
Pathogenesis of Alkali Injury-Induced Limbal Stem Cell Deficiency: A Literature Survey of Animal Models
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
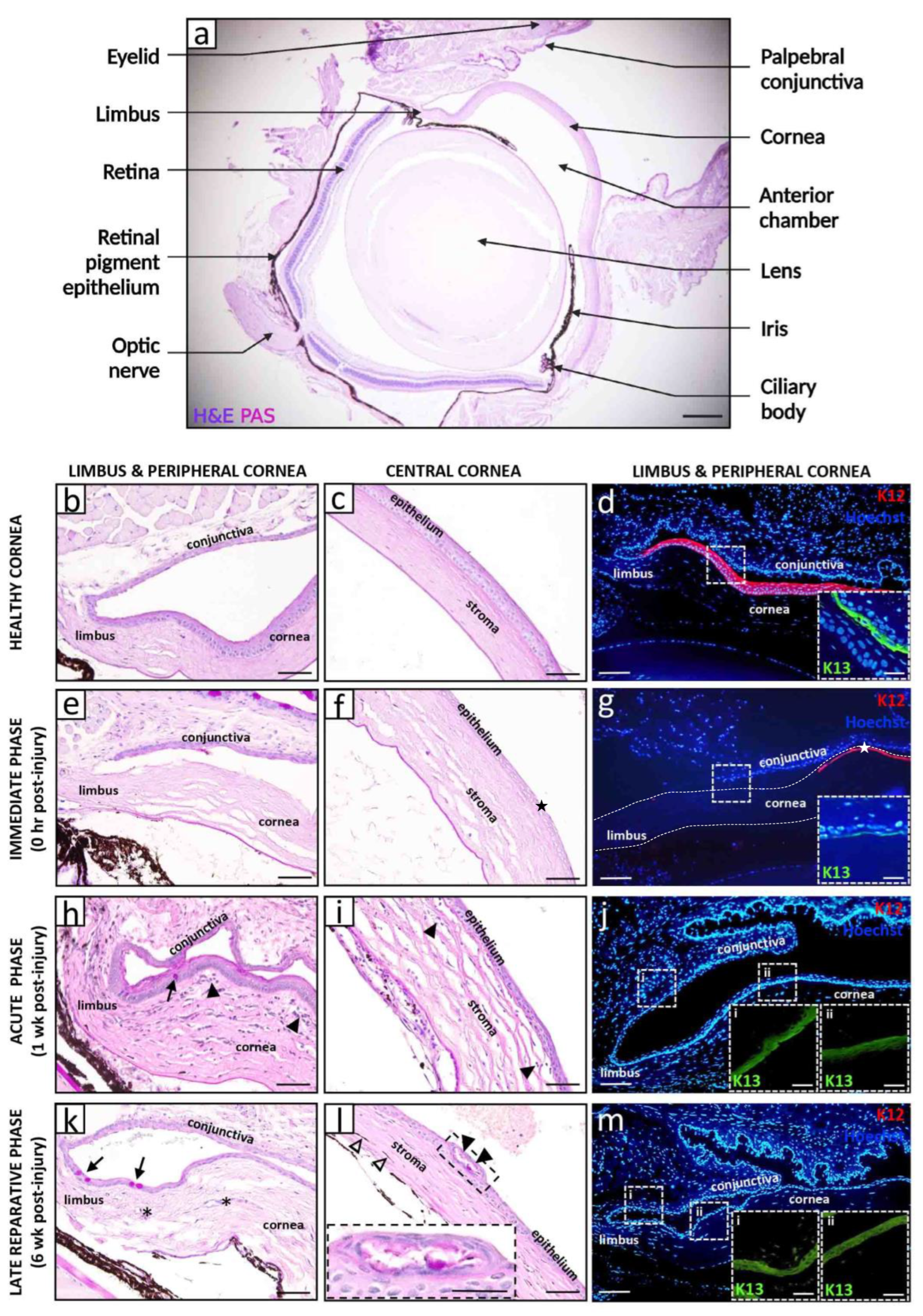
2. Models of Corneal Alkali Injury and LSCD
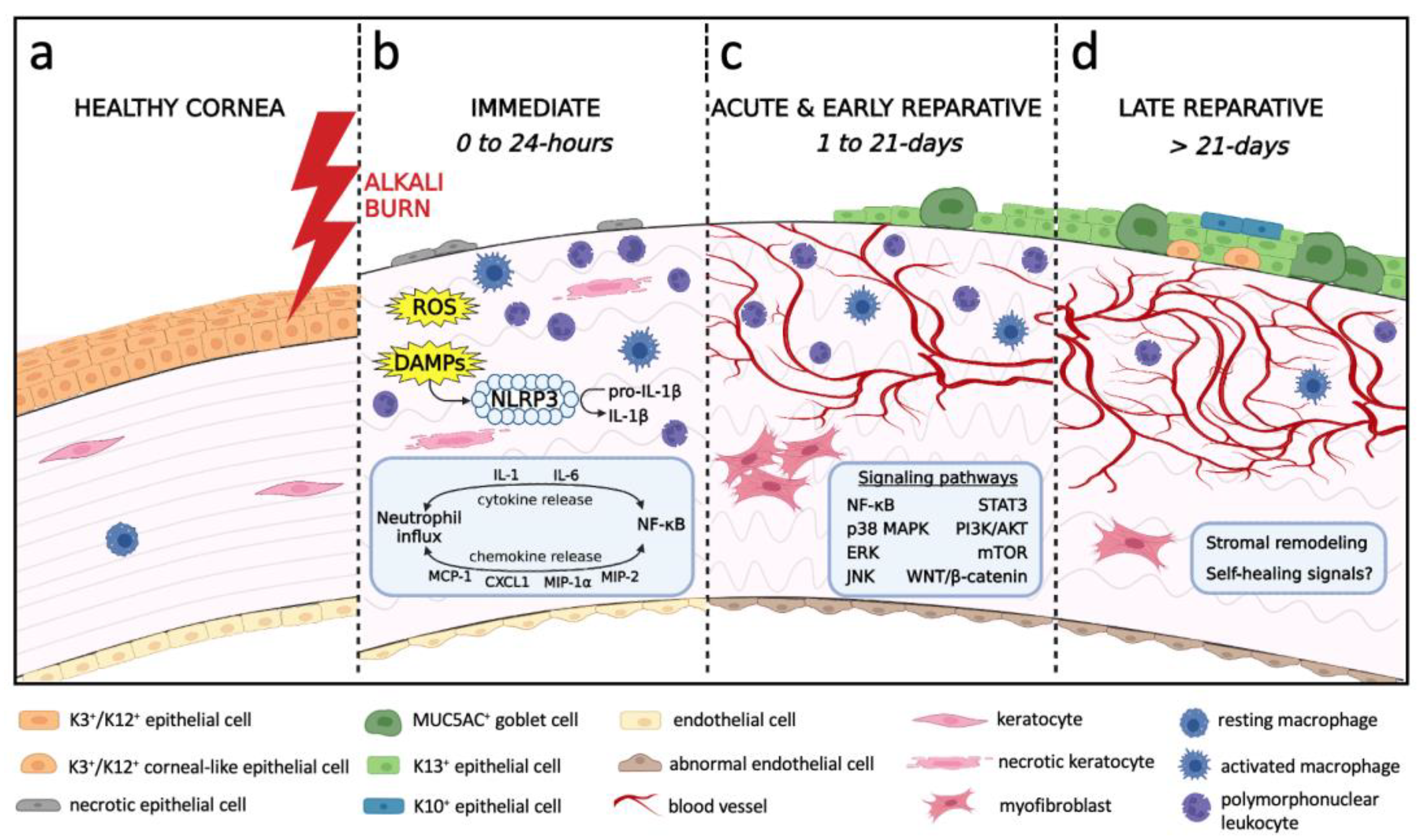
3. Natural History and Pathophysiology of Alkali-Induced LSCD
3.1. Immediate Phase and Induction of Inflammation
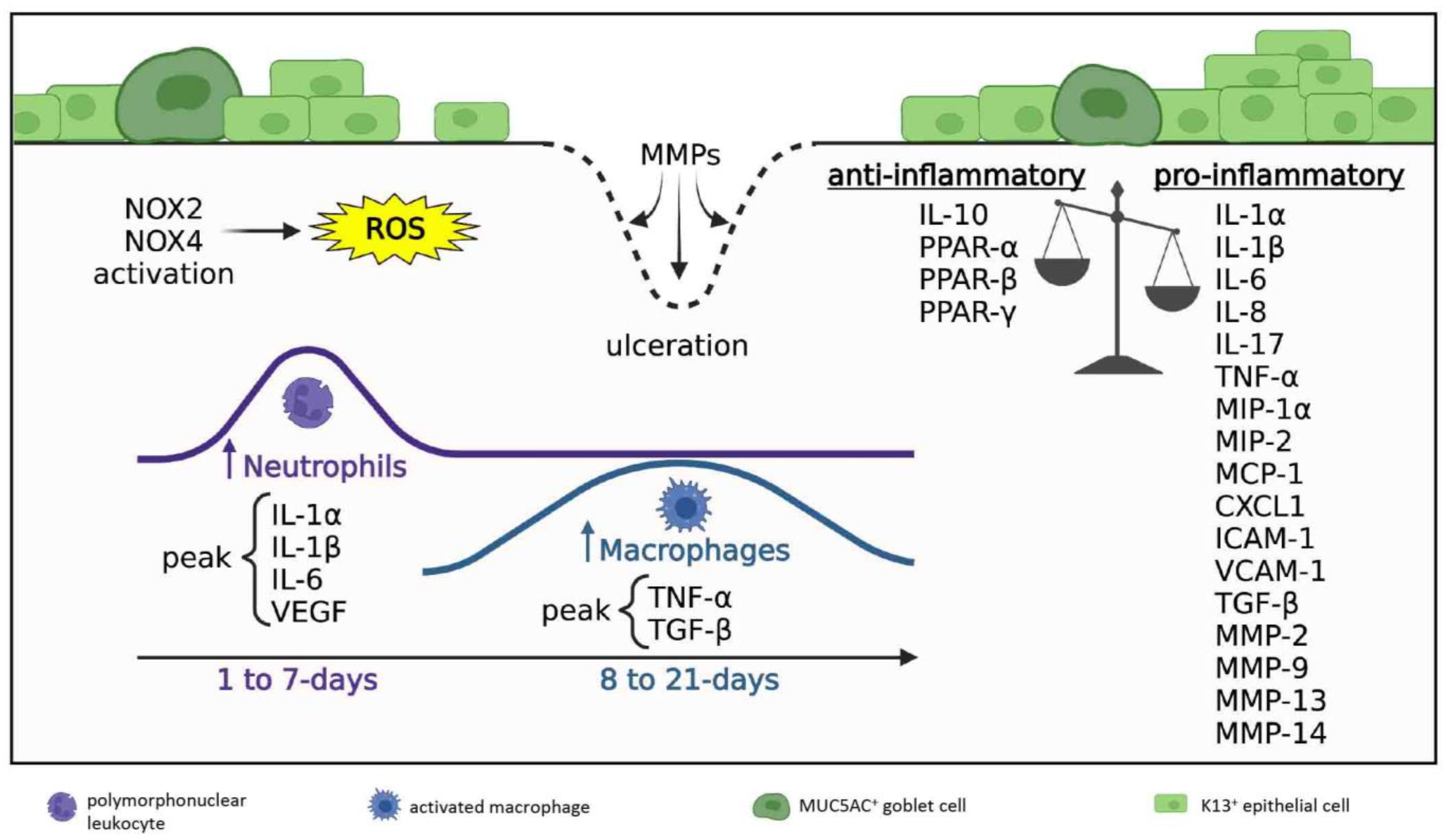
3.2. Acute and Early Reparative Phases
3.2.1. Inflammatory Milieu
3.2.2. Neovascularization
3.2.3. Re-Epithelialization
3.2.4. Innervation
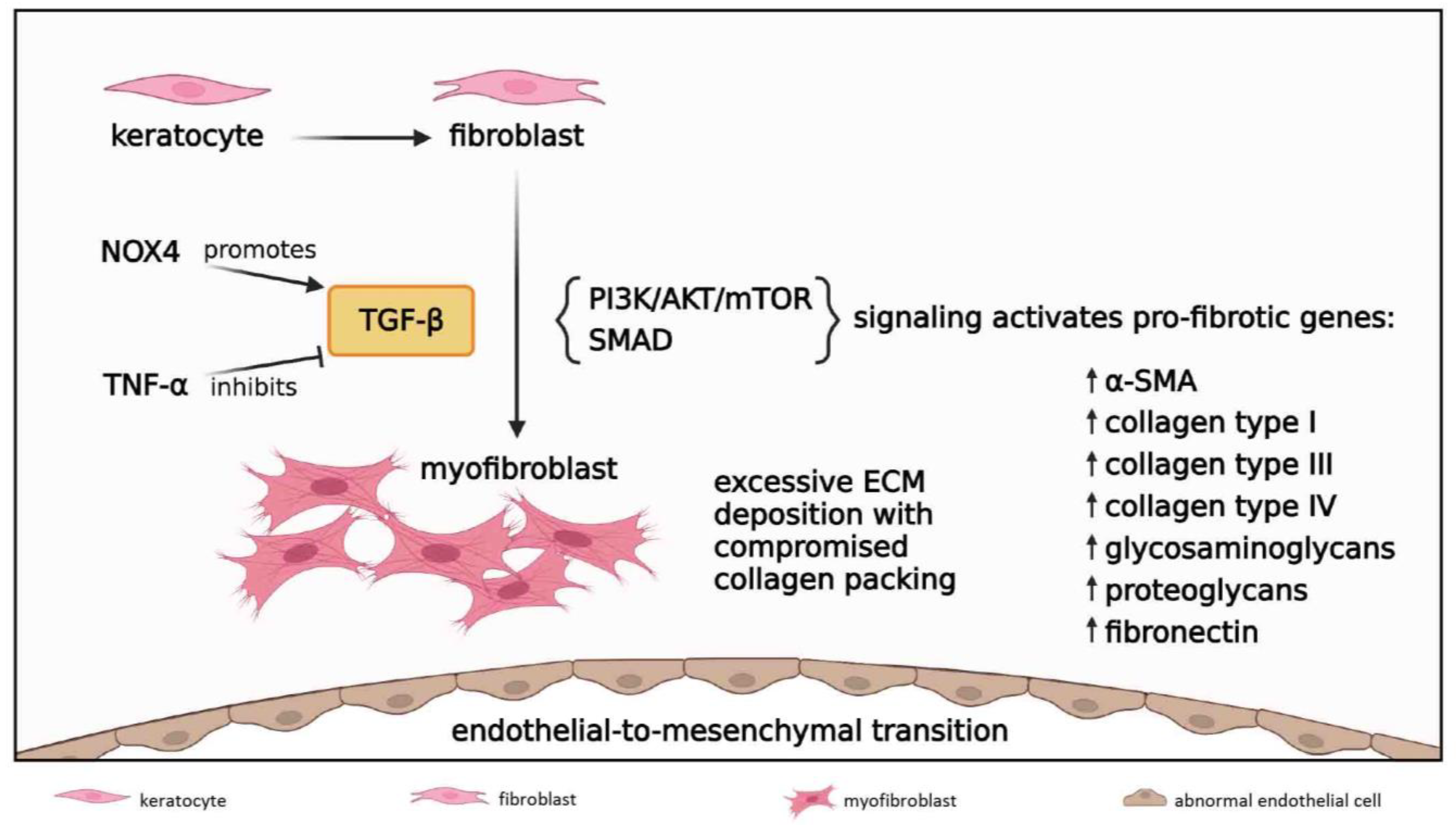
3.2.5. Fibrosis
3.3. Late Reparative Phase
4. Conclusions and Future Directions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ADAMTS | anti-angiogenic disintegrin and metalloproteinase with thrombospondin motifs |
| AKT | protein kinase B |
| α-SMA | α-smooth muscle actin |
| BDNF | brain-derived neurotrophic factor |
| BM | basement membrane |
| CAB | central alkali burn |
| CGRP | calcitonin gene-related peptide |
| CNV | corneal neovascularization |
| CXCL | C-X-C motif ligand |
| DAMP | danger-associated molecular pattern |
| ECM | extracellular matrix |
| EGF | epidermal growth factor |
| ERK | extracellular signal-regulated kinase |
| FGF | fibroblast growth factor |
| GC | goblet cell |
| GDNF | glial cell line-derived neurotrophic factor |
| GF | growth factor |
| ICAM | intercellular adhesion molecule |
| ICBN | intraepithelial corneal basal nerve |
| IL | interleukin |
| IL-1RI | IL-1 receptor I |
| JNK | c-Jun N-terminal kinase |
| K | cytokeratin |
| KGF | keratinocyte growth factor |
| LAB | limbal alkali burn |
| LESC | limbal epithelial stem cell |
| LNC | limbal niche cell |
| LRG1 | leucine-rich α-2-glycoprotein-1 |
| LSCD | limbal stem cell deficiency |
| MAPK | mitogen-activated protein kinase |
| MCP | monocyte chemoattractant protein |
| MSC | mesenchymal stem cell |
| MIP | macrophage inflammatory protein |
| MK2 | mitogen-activated protein kinase–activated protein kinase-2 |
| MMP | matrix metalloproteinase |
| MUC | mucin |
| mTOR | mammalian target of rapamycin |
| NF-κB | nuclear factor-kappa B |
| NGF | nerve growth factor |
| NK-1R | neurokinin-1 receptor |
| NLRP3 | nucleotide-binding oligomerization domain, leucine-rich repeat and pyrin domain containing protein 3 |
| NOX | NADPH oxidases |
| NT | neurotrophin |
| PDGF | platelet-derived growth factor |
| PEDF | pigment epithelium-derived growth factor |
| PI3K | phosphoinositide 3-kinase |
| PPAR | peroxisome proliferator-activated receptor |
| ROS | reactive oxygen species |
| SDF-1α | stromal derived factor-1α |
| SLURP1 | secreted Ly6/uPAR related protein-1 |
| STAT3 | signal transducer and activator of transcription 3 |
| TGF | transforming growth factor |
| TNF | tumor necrosis factor |
| TSP | thrombospondin |
| VCAM | vascular cell adhesion molecule |
| VEGF | vascular endothelial growth factor |
| VEGFR | vascular endothelial growth factor receptor |
| YAP | yes-associated protein |
References
- Di Girolamo, N. Moving epithelia: Tracking the fate of mammalian limbal epithelial stem cells. Prog. Retin. Eye Res. 2015, 48, 203–225. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.X.; Borderie, V.; Chan, C.C.; Dana, R.; Figueiredo, F.C.; Gomes, J.A.P.; Pellegrini, G.; Shimmura, S.; Kruse, F.E.; The International Limbal Stem Cell Deficiency Working Group. Global consensus on definition, classification, diagnosis, and staging of limbal stem cell deficiency. Cornea 2019, 38, 364–375. [Google Scholar] [CrossRef] [PubMed]
- Dua, H.S.; Ting, D.S.J.; Al Saadi, A.; Said, D.G. Chemical eye injury: Pathophysiology, assessment and management. Eye 2020, 34, 2001–2019. [Google Scholar] [CrossRef]
- Kethiri, A.R.; Singh, V.K.; Damala, M.; Basu, S.; Rao, C.M.; Bokara, K.K.; Singh, V. Long term observation of ocular surface alkali burn in rabbit models: Quantitative analysis of corneal haze, vascularity and self-recovery. Exp. Eye Res. 2021, 205, 108526. [Google Scholar] [CrossRef] [PubMed]
- Saghizadeh, M.; Kramerov, A.A.; Svendsen, C.N.; Ljubimov, A.V. Concise Review: Stem Cells for Corneal Wound Healing. Stem Cells 2017, 35, 2105–2114. [Google Scholar] [CrossRef]
- Deng, S.X.; Kruse, F.; Gomes, J.A.P.; Chan, C.C.; Daya, S.; Dana, R.; Figueiredo, F.C.; Kinoshita, S.; Rama, P.; Sangwan, V.; et al. Global consensus on the management of limbal stem cell deficiency. Cornea 2020, 39, 1291–1302. [Google Scholar] [CrossRef]
- Wilson, S.E. Corneal wound healing. Exp. Eye Res. 2020, 197, 108089. [Google Scholar] [CrossRef] [PubMed]
- Gulias-Canizo, R.; Lagunes-Guillen, A.; Gonzalez-Robles, A.; Sanchez-Guzman, E.; Castro-Munozledo, F. (-)-Epigallocatechin-3-gallate, reduces corneal damage secondary from experimental grade II alkali burns in mice. Burns 2019, 45, 398–412. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, M.; Shimizu, A.; Masuda, Y.; Nagasaka, S.; Fukuda, Y.; Takahashi, H. An ophthalmic solution of a peroxisome proliferator-activated receptor gamma agonist prevents corneal inflammation in a rat alkali burn model. Mol. Vis. 2013, 19, 2135–2150. [Google Scholar] [PubMed]
- Nakano, Y.; Uchiyama, M.; Arima, T.; Nagasaka, S.; Igarashi, T.; Shimizu, A.; Takahashi, H. PPARalpha agonist suppresses inflammation after corneal alkali burn by suppressing proinflammatory cytokines, MCP-1, and nuclear translocation of NF-kappaB. Molecules 2018, 24, 114. [Google Scholar] [CrossRef]
- Yi, Q.; Zou, W.J. The wound healing effect of doxycycline after corneal alkali burn in rats. J. Ophthalmol. 2019, 2019, 5168652. [Google Scholar] [CrossRef]
- Saika, S.; Miyamoto, T.; Yamanaka, O.; Kato, T.; Ohnishi, Y.; Flanders, K.C.; Ikeda, K.; Nakajima, Y.; Kao, W.W.; Sato, M.; et al. Therapeutic effect of topical administration of SN50, an inhibitor of nuclear factor-kappaB, in treatment of corneal alkali burns in mice. Am. J. Pathol. 2005, 166, 1393–1403. [Google Scholar] [CrossRef]
- Shin, Y.J.; Hyon, J.Y.; Choi, W.S.; Yi, K.; Chung, E.S.; Chung, T.Y.; Wee, W.R. Chemical injury-induced corneal opacity and neovascularization reduced by rapamycin via TGF-beta1/ERK pathways regulation. Investig. Ophthalmol. Vis. Sci. 2013, 54, 4452–4458. [Google Scholar] [CrossRef]
- Zhang, Q.Y.; Tao, S.Y.; Lu, C.; Li, J.J.; Li, X.M.; Yao, J.; Jiang, Q.; Yan, B. SKLB1002, a potent inhibitor of VEGF receptor 2 signaling, inhibits endothelial angiogenic function in vitro and ocular angiogenesis in vivo. Mol. Med. Rep. 2020, 21, 2571–2579. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Shu, Y.; Yin, L.; Xie, T.; Zou, J.; Zhan, P.; Wang, Y.; Wei, T.; Zhu, L.; Yang, X.; et al. Protective roles of the TIR/BB-loop mimetic AS-1 in alkali-induced corneal neovascularization by inhibiting ERK phosphorylation. Exp. Eye Res. 2021, 207, 108568. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, W.; Zhang, X.; Yang, S.; Peng, G.; Wu, T.; Zhou, Y.; Huang, C.; Reinach, P.S.; Li, W.; et al. MK2 inhibitor reduces alkali burn-induced inflammation in rat cornea. Sci. Rep. 2016, 6, 28145. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.; Zhou, X.Z.; Ye, L.; Yuan, Q.; Shi, C.; Zhu, P.W.; Jiang, N.; Ma, M.Y.; Yang, Q.C.; Shao, Y. Xanthatin inhibits corneal neovascularization by inhibiting the VEGFR2mediated STAT3/PI3K/Akt signaling pathway. Int. J. Mol. Med. 2018, 42, 769–778. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Jeong, H.; Yang, M.S.; Lim, C.W.; Kim, B. Therapeutic effects of zerumbone in an alkali-burned corneal wound healing model. Int. Immunopharmacol. 2017, 48, 126–134. [Google Scholar] [CrossRef]
- Wang, Y.; Gao, Y.; Huang, Y.; Pan, Y.; Yu, Y.; Zhou, Y.; Wan, S.S.; Yang, Y.N. The potential protective effects of miR-497 on corneal neovascularization are mediated via macrophage through the IL-6/STAT3/VEGF signaling pathway. Int. Immunopharmacol. 2021, 96, 107745. [Google Scholar] [CrossRef]
- Li, J.; Du, S.; Shi, Y.; Han, J.; Niu, Z.; Wei, L.; Yang, P.; Chen, L.; Tian, H.; Gao, L. Rapamycin ameliorates corneal injury after alkali burn through methylation modification in mouse TSC1 and mTOR genes. Exp. Eye Res. 2021, 203, 108399. [Google Scholar] [CrossRef]
- Wang, Y.; Gao, G.; Wu, Y.; Wang, Y.; Wu, X.; Zhou, Q. S100A4 silencing facilitates corneal wound healing after alkali burns by promoting autophagy via blocking the PI3K/Akt/mTOR signaling pathway. Investig. Ophthalmol. Vis. Sci. 2020, 61, 19. [Google Scholar] [CrossRef]
- Qiu, F.; Shin, Y.; Chen, D.; Cheng, R.; Chen, Q.; Zhou, K.; Larrick, J.W.; Mendelson, A.R.; Ma, J.X. Anti-angiogenic effect of a humanized antibody blocking the Wnt/beta-catenin signaling pathway. Microvasc. Res. 2018, 119, 29–37. [Google Scholar] [CrossRef]
- Khorolskaya, J.I.; Perepletchikova, D.A.; Kachkin, D.V.; Zhurenkov, K.E.; Alexander-Sinkler, E.I.; Ivanova, J.S.; Mikhailova, N.A.; Blinova, M.I. Derivation and characterization of EGFP-labeled rabbit limbal mesenchymal stem cells and their potential for research in regenerative ophthalmology. Biomedicines 2021, 9, 1134. [Google Scholar] [CrossRef]
- Altshuler, A.; Amitai-Lange, A.; Tarazi, N.; Dey, S.; Strinkovsky, L.; Hadad-Porat, S.; Bhattacharya, S.; Nasser, W.; Imeri, J.; Ben-David, G.; et al. Discrete limbal epithelial stem cell populations mediate corneal homeostasis and wound healing. Cell Stem Cell 2021, 28, 1248–1261.e1248. [Google Scholar] [CrossRef]
- Polisetti, N.; Giessl, A.; Zenkel, M.; Heger, L.; Dudziak, D.; Naschberger, E.; Stich, L.; Steinkasserer, A.; Kruse, F.E.; Schlotzer-Schrehardt, U. Melanocytes as emerging key players in niche regulation of limbal epithelial stem cells. Ocul. Surf. 2021, 22, 172–189. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Zhang, Y.; Cai, S.; Sun, M.; Wang, J.; Li, S.; Li, X.; Tighe, S.; Chen, S.; Xie, H.; et al. Human limbal niche cells are a powerful regenerative source for the prevention of limbal stem cell deficiency in a rabbit model. Sci. Rep. 2018, 8, 6566. [Google Scholar] [CrossRef] [PubMed]
- Delic, N.C.; Cai, J.R.; Watson, S.L.; Downie, L.E.; Di Girolamo, N. Evaluating the clinical translational relevance of animal models for limbal stem cell deficiency: A systematic review. Ocul. Surf. 2022, 23, 169–183. [Google Scholar] [CrossRef]
- McCulley, J. Chemical injuries. In The Cornea: Scientific Foundation and Clinical Practice; Little Brown and Co.: Boston, MA, USA, 1987; pp. 527–542. [Google Scholar]
- Choi, H.; Phillips, C.; Oh, J.Y.; Stock, E.M.; Kim, D.K.; Won, J.K.; Fulcher, S. Comprehensive modeling of corneal alkali injury in the rat eye. Curr. Eye Res. 2017, 42, 1348–1357. [Google Scholar] [CrossRef] [PubMed]
- Tobita, Y.; Arima, T.; Nakano, Y.; Uchiyama, M.; Shimizu, A.; Takahashi, H. Effects of selective peroxisome proliferator activated receptor agonists on corneal epithelial wound healing. Pharmaceuticals 2021, 14, 88. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.E.; McMullen, T.C.; Campbell, I.L.; Rohan, R.; Kaji, Y.; Afshari, N.A.; Usui, T.; Archer, D.B.; Adamis, A.P. The inflammatory milieu associated with conjunctivalized cornea and its alteration with IL-1 RA gene therapy. Investig. Ophthalmol. Vis. Sci. 2002, 43, 2905–2915. [Google Scholar]
- Kubota, M.; Shimmura, S.; Kubota, S.; Miyashita, H.; Kato, N.; Noda, K.; Ozawa, Y.; Usui, T.; Ishida, S.; Umezawa, K.; et al. Hydrogen and N-acetyl-L-cysteine rescue oxidative stress-induced angiogenesis in a mouse corneal alkali-burn model. Investig. Ophthalmol. Vis. Sci. 2011, 52, 427–433. [Google Scholar] [CrossRef]
- Kabe, Y.; Ando, K.; Hirao, S.; Yoshida, M.; Handa, H. Redox regulation of NF-kappaB activation: Distinct redox regulation between the cytoplasm and the nucleus. Antioxid Redox Signal. 2005, 7, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-kappaB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Yu, H.; Lin, L.; Zhang, Z.; Zhang, H.; Hu, H. Targeting NF-kappaB pathway for the therapy of diseases: Mechanism and clinical study. Signal Transduct. Target. Ther. 2020, 5, 209. [Google Scholar] [CrossRef]
- Oguido, A.; Hohmann, M.S.N.; Pinho-Ribeiro, F.A.; Crespigio, J.; Domiciano, T.P.; Verri, W.A., Jr.; Casella, A.M.B. Naringenin eye drops inhibit corneal neovascularization by anti-inflammatory and antioxidant mechanisms. Investig. Ophthalmol. Vis. Sci. 2017, 58, 5764–5776. [Google Scholar] [CrossRef]
- Sotozono, C.; He, J.; Matsumoto, Y.; Kita, M.; Imanishi, J.; Kinoshita, S. Cytokine expression in the alkali-burned cornea. Curr. Eye Res. 1997, 16, 670–676. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Jeong, H.J.; Kim, M.K.; Wee, W.R. Bone marrow-derived mesenchymal stem cells affect immunologic profiling of interleukin-17-secreting cells in a chemical burn mouse model. Korean J. Ophthalmol. 2014, 28, 246–256. [Google Scholar] [CrossRef] [PubMed]
- Bian, F.; Xiao, Y.; Zaheer, M.; Volpe, E.A.; Pflugfelder, S.C.; Li, D.Q.; de Paiva, C.S. Inhibition of NLRP3 inflammasome pathway by butyrate improves corneal wound healing in corneal alkali burn. Int. J. Mol. Sci. 2017, 18, 562. [Google Scholar] [CrossRef]
- Shimizu, H.; Sakimoto, T.; Yamagami, S. Pro-inflammatory role of NLRP3 inflammasome in experimental sterile corneal inflammation. Sci. Rep. 2019, 9, 9596. [Google Scholar] [CrossRef] [PubMed]
- Arima, T.; Uchiyama, M.; Nakano, Y.; Nagasaka, S.; Kang, D.; Shimizu, A.; Takahashi, H. Peroxisome proliferator-activated receptor alpha agonist suppresses neovascularization by reducing both vascular endothelial growth factor and angiopoietin-2 in corneal alkali burn. Sci. Rep. 2017, 7, 17763. [Google Scholar] [CrossRef]
- Xiao, O.; Xie, Z.L.; Lin, B.W.; Yin, X.F.; Pi, R.B.; Zhou, S.Y. Minocycline inhibits alkali burn-induced corneal neovascularization in mice. PLoS ONE 2012, 7, e41858. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Li, L.; Kuno, K.; Wu, Y.; Baba, T.; Li, Y.Y.; Zhang, X.; Mukaida, N. Protective roles of the fractalkine/CX3CL1-CX3CR1 interactions in alkali-induced corneal neovascularization through enhanced antiangiogenic factor expression. J. Immunol. 2008, 180, 4283–4291. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.W.; Lee, S.M.; Oh, K.H.; Park, S.G.; Choi, I.W.; Seo, S.K. Effects of topical chondrocyte-derived extracellular matrix treatment on corneal wound healing, following an alkali burn injury. Mol. Med. Rep. 2015, 11, 461–467. [Google Scholar] [CrossRef]
- Lu, P.; Li, L.; Mukaida, N.; Zhang, X. Alkali-induced corneal neovascularization is independent of CXCR2-mediated neutrophil infiltration. Cornea 2007, 26, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Shao, Y.; Lin, Z.; Qu, Y.L.; Wang, H.; Zhou, Y.; Chen, W.; Chen, Y.; Chen, W.L.; Hu, F.R.; et al. Netrin-1 simultaneously suppresses corneal inflammation and neovascularization. Investig. Ophthalmol. Vis. Sci. 2012, 53, 1285–1295. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Long, D.; Hsu, C.C.; Liu, H.; Chen, L.; Slavin, B.; Lin, H.; Li, X.; Tang, J.; Yiu, S.; et al. Nanofiber-reinforced decellularized amniotic membrane improves limbal stem cell transplantation in a rabbit model of corneal epithelial defect. Acta Biomater 2019, 97, 310–320. [Google Scholar] [CrossRef]
- Lee, C.M.; Jung, W.K.; Na, G.; Lee, D.S.; Park, S.G.; Seo, S.K.; Yang, J.W.; Yea, S.S.; Lee, Y.M.; Park, W.S.; et al. Inhibitory effects of the platelet-activating factor receptor antagonists, CV-3988 and Ginkgolide B, on alkali burn-induced corneal neovascularization. Cutan. Ocul. Toxicol. 2015, 34, 53–60. [Google Scholar] [CrossRef]
- Chen, L.; Zhong, J.; Li, S.; Li, W.; Wang, B.; Deng, Y.; Yuan, J. The long-term effect of tacrolimus on alkali burn-induced corneal neovascularization and inflammation surpasses that of anti-vascular endothelial growth factor. Drug Des. Devel. Ther. 2018, 12, 2959–2969. [Google Scholar] [CrossRef]
- Lu, P.; Li, L.; Wu, Y.; Mukaida, N.; Zhang, X. Essential contribution of CCL3 to alkali-induced corneal neovascularization by regulating vascular endothelial growth factor production by macrophages. Mol. Vis. 2008, 14, 1614–1622. [Google Scholar]
- Hakami, N.Y.; Dusting, G.J.; Chan, E.C.; Shah, M.H.; Peshavariya, H.M. Wound healing after alkali burn injury of the cornea involves Nox4-type NADPH oxidase. Investig. Ophthalmol. Vis. Sci. 2020, 61, 20. [Google Scholar] [CrossRef]
- Kim, D.H.; Im, S.T.; Yoon, J.Y.; Kim, S.; Kim, M.K.; Chung, M.H.; Park, C.K. Comparison of therapeutic effects between topical 8-oxo-2’-deoxyguanosine and corticosteroid in ocular alkali burn model. Sci. Rep. 2021, 11, 6909. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.J.; Liu, X.; Chen, Y.Y.; Zhao, Y.; Xu, M.; Han, X.J.; Liu, Q.P.; Yi, J.L.; Li, J.M. Involvement of NADPH oxidases in alkali burn-induced corneal injury. Int. J. Mol. Med. 2016, 38, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Di, G.H.; Qi, X.; Xu, J.; Yu, C.Q.; Cao, Q.L.; Xing, Z.J.; Li, Z.C. Therapeutic effect of secretome from TNF-alpha stimulated mesenchymal stem cells in an experimental model of corneal limbal stem cell deficiency. Int J. Ophthalmol. 2021, 14, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Joussen, A.M.; Beecken, W.D.; Moromizato, Y.; Schwartz, A.; Kirchhof, B.; Poulaki, V. Inhibition of inflammatory corneal angiogenesis by TNP-470. Investig. Ophthalmol. Vis. Sci. 2001, 42, 2510–2516. [Google Scholar]
- Lu, P.; Li, L.; Liu, G.; van Rooijen, N.; Mukaida, N.; Zhang, X. Opposite roles of CCR2 and CX3CR1 macrophages in alkali-induced corneal neovascularization. Cornea 2009, 28, 562–569. [Google Scholar] [CrossRef]
- Jiang, L.; He, W.; Tang, F.; Tang, N.; Huang, G.; Huang, W.; Wu, X.; Guan, J.; Zeng, S.; Li, M.; et al. Epigenetic landscape analysis of the long non-coding RNA and messenger RNA in a mouse model of corneal alkali burns. Investig. Ophthalmol. Vis. Sci. 2021, 62, 28. [Google Scholar] [CrossRef] [PubMed]
- Joussen, A.M.; Poulaki, V.; Mitsiades, N.; Stechschulte, S.U.; Kirchhof, B.; Dartt, D.A.; Fong, G.H.; Rudge, J.; Wiegand, S.J.; Yancopoulos, G.D.; et al. VEGF-dependent conjunctivalization of the corneal surface. Investig. Ophthalmol. Vis. Sci. 2003, 44, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, M.W.; Xu, J.J.; Wu, X.Y.; Yao, J. Gelatin methacryloyl hydrogel eye pad loaded with amniotic extract prevents symblepharon in rabbit eyes. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 10134–10142. [Google Scholar] [CrossRef]
- Muether, P.S.; Dell, S.; Kociok, N.; Zahn, G.; Stragies, R.; Vossmeyer, D.; Joussen, A.M. The role of integrin alpha5beta1 in the regulation of corneal neovascularization. Exp. Eye Res. 2007, 85, 356–365. [Google Scholar] [CrossRef]
- Bignami, F.; Lorusso, A.; Rama, P.; Ferrari, G. Growth inhibition of formed corneal neovascularization following Fosaprepitant treatment. Acta Ophthalmol. 2017, 95, e641–e648. [Google Scholar] [CrossRef]
- Poon, M.W.; Jiang, D.; Qin, P.; Zhang, Y.; Qiu, B.; Chanda, S.; Tergaonkar, V.; Li, Q.; Wong, I.Y.; Yu, Z.; et al. Inhibition of NUCKS facilitates corneal recovery following alkali burn. Sci. Rep. 2017, 7, 41224. [Google Scholar] [CrossRef]
- Han, Y.; Shao, Y.; Liu, T.; Qu, Y.L.; Li, W.; Liu, Z. Therapeutic effects of topical netrin-4 inhibits corneal neovascularization in alkali-burn rats. PLoS ONE 2015, 10, e0122951. [Google Scholar] [CrossRef] [PubMed]
- Swamynathan, S.; Loughner, C.L.; Swamynathan, S.K. Inhibition of HUVEC tube formation via suppression of NFkappaB suggests an anti-angiogenic role for SLURP1 in the transparent cornea. Exp. Eye Res. 2017, 164, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Cheng, J.; Yu, B.J.; Zhou, L.; Xu, H.F.; Yang, L.L. LRG1 promotes corneal angiogenesis and lymphangiogenesis in a corneal alkali burn mouse model. Int. J. Ophthalmol. 2020, 13, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Wu, H.; Ren, C.; Liu, G.; Zhang, W.; Liu, W.; Lu, P. Inhibition of PDGF-BB reduces alkali-induced corneal neovascularization in mice. Mol. Med. Rep. 2021, 23, 238. [Google Scholar] [CrossRef]
- Liu, G.; Lu, P.; Li, L.; Jin, H.; He, X.; Mukaida, N.; Zhang, X. Critical role of SDF-1alpha-induced progenitor cell recruitment and macrophage VEGF production in the experimental corneal neovascularization. Mol. Vis. 2011, 17, 2129–2138. [Google Scholar] [PubMed]
- Lim, P.; Fuchsluger, T.A.; Jurkunas, U.V. Limbal stem cell deficiency and corneal neovascularization. Semin. Ophthalmol. 2009, 24, 139–148. [Google Scholar] [CrossRef]
- Wang, X.; Tang, L.; Zhang, Z.; Li, W.; Chen, Y. Keratocytes promote corneal neovascularization through VEGFr3 induced by PPARalpha-inhibition. Exp. Eye Res. 2020, 193, 107982. [Google Scholar] [CrossRef]
- Ma, J.; Zhou, D.; Fan, M.; Wang, H.; Huang, C.; Zhang, Z.; Wu, Y.; Li, W.; Chen, Y.; Liu, Z. Keratocytes create stromal spaces to promote corneal neovascularization via MMP13 expression. Investig. Ophthalmol. Vis. Sci. 2014, 55, 6691–6703. [Google Scholar] [CrossRef]
- Su, W.; Li, Z.; Li, Y.; Lin, M.; Yao, L.; Liu, Y.; He, Z.; Wu, C.; Liang, D. Doxycycline enhances the inhibitory effects of bevacizumab on corneal neovascularization and prevents its side effects. Investig. Ophthalmol. Vis. Sci. 2011, 52, 9108–9115. [Google Scholar] [CrossRef]
- Fujita, K.; Miyamoto, T.; Saika, S. Sonic hedgehog: Its expression in a healing cornea and its role in neovascularization. Mol. Vis. 2009, 15, 1036–1044. [Google Scholar] [PubMed]
- Ishii, R.; Yanagisawa, H.; Sada, A. Defining compartmentalized stem cell populations with distinct cell division dynamics in the ocular surface epithelium. Development 2020, 147, dev197590. [Google Scholar] [CrossRef]
- Nasser, W.; Amitai-Lange, A.; Soteriou, D.; Hanna, R.; Tiosano, B.; Fuchs, Y.; Shalom-Feuerstein, R. Corneal-committed cells restore the stem cell pool and tissue boundary following injury. Cell Rep. 2018, 22, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Gouveia, R.M.; Lepert, G.; Gupta, S.; Mohan, R.R.; Paterson, C.; Connon, C.J. Assessment of corneal substrate biomechanics and its effect on epithelial stem cell maintenance and differentiation. Nat. Commun. 2019, 10, 1496. [Google Scholar] [CrossRef]
- Saika, S.; Ikeda, K.; Yamanaka, O.; Flanders, K.C.; Nakajima, Y.; Miyamoto, T.; Ohnishi, Y.; Kao, W.W.; Muragaki, Y.; Ooshima, A. Therapeutic effects of adenoviral gene transfer of bone morphogenic protein-7 on a corneal alkali injury model in mice. Lab. Investig. 2005, 85, 474–486. [Google Scholar] [CrossRef] [PubMed]
- Saika, S.; Ikeda, K.; Yamanaka, O.; Miyamoto, T.; Ohnishi, Y.; Sato, M.; Muragaki, Y.; Ooshima, A.; Nakajima, Y.; Kao, W.W.; et al. Expression of Smad7 in mouse eyes accelerates healing of corneal tissue after exposure to alkali. Am. J. Pathol. 2005, 166, 1405–1418. [Google Scholar] [CrossRef]
- Holan, V.; Trosan, P.; Cejka, C.; Javorkova, E.; Zajicova, A.; Hermankova, B.; Chudickova, M.; Cejkova, J. A comparative study of the therapeutic potential of mesenchymal stem cells and limbal epithelial stem cells for ocular surface reconstruction. Stem Cells Transl. Med. 2015, 4, 1052–1063. [Google Scholar] [CrossRef]
- Xiao, Y.T.; Xie, H.T.; Liu, X.; Duan, C.Y.; Qu, J.Y.; Zhang, M.C.; Zhao, X.Y. Subconjunctival injection of transdifferentiated oral mucosal epithelial cells for limbal stem cell deficiency in rats. J. Histochem. Cytochem. 2021, 69, 177–190. [Google Scholar] [CrossRef]
- Yan, D.; Yu, F.; Chen, L.; Yao, Q.; Yan, C.; Zhang, S.; Wu, N.; Gong, D.; Sun, H.; Fu, Y.; et al. Subconjunctival injection of regulatory T cells potentiates corneal healing via orchestrating inflammation and tissue repair after acute alkali burn. Investig. Ophthalmol. Vis. Sci. 2020, 61, 22. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Zhou, Q.; Wang, Z.; Guo, R.; Yang, R.; Yang, X.; Li, W.; Ahmad, N.; Chen, Q.; Hui, Q.; et al. Comparative analysis of KGF-2 and bFGF in prevention of excessive wound healing and scar formation in a corneal alkali burn model. Cornea 2019, 38, 1430–1437. [Google Scholar] [CrossRef]
- Ebihara, N.; Matsuda, A.; Nakamura, S.; Matsuda, H.; Murakami, A. Role of the IL-6 classic- and trans-signaling pathways in corneal sterile inflammation and wound healing. Investig. Ophthalmol. Vis. Sci. 2011, 52, 8549–8557. [Google Scholar] [CrossRef] [PubMed]
- Stepp, M.A.; Pal-Ghosh, S.; Downie, L.E.; Zhang, A.C.; Chinnery, H.R.; Machet, J.; Di Girolamo, N. Corneal epithelial “neuromas”: A case of mistaken identity? Cornea 2020, 39, 930–934. [Google Scholar] [CrossRef] [PubMed]
- Lasagni Vitar, R.M.; Rama, P.; Ferrari, G. The two-faced effects of nerves and neuropeptides in corneal diseases. Prog. Retin. Eye Res. 2022, 86, 100974. [Google Scholar] [CrossRef]
- Tuck, H.; Park, M.; Carnell, M.; Machet, J.; Richardson, A.; Jukic, M.; Di Girolamo, N. Neuronal-epithelial cell alignment: A determinant of health and disease status of the cornea. Ocul. Surf. 2021, 21, 257–270. [Google Scholar] [CrossRef]
- Ferrari, G.; Hajrasouliha, A.R.; Sadrai, Z.; Ueno, H.; Chauhan, S.K.; Dana, R. Nerves and neovessels inhibit each other in the cornea. Investig. Ophthalmol. Vis. Sci. 2013, 54, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.; Bell, N.; Botzet, G.; Vora, P.; Fowler, B.J.; Donahue, R.; Bush, H.; Taylor, B.K.; Albuquerque, R.J.C. Latent sensitization in a mouse model of ocular neuropathic pain. Transl. Vis. Sci. Technol. 2019, 8, 6. [Google Scholar] [CrossRef] [PubMed]
- Bignami, F.; Giacomini, C.; Lorusso, A.; Aramini, A.; Rama, P.; Ferrari, G. NK1 receptor antagonists as a new treatment for corneal neovascularization. Investig. Ophthalmol. Vis. Sci. 2014, 55, 6783–6794. [Google Scholar] [CrossRef]
- Barbariga, M.; Fonteyne, P.; Ostadreza, M.; Bignami, F.; Rama, P.; Ferrari, G. Substance P modulation of human and murine corneal neovascularization. Investig. Ophthalmol. Vis. Sci. 2018, 59, 1305–1312. [Google Scholar] [CrossRef] [PubMed]
- Kallinikos, P.; Berhanu, M.; O’Donnell, C.; Boulton, A.J.; Efron, N.; Malik, R.A. Corneal nerve tortuosity in diabetic patients with neuropathy. Investig. Ophthalmol. Vis. Sci. 2004, 45, 418–422. [Google Scholar] [CrossRef]
- Roszkowska, A.M.; Wylegala, A.; Gargano, R.; Spinella, R.; Inferrera, L.; Orzechowska-Wylegala, B.; Aragona, P. Impact of corneal parameters, refractive error and age on density and morphology of the subbasal nerve plexus fibers in healthy adults. Sci. Rep. 2021, 11, 6076. [Google Scholar] [CrossRef]
- Pham, T.L.; Kakazu, A.H.; He, J.; Jun, B.; Bazan, N.G.; Bazan, H.E.P. Novel RvD6 stereoisomer induces corneal nerve regeneration and wound healing post-injury by modulating trigeminal transcriptomic signature. Sci. Rep. 2020, 10, 4582. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Hu, X.; Qi, X.; Di, G.; Zhang, Y.; Wang, Q.; Zhou, Q. Resolvin D1 promotes corneal epithelial wound healing and restoration of mechanical sensation in diabetic mice. Mol. Vis. 2018, 24, 274–285. [Google Scholar] [PubMed]
- Mallone, F.; Costi, R.; Marenco, M.; Plateroti, R.; Minni, A.; Attanasio, G.; Artico, M.; Lambiase, A. Understanding drivers of ocular fibrosis: Current and future therapeutic perspectives. Int. J. Mol. Sci. 2021, 22, 11748. [Google Scholar] [CrossRef]
- Okada, Y.; Sumioka, T.; Reinach, P.S.; Miyajima, M.; Saika, S. Roles of epithelial and mesenchymal TRP channels in mediating inflammatory fibrosis. Front. Immunol. 2021, 12, 731674. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.E. Corneal myofibroblasts and fibrosis. Exp. Eye Res. 2020, 201, 108272. [Google Scholar] [CrossRef] [PubMed]
- Villabona-Martinez, V.; Sampaio, L.P.; Shiju, T.M.; Wilson, S.E. Standardization of corneal alkali burn methodology in rabbits. Exp. Eye Res. 2023, 230, 109443. [Google Scholar] [CrossRef] [PubMed]
- Nuwormegbe, S.A.; Kim, S.W. AMPK activation by 5-amino-4-imidazole carboxamide riboside-1-beta-D-ribofuranoside attenuates alkali injury-induced corneal fibrosis. Investig. Ophthalmol. Vis. Sci. 2020, 61, 43. [Google Scholar] [CrossRef]
- Joung, C.; Noh, H.; Jung, J.; Song, H.Y.; Bae, H.; Pahk, K.; Kim, W.K. A novel CD147 inhibitor, SP-8356, attenuates pathological fibrosis in alkali-burned rat cornea. Int. J. Mol. Sci. 2020, 21, 2990. [Google Scholar] [CrossRef]
- Lorenzo-Martin, E.; Gallego-Munoz, P.; Mar, S.; Fernandez, I.; Cidad, P.; Martinez-Garcia, M.C. Dynamic changes of the extracellular matrix during corneal wound healing. Exp. Eye Res. 2019, 186, 107704. [Google Scholar] [CrossRef]
- Wilson, S.E. The Yin and Yang of Mesenchymal Cells in the Corneal Stromal Fibrosis Response to Injury: The Cornea as a Model of Fibrosis in Other Organs. Biomolecules 2022, 13, 87. [Google Scholar] [CrossRef]
- Wilson, S.E. Interleukin-1 and Transforming Growth Factor Beta: Commonly Opposing, but Sometimes Supporting, Master Regulators of the Corneal Wound Healing Response to Injury. Investig. Ophthalmol. Vis. Sci. 2021, 62, 8. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.E. TGF beta -1, -2 and -3 in the modulation of fibrosis in the cornea and other organs. Exp. Eye Res. 2021, 207, 108594. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, T.; Sumioka, T.; Saika, S. Endothelial mesenchymal transition: A therapeutic target in retrocorneal membrane. Cornea 2010, 29 (Suppl. S1), S52–S56. [Google Scholar] [CrossRef]
- Ishizaki, M.; Zhu, G.; Haseba, T.; Shafer, S.S.; Kao, W.W. Expression of collagen I, smooth muscle alpha-actin, and vimentin during the healing of alkali-burned and lacerated corneas. Investig. Ophthalmol. Vis. Sci. 1993, 34, 3320–3328. [Google Scholar]
- Kethiri, A.R.; Raju, E.; Bokara, K.K.; Mishra, D.K.; Basu, S.; Rao, C.M.; Sangwan, V.S.; Singh, V. Inflammation, vascularization and goblet cell differences in LSCD: Validating animal models of corneal alkali burns. Exp. Eye Res. 2019, 185, 107665. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Robert, M.C.; Kapoulea, V.; Lei, F.; Stagner, A.M.; Jakobiec, F.A.; Dohlman, C.H.; Paschalis, E.I. Sustained subconjunctival delivery of infliximab protects the cornea and retina following alkali burn to the eye. Investig. Ophthalmol. Vis. Sci. 2017, 58, 96–105. [Google Scholar] [CrossRef]
- Di Girolamo, N.; Park, M. Cell identity changes in ocular surface epithelia. Prog. Retin. Eye Res. 2022, 101148. [Google Scholar] [CrossRef]
- Park, M.; Zhang, R.; Pandzic, E.; Sun, M.; Coulson-Thomas, V.J.; Di Girolamo, N. Plasticity of ocular surface epithelia: Using a murine model of limbal stem cell deficiency to delineate metaplasia and transdifferentiation. Stem Cell Rep. 2022, 17, 2451–2466. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sprogyte, L.; Park, M.; Di Girolamo, N. Pathogenesis of Alkali Injury-Induced Limbal Stem Cell Deficiency: A Literature Survey of Animal Models. Cells 2023, 12, 1294. https://doi.org/10.3390/cells12091294
Sprogyte L, Park M, Di Girolamo N. Pathogenesis of Alkali Injury-Induced Limbal Stem Cell Deficiency: A Literature Survey of Animal Models. Cells. 2023; 12(9):1294. https://doi.org/10.3390/cells12091294
Chicago/Turabian StyleSprogyte, Lina, Mijeong Park, and Nick Di Girolamo. 2023. "Pathogenesis of Alkali Injury-Induced Limbal Stem Cell Deficiency: A Literature Survey of Animal Models" Cells 12, no. 9: 1294. https://doi.org/10.3390/cells12091294