151–5162. [Google Scholar] [CrossRef]
- Miao, Y.; Zhao, G.L.; Cheng, S.; Wang, Z.; Yang, X.L. Activation of retinal glial cells contributes to the degeneration of ganglion cells in experimental glaucoma. Prog. Retin. Eye Res. 2023, 93, 101169. [Google Scholar] [CrossRef] [PubMed]
- Vecino, E.; Rodriguez, F.D.; Ruzafa, N.; Pereiro, X.; Sharma, S.C. Glia-neuron interactions in the mammalian retina. Prog. Retin. Eye Res. 2016, 51, 1–40. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.K.W.; Tang, F.; Tham, C.C.Y.; Young, A.L.; Cheung, C.Y. Retinal vasculature in glaucoma: A review. BMJ Open Ophthalmol. 2017, 1, e000032. [Google Scholar] [CrossRef] [PubMed]
- Tezel, G. Molecular regulation of neuroinflammation in glaucoma: Current knowledge and the ongoing search for new treatment targets. Prog. Retin. Eye Res. 2022, 87, 100998. [Google Scholar] [CrossRef]
- Guttenplan, K.A.; Stafford, B.K.; El-Danaf, R.N.; Adler, D.I.; Münch, A.E.; Weigel, M.K.; Huberman, A.D.; Liddelow, S.A. Neurotoxic Reactive Astrocytes Drive Neuronal Death after Retinal Injury. Cell Rep. 2020, 31, 107776. [Google Scholar] [CrossRef]
- Quillen, S.; Schaub, J.; Quigley, H.; Pease, M.; Korneva, A.; Kimball, E. Astrocyte responses to experimental glaucoma in mouse optic nerve head. PLoS ONE 2020, 15, e0238104. [Google Scholar] [CrossRef]
- Bolz, S.; Schuettauf, F.; Fries, J.E.; Thaler, S.; Reichenbach, A.; Pannicke, T. K+ currents fail to change in reactive retinal glial cells in a mouse model of glaucoma. Graefes Arch. Clin. Exp. Ophthalmol. 2008, 246, 1249–1254. [Google Scholar] [CrossRef]
- Jakobs, T.C.; Libby, R.T.; Ben, Y.; John, S.W.; Masland, R.H. Retinal ganglion cell degeneration is topological but not cell type specific in DBA/2J mice. J. Cell Biol. 2005, 171, 313–325. [Google Scholar] [CrossRef]
- Bringmann, A.; Iandiev, I.; Pannicke, T.; Wurm, A.; Hollborn, M.; Wiedemann, P.; Osborne, N.N.; Reichenbach, A. Cellular signaling and factors involved in Müller cell gliosis: Neuroprotective and detrimental effects. Prog. Retin. Eye Res. 2009, 28, 423–451. [Google Scholar] [CrossRef]
- Evangelho, K.; Mogilevskaya, M.; Losada-Barragan, M.; Vargas-Sanchez, J.K. Pathophysiology of primary open-angle glaucoma from a neuroinflammatory and neurotoxicity perspective: A review of the literature. Int. Ophthalmol. 2019, 39, 259–271. [Google Scholar] [CrossRef]
- Lebrun-Julien, F.; Duplan, L.; Pernet, V.; Osswald, I.; Sapieha, P.; Bourgeois, P.; Dickson, K.; Bowie, D.; Barker, P.A.; Di Polo, A. Excitotoxic death of retinal neurons in vivo occurs via a non-cell-autonomous mechanism. J. Neurosci. 2009, 29, 5536–5545. [Google Scholar] [CrossRef]
- Fischer, R.A.; Roux, A.L.; Wareham, L.K.; Sappington, R.M. Pressure-dependent modulation of inward-rectifying K+ channels: Implications for cation homeostasis and K+ dynamics in glaucoma. Am. J. Physiol. Cell. Physiol. 2019, 317, C375–C389. [Google Scholar] [CrossRef] [PubMed]
- Frishman, L.J.; Yamamoto, F.; Bogucka, J.; Steinberg, R.H. Light-evoked changes in [K+]o in proximal portion of light-adapted cat retina. J. Neurophysiol. 1992, 67, 1201–1212. [Google Scholar] [CrossRef] [PubMed]
- Sieving, P.A.; Steinberg, R.H. Contribution from proximal retina to intraretinal pattern ERG: The M-wave. Investig. Ophthalmol. Vis. Sci. 1985, 26, 1642–1647. [Google Scholar]
- Ergorul, C.; Ray, A.; Huang, W.; Wang, D.Y.; Ben, Y.; Cantuti-Castelvetri, I.; Grosskreutz, C.L. Hypoxia inducible factor-1α (HIF-1α) and some HIF-1 target genes are elevated in experimental glaucoma. J. Mol. Neurosci. 2010, 42, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, H.; Yu, A.Y.; Della, N.; Ozaki, K.; Luna, J.D.; Yamada, H.; Hackett, S.F.; Okamoto, N.; Zack, D.J.; Semenza, G.L.; et al. Hypoxia inducible factor-1alpha is increased in ischemic retina: Temporal and spatial correlation with VEGF expression. Investig. Ophthalmol. Vis. Sci. 1999, 40, 182–189. [Google Scholar]
- Pisani, F.; Cammalleri, M.; Dal Monte, M.; Locri, F.; Mola, M.G.; Nicchia, G.P.; Frigeri, A.; Bagnoli, P.; Svelto, M. Potential role of the methylation of VEGF gene promoter in response to hypoxia in oxygen-induced retinopathy: Beneficial effect of the absence of AQP4. J. Cell. Mol. Med. 2018, 22, 613–627. [Google Scholar] [CrossRef] [PubMed]
- Foxton, R.H.; Finkelstein, A.; Vijay, S.; Dahlmann-Noor, A.; Khaw, P.T.; Morgan, J.E.; Shima, D.T.; Ng, Y.S. VEGF-A is necessary and sufficient for retinal neuroprotection in models of experimental glaucoma. Am. J. Pathol. 2013, 182, 1379–1390. [Google Scholar] [CrossRef]
- Wang, J.; Xu, X.; Elliott, M.H.; Zhu, M.; Le, Y.Z. Müller cell-derived VEGF is essential for diabetes-induced retinal inflammation and vascular leakage. Diabetes. 2010, 59, 2297–2305. [Google Scholar] [CrossRef]
- Ou, K.; Mertsch, S.; Theodoropoulou, S.; Wu, J.; Liu, J.; Copland, D.A.; Schrader, S.; Liu, L.; Dick, A.D. Restoring retinal neurovascular health via substance P. Exp. Cell. Res. 2019, 380, 115–123. [Google Scholar] [CrossRef]
- Fudalej, E.; Justyniarska, M.; Kasarełło, K.; Dziedziak, J.; Szaflik, J.P.; Cudnoch-Jędrzejewska, A. Neuroprotective Factors of the Retina and Their Role in Promoting Survival of Retinal Ganglion Cells: A Review. Ophthalmic Res. 2021, 64, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Dimtsas, G.S.; Tsiogka, A.; Moschos, M.M. VEGF levels in the aqueous humor of patients with primary open angle glaucoma: A systematic review and a meta-analysis. Eur. J. Ophthalmol. 2023. advance online publication. [Google Scholar] [CrossRef] [PubMed]
- Qiu, A.W.; Huang, D.R.; Li, B.; Fang, Y.; Zhang, W.W.; Liu, Q.H. IL-17A injury to retinal ganglion cells is mediated by retinal Müller cells in diabetic retinopathy. Cell Death Dis. 2021, 12, 1057. [Google Scholar] [CrossRef] [PubMed]
- Devoldere, J.; Peynshaert, K.; De Smedt, S.C.; Remaut, K. Müller cells as a target for retinal therapy. Drug. Discov. Today 2019, 24, 1483–1498. [Google Scholar] [CrossRef] [PubMed]
- O’Carroll, S.J.; Cook, W.H.; Young, D. AAV Targeting of Glial Cell Types in the Central and Peripheral Nervous System and Relevance to Human Gene Therapy. Front. Mol. Neurosci. 2021, 13, 618020. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, D.; Do, H.; Mazo, K.W.; Chopra, M.; Wahlin, K.J. Restoring vision and rebuilding the retina by Müller glial cell reprogramming. Stem Cell Res. 2023, 66, 103006. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amato, R.; Cammalleri, M.; Melecchi, A.; Bagnoli, P.; Porciatti, V. Natural History of Glaucoma Progression in the DBA/2J Model: Early Contribution of Müller Cell Gliosis. Cells 2023, 12, 1272. https://doi.org/10.3390/cells12091272
Amato R, Cammalleri M, Melecchi A, Bagnoli P, Porciatti V. Natural History of Glaucoma Progression in the DBA/2J Model: Early Contribution of Müller Cell Gliosis. Cells. 2023; 12(9):1272. https://doi.org/10.3390/cells12091272
Chicago/Turabian StyleAmato, Rosario, Maurizio Cammalleri, Alberto Melecchi, Paola Bagnoli, and Vittorio Porciatti. 2023. "Natural History of Glaucoma Progression in the DBA/2J Model: Early Contribution of Müller Cell Gliosis" Cells 12, no. 9: 1272. https://doi.org/10.3390/cells12091272
APA StyleAmato, R., Cammalleri, M., Melecchi, A., Bagnoli, P., & Porciatti, V. (2023). Natural History of Glaucoma Progression in the DBA/2J Model: Early Contribution of Müller Cell Gliosis. Cells, 12(9), 1272. https://doi.org/10.3390/cells12091272