
Pathophysiological Effects of Autoantibodies in Autoimmune Encephalitides
 ,
,  , and
, and
Abstract
:1. Introduction
- -
- Autoantibodies targeting specific extracellular or intracellular synaptic antigens are present, and preferably identified in the affected organ
- -
- In vitro experiments have demonstrated pathogenic mechanisms for the autoantibodies, which have been validated by an independent research group
- -
- In vivo studies have reproduced the autoimmune disease either by passively transferring patient serum or purified autoantibodies, or through active immunization with the antigen. The studies have been validated by an independent research group
- -
- Autoantibodies targeting specific extracellular or intracellular synaptic antigens are present, and preferably identified in the affected organ
- -
- In vitro or in vivo experiments have indicated a pathogenic mechanism for the autoantibody
- -
- Results have not yet been validated by an independent research group
- -
- Autoantibodies targeting specific extracellular or intracellular synaptic antigens are present, and preferably identified in the affected organ
- -
- Pathogenic mechanisms are unknown, as no in vitro or in vivo studies have been reported
2. Autoantibodies with Extracellular Targets
2.1. Class I: Autoantibodies with Confirmed Pathogenicity
2.1.1. NMDAR Autoantibodies
2.1.2. Leucine Rich Glioma-Inactivated 1 (LGI1) Autoantibodies
2.1.3. CASPR2 Autoantibodies
2.1.4. AMPAR Autoantibodies
2.1.5. Aminobutyric Acid (GABA) Type A Receptor Autoantibodies
2.1.6. Dipeptidyl-Peptidase-like Protein-6 (DPPX) Autoantibodies
2.1.7. IgLON5 Autoantibodies
2.1.8. Glycine Receptor (GlyR) Autoantibodies
2.2. Class II: Autoantibodies with Highly Suspected Pathogenicity
2.2.1. GABA Type B Receptor (GABABR) Autoantibodies
2.2.2. Metabotropic Glutamate Receptor 1 (mGluR1) Autoantibodies
2.2.3. Metabotropic Glutamate Receptor 5 (mGluR5) Autoantibodies
2.2.4. Neurexin-3α Autoantibodies
2.2.5. Kainate Receptor Subunit 2 (GluK2) Autoantibodies
2.2.6. CaVα2δ Autoantibodies
2.3. Class III: Autoantibodies without Established Pathogenicity
3. Autoantibodies with Intracellular Synaptic Targets
3.1. Class I: Autoantibodies with Confirmed Pathogenicity
Glutamic Acid Decarboxylase 65 (GAD65) Autoantibodies
3.2. Class II: Autoantibodies without Established Pathogenicity
3.2.1. Amphiphysin Autoantibodies
3.2.2. Drebrin Autoantibodies
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pruss, H. Autoantibodies in neurological disease. Nat. Rev. Immunol. 2021, 21, 798–813. [Google Scholar] [CrossRef] [PubMed]
- Alexopoulos, H.; Dalakas, M.C. The immunobiology of autoimmune encephalitides. J. Autoimmun. 2019, 104, 102339. [Google Scholar] [CrossRef] [PubMed]
- Koneczny, I.; Tzartos, J.; Mane-Damas, M.; Yilmaz, V.; Huijbers, M.G.; Lazaridis, K.; Hoftberger, R.; Tuzun, E.; Martinez-Martinez, P.; Tzartos, S.; et al. IgG4 Autoantibodies in Organ-Specific Autoimmunopathies: Reviewing Class Switching, Antibody-Producing Cells, and Specific Immunotherapies. Front. Immunol. 2022, 13, 834342. [Google Scholar] [CrossRef] [PubMed]
- Oostindie, S.C.; Lazar, G.A.; Schuurman, J.; Parren, P. Avidity in antibody effector functions and biotherapeutic drug design. Nat. Rev. Drug. Discov. 2022, 21, 715–735. [Google Scholar] [CrossRef]
- Vidarsson, G.; Dekkers, G.; Rispens, T. IgG subclasses and allotypes: From structure to effector functions. Front. Immunol. 2014, 5, 520. [Google Scholar] [CrossRef]
- Volkov, M.; Coppola, M.; Huizinga, R.; Eftimov, F.; Huizinga, T.W.J.; van der Kooi, A.J.; Oosten, L.E.M.; Raaphorst, J.; Rispens, T.; Sciarrillo, R.; et al. Comprehensive overview of autoantibody isotype and subclass distribution. J. Allergy Clin. Immunol. 2022, 150, 999–1010. [Google Scholar] [CrossRef]
- Valeich, J.; Boyd, D.; Kanwar, M.; Stenzel, D.; De Ghosh, D.; Ebrahimi, A.; Woo, J.; Wang, J.; Ambrogelly, A. Taking the Hinge off: An Approach to Effector-Less Monoclonal Antibodies. Antibodies 2020, 9, 50. [Google Scholar] [CrossRef]
- Napodano, C.; Marino, M.; Stefanile, A.; Pocino, K.; Scatena, R.; Gulli, F.; Rapaccini, G.L.; Delli Noci, S.; Capozio, G.; Rigante, D.; et al. Immunological Role of IgG Subclasses. Immunol. Investig. 2021, 50, 427–444. [Google Scholar] [CrossRef]
- Rowley, M.J.; Whittingham, S.F. The Role of Pathogenic Autoantibodies in Autoimmunity. Antibodies 2015, 4, 314–353. [Google Scholar] [CrossRef]
- Burbelo, P.D.; Iadarola, M.J.; Keller, J.M.; Warner, B.M. Autoantibodies Targeting Intracellular and Extracellular Proteins in Autoimmunity. Front. Immunol. 2021, 12, 548469. [Google Scholar] [CrossRef]
- Okun, E.; Mattson, M.P.; Arumugam, T.V. Involvement of Fc receptors in disorders of the central nervous system. Neuromolecular. Med. 2010, 12, 164–178. [Google Scholar] [CrossRef]
- Baumgartner, A.; Rauer, S.; Hottenrott, T.; Leypoldt, F.; Ufer, F.; Hegen, H.; Pruss, H.; Lewerenz, J.; Deisenhammer, F.; Stich, O. Admission diagnoses of patients later diagnosed with autoimmune encephalitis. J. Neurol. 2019, 266, 124–132. [Google Scholar] [CrossRef]
- Rossling, R.; Pruss, H. SOP: Antibody-associated autoimmune encephalitis. Neurol. Res. Pract. 2020, 2, 1. [Google Scholar] [CrossRef] [PubMed]
- Endres, D.; Maier, V.; Leypoldt, F.; Wandinger, K.P.; Lennox, B.; Pollak, T.A.; Nickel, K.; Maier, S.; Feige, B.; Domschke, K.; et al. Autoantibody-associated psychiatric syndromes: A systematic literature review resulting in 145 cases. Psychol. Med. 2022, 52, 1135–1146. [Google Scholar] [CrossRef] [PubMed]
- Bien, C.G.; Mirzadjanova, Z.; Baumgartner, C.; Onugoren, M.D.; Grunwald, T.; Holtkamp, M.; Isenmann, S.; Kermer, P.; Melzer, N.; Naumann, M.; et al. Anti-contactin-associated protein-2 encephalitis: Relevance of antibody titers, presentation and outcome. Eur. J. Neurol. 2017, 24, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Gresa-Arribas, N.; Titulaer, M.J.; Torrents, A.; Aguilar, E.; McCracken, L.; Leypoldt, F.; Gleichman, A.J.; Balice-Gordon, R.; Rosenfeld, M.R.; Lynch, D.; et al. Antibody titers at diagnosis and during follow-up of anti-NMDA receptor encephalitis: A retrospective study. Lancet Neurol. 2014, 13, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Zandi, M.S.; Paterson, R.W.; Ellul, M.A.; Jacobson, L.; Al-Diwani, A.; Jones, J.L.; Cox, A.L.; Lennox, B.; Stamelou, M.; Bhatia, K.P.; et al. Clinical relevance of serum antibodies to extracellular N-methyl-D-aspartate receptor epitopes. J. Neurol. Neurosurg. Psychiatry 2015, 86, 708–713. [Google Scholar] [CrossRef] [PubMed]
- Hammer, C.; Stepniak, B.; Schneider, A.; Papiol, S.; Tantra, M.; Begemann, M.; Siren, A.L.; Pardo, L.A.; Sperling, S.; Mohd Jofrry, S.; et al. Neuropsychiatric disease relevance of circulating anti-NMDA receptor autoantibodies depends on blood-brain barrier integrity. Mol. Psychiatry 2014, 19, 1143–1149. [Google Scholar] [CrossRef]
- Hummert, M.W.; Jendretzky, K.F.; Fricke, K.; Gingele, M.; Ratuszny, D.; Mohn, N.; Trebst, C.; Skripuletz, T.; Gingele, S.; Suhs, K.W. The relevance of NMDA receptor antibody-specific index for diagnosis and prognosis in patients with anti-NMDA receptor encephalitis. Sci. Rep. 2023, 13, 12696. [Google Scholar] [CrossRef]
- Lang, K.; Pruss, H. Frequencies of neuronal autoantibodies in healthy controls: Estimation of disease specificity. Neurol. Neuroimmunol. Neuroinflamm. 2017, 4, e386. [Google Scholar] [CrossRef]
- Ricken, G.; Schwaiger, C.; De Simoni, D.; Pichler, V.; Lang, J.; Glatter, S.; Macher, S.; Rommer, P.S.; Scholze, P.; Kubista, H.; et al. Detection Methods for Autoantibodies in Suspected Autoimmune Encephalitis. Front. Neurol. 2018, 9, 841. [Google Scholar] [CrossRef] [PubMed]
- Abboud, H.; Probasco, J.C.; Irani, S.; Ances, B.; Benavides, D.R.; Bradshaw, M.; Christo, P.P.; Dale, R.C.; Fernandez-Fournier, M.; Flanagan, E.P.; et al. Autoimmune encephalitis: Proposed best practice recommendations for diagnosis and acute management. J. Neurol. Neurosurg. Psychiatry 2021, 92, 757–768. [Google Scholar] [CrossRef] [PubMed]
- Witebsky, E.; Rose, N.R.; Terplan, K.; Paine, J.R.; Egan, R.W. Chronic thyroiditis and autoimmunization. J. Am. Med. Assoc. 1957, 164, 1439–1447. [Google Scholar] [CrossRef] [PubMed]
- Rose, N.R.; Bona, C. Defining criteria for autoimmune diseases (Witebsky’s postulates revisited). Immunol. Today 1993, 14, 426–430. [Google Scholar] [CrossRef] [PubMed]
- Naparstek, Y.; Plotz, P.H. The role of autoantibodies in autoimmune disease. Annu. Rev. Immunol. 1993, 11, 79–104. [Google Scholar] [CrossRef]
- Koneczny, I. A New Classification System for IgG4 Autoantibodies. Front. Immunol. 2018, 9, 97. [Google Scholar] [CrossRef]
- Uy, C.E.; Binks, S.; Irani, S.R. Autoimmune encephalitis: Clinical spectrum and management. Pract. Neurol. 2021, 21, 412–423. [Google Scholar] [CrossRef]
- van Sonderen, A.; Thijs, R.D.; Coenders, E.C.; Jiskoot, L.C.; Sanchez, E.; de Bruijn, M.A.; van Coevorden-Hameete, M.H.; Wirtz, P.W.; Schreurs, M.W.; Sillevis Smitt, P.A.; et al. Anti-LGI1 encephalitis: Clinical syndrome and long-term follow-up. Neurology 2016, 87, 1449–1456. [Google Scholar] [CrossRef]
- Nosadini, M.; Eyre, M.; Molteni, E.; Thomas, T.; Irani, S.R.; Dalmau, J.; Dale, R.C.; Lim, M.; International, N.A.E.C.G.; Anlar, B.; et al. Use and Safety of Immunotherapeutic Management of N-Methyl-d-Aspartate Receptor Antibody Encephalitis: A Meta-analysis. JAMA Neurol. 2021, 78, 1333–1344. [Google Scholar] [CrossRef]
- Nissen, M.S.; Ryding, M.; Meyer, M.; Blaabjerg, M. Autoimmune encephalitis: Current knowledge on subtypes, disease mechanisms and treatment. CNS Neurol. Disord. Drug Targets 2020, 19, 584–598. [Google Scholar] [CrossRef]
- Dalmau, J.; Gleichman, A.J.; Hughes, E.G.; Rossi, J.E.; Peng, X.; Lai, M.; Dessain, S.K.; Rosenfeld, M.R.; Balice-Gordon, R.; Lynch, D.R. Anti-NMDA-receptor encephalitis: Case series and analysis of the effects of antibodies. Lancet Neurol. 2008, 7, 1091–1098. [Google Scholar] [CrossRef] [PubMed]
- Hughes, E.G.; Peng, X.; Gleichman, A.J.; Lai, M.; Zhou, L.; Tsou, R.; Parsons, T.D.; Lynch, D.R.; Dalmau, J.; Balice-Gordon, R.J. Cellular and synaptic mechanisms of anti-NMDA receptor encephalitis. J. Neurosci. 2010, 30, 5866–5875. [Google Scholar] [CrossRef] [PubMed]
- Moscato, E.H.; Peng, X.; Jain, A.; Parsons, T.D.; Dalmau, J.; Balice-Gordon, R.J. Acute mechanisms underlying antibody effects in anti-N-methyl-D-aspartate receptor encephalitis. Ann. Neurol. 2014, 76, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Mikasova, L.; De Rossi, P.; Bouchet, D.; Georges, F.; Rogemond, V.; Didelot, A.; Meissirel, C.; Honnorat, J.; Groc, L. Disrupted surface cross-talk between NMDA and Ephrin-B2 receptors in anti-NMDA encephalitis. Brain 2012, 135, 1606–1621. [Google Scholar] [CrossRef] [PubMed]
- Planaguma, J.; Haselmann, H.; Mannara, F.; Petit-Pedrol, M.; Grunewald, B.; Aguilar, E.; Ropke, L.; Martin-Garcia, E.; Titulaer, M.J.; Jercog, P.; et al. Ephrin-B2 prevents N-methyl-D-aspartate receptor antibody effects on memory and neuroplasticity. Ann. Neurol. 2016, 80, 388–400. [Google Scholar] [CrossRef] [PubMed]
- Jantzen, S.U.; Ferrea, S.; Wach, C.; Quasthoff, K.; Illes, S.; Scherfeld, D.; Hartung, H.P.; Seitz, R.J.; Dihne, M. In vitro neuronal network activity in NMDA receptor encephalitis. BMC Neurosci. 2013, 14, 17. [Google Scholar] [CrossRef] [PubMed]
- Wurdemann, T.; Kersten, M.; Tokay, T.; Guli, X.; Kober, M.; Rohde, M.; Porath, K.; Sellmann, T.; Bien, C.G.; Kohling, R.; et al. Stereotactic injection of cerebrospinal fluid from anti-NMDA receptor encephalitis into rat dentate gyrus impairs NMDA receptor function. Brain Res. 2016, 1633, 10–18. [Google Scholar] [CrossRef]
- Matute, C.; Palma, A.; Serrano-Regal, M.P.; Maudes, E.; Barman, S.; Sanchez-Gomez, M.V.; Domercq, M.; Goebels, N.; Dalmau, J. N-Methyl-D-Aspartate Receptor Antibodies in Autoimmune Encephalopathy Alter Oligodendrocyte Function. Ann. Neurol. 2020, 87, 670–676. [Google Scholar] [CrossRef]
- Grea, H.; Bouchet, D.; Rogemond, V.; Hamdani, N.; Le Guen, E.; Tamouza, R.; Darrau, E.; Passerieux, C.; Honnorat, J.; Leboyer, M.; et al. Human Autoantibodies Against N-Methyl-D-Aspartate Receptor Modestly Alter Dopamine D1 Receptor Surface Dynamics. Front. Psychiatry 2019, 10, 670. [Google Scholar] [CrossRef]
- Carceles-Cordon, M.; Mannara, F.; Aguilar, E.; Castellanos, A.; Planaguma, J.; Dalmau, J. NMDAR Antibodies Alter Dopamine Receptors and Cause Psychotic Behavior in Mice. Ann. Neurol. 2020, 88, 603–613. [Google Scholar] [CrossRef]
- Wang, X.; Ma, C.; Liu, C.Y.; Li, G.J.; Zhao, D.; Han, D.F. Neuronal NMDAR Currents of the Hippocampus and Learning Performance in Autoimmune Anti-NMDAR Encephalitis and Involvement of TNF-alpha and IL-6. Front. Neurol. 2019, 10, 684. [Google Scholar] [CrossRef] [PubMed]
- Planaguma, J.; Leypoldt, F.; Mannara, F.; Gutierrez-Cuesta, J.; Martin-Garcia, E.; Aguilar, E.; Titulaer, M.J.; Petit-Pedrol, M.; Jain, A.; Balice-Gordon, R.; et al. Human N-methyl D-aspartate receptor antibodies alter memory and behaviour in mice. Brain 2015, 138, 94–109. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Z.; Tanaka, K.; Wang, L.; Ishigakil, Y.; Kato, N. Induction of Memory Deficit in Mice with Chronic Exposure to Cerebrospinal Fluid from Patients with Anti-N-Methyl-D-Aspartate Receptor Encephalitis. Tohoku J. Exp. Med. 2015, 237, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Jones, B.E.; Tovar, K.R.; Goehring, A.; Jalali-Yazdi, F.; Okada, N.J.; Gouaux, E.; Westbrook, G.L. Autoimmune receptor encephalitis in mice induced by active immunization with conformationally stabilized holoreceptors. Sci. Transl. Med. 2019, 11, eaaw0044. [Google Scholar] [CrossRef] [PubMed]
- Taraschenko, O.; Fox, H.S.; Pittock, S.J.; Zekeridou, A.; Gafurova, M.; Eldridge, E.; Liu, J.X.; Dravid, S.M.; Dingledine, R. A mouse model of seizures in anti-N-methyl-d-aspartate receptor encephalitis. Epilepsia 2019, 60, 452–463. [Google Scholar] [CrossRef]
- Ohkawa, T.; Fukata, Y.; Yamasaki, M.; Miyazaki, T.; Yokoi, N.; Takashima, H.; Watanabe, M.; Watanabe, O.; Fukata, M. Autoantibodies to Epilepsy-Related LGI1 in Limbic Encephalitis Neutralize LGI1-ADAM22 Interaction and Reduce Synaptic AMPA Receptors. J. Neurosci. 2013, 33, 18161–18174. [Google Scholar] [CrossRef]
- Petit-Pedrol, M.; Sell, J.; Planaguma, J.; Mannara, F.; Radosevic, M.; Haselmann, H.; Ceanga, M.; Sabater, L.; Spatola, M.; Soto, D.; et al. LG antibodies alter K-v I.I and AMPA receptors changing synaptic excitability, plasticity and memory. Brain 2018, 141, 3144–3159. [Google Scholar] [CrossRef]
- Kornau, H.C.; Kreye, J.; Stumpf, A.; Fukata, Y.; Parthier, D.; Sammons, R.P.; Imbrosci, B.; Kurpjuweit, S.; Kowski, A.B.; Fukata, M.; et al. Human Cerebrospinal Fluid Monoclonal LGI1 Autoantibodies Increase Neuronal Excitability. Ann. Neurol. 2020, 87, 405–418. [Google Scholar] [CrossRef]
- Ramberger, M.; Berretta, A.; Tan, J.M.M.; Sun, B.; Michael, S.; Yeo, T.; Theorell, J.; Bashford-Rogers, R.; Paneva, S.; O’Dowd, V.; et al. Distinctive binding properties of human monoclonal LGI1 autoantibodies determine pathogenic mechanisms. Brain 2020, 143, 1731–1745. [Google Scholar] [CrossRef]
- Romoli, M.; Krashia, P.; Sen, A.; Franciotta, D.; Gastaldi, M.; Nobili, A.; Mancini, A.; Cesarini, E.N.; Nigro, P.; Tambasco, N.; et al. Hippocampal epileptogenesis in autoimmune encephalitis. Ann. Clin. Transl. Neur. 2019, 6, 2261–2269. [Google Scholar] [CrossRef]
- Troscher, A.R.; Klang, A.; French, M.; Quemada-Garrido, L.; Kneissl, S.M.; Bien, C.G.; Pakozdy, A.; Bauer, J. Selective Limbic Blood-Brain Barrier Breakdown in a Feline Model of Limbic Encephalitis with LGI1 Antibodies. Front. Immunol. 2017, 8, 1364. [Google Scholar] [CrossRef] [PubMed]
- Irani, S.R.; Alexander, S.; Waters, P.; Kleopa, K.A.; Pettingill, P.; Zuliani, L.; Peles, E.; Buckley, C.; Lang, B.; Vincent, A. Antibodies to Kv1 potassium channel-complex proteins leucine-rich, glioma inactivated 1 protein and contactin-associated protein-2 in limbic encephalitis, Morvan’s syndrome and acquired neuromyotonia. Brain 2010, 133, 2734–2748. [Google Scholar] [CrossRef] [PubMed]
- Patterson, K.R.; Dalmau, J.; Lancaster, E. Mechanisms of Caspr2 Antibodies in Autoimmune Encephalitis and Neuromyotonia. Ann. Neurol. 2018, 83, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Saint-Martin, M.; Pieters, A.; Dechelotte, B.; Malleval, C.; Pinatel, D.; Pascual, O.; Karagogeos, D.; Honnorat, J.; Pellier-Monnin, V.; Noraz, N. Impact of anti-CASPR2 autoantibodies from patients with autoimmune encephalitis on CASPR2/TAG-1 interaction and Kv1 expression. J. Autoimmun. 2019, 103, 102284. [Google Scholar] [CrossRef]
- Giannoccaro, M.P.; Menassa, D.A.; Jacobson, L.; Coutinho, E.; Prota, G.; Lang, B.; Leite, M.I.; Cerundolo, V.; Liguori, R.; Vincent, A. Behaviour and neuropathology in mice injected with human contactin-associated protein 2 antibodies. Brain 2019, 142, 2000–2012. [Google Scholar] [CrossRef]
- Dawes, J.M.; Weir, G.A.; Middleton, S.J.; Patel, R.; Chisholm, K.I.; Pettingill, P.; Peck, L.J.; Sheridan, J.; Shakir, A.; Jacobson, L.; et al. Immune or Genetic-Mediated Disruption of CASPR2 Causes Pain Hypersensitivity Due to Enhanced Primary Afferent Excitability. Neuron 2018, 97, 806–822.e10. [Google Scholar] [CrossRef]
- Joubert, B.; Petit-Pedrol, M.; Planaguma, J.; Mannara, F.; Radosevic, M.; Marsal, M.; Maudes, E.; Garcia-Serra, A.; Aguilar, E.; Andres-Bilbe, A.; et al. Human CASPR2 Antibodies Reversibly Alter Memory and the CASPR2 Protein Complex. Ann. Neurol. 2022, 91, 801–813. [Google Scholar] [CrossRef]
- Lai, M.Z.; Hughes, E.G.; Peng, X.Y.; Zhou, L.; Gleichman, A.J.; Shu, H.; Mata, S.; Kremens, D.; Vitaliani, R.; Geschwind, M.D.; et al. AMPA Receptor Antibodies in Limbic Encephalitis Alter Synaptic Receptor Location. Ann. Neurol. 2009, 65, 424–434. [Google Scholar] [CrossRef]
- Peng, X.Y.; Hughes, E.G.; Moscato, E.H.; Parsons, T.D.; Dalmau, J.; Balice-Gordon, R.J. Cellular Plasticity Induced by Anti-alpha-Amino-3-Hydroxy-5-Methyl-4-Isoxazolepropionic Acid (AMPA) Receptor Encephalitis Antibodies. Ann. Neurol. 2015, 77, 381–398. [Google Scholar] [CrossRef]
- Haselmann, H.; Mannara, F.; Werner, C.; Planaguma, J.; Miguez-Cabello, F.; Schmidl, L.; Grunewald, B.; Petit-Pedrol, M.; Kirmse, K.; Classen, J.; et al. Human Autoantibodies against the AMPA Receptor Subunit GluA2 Induce Receptor Reorganization and Memory Dysfunction. Neuron 2018, 100, 91–105.e9. [Google Scholar] [CrossRef]
- Gleichman, A.J.; Panzer, J.A.; Baumann, B.H.; Dalmau, J.; Lynch, D.R. Antigenic and mechanistic characterization of anti-AMPA receptor encephalitis. Ann. Clin. Transl. Neurol. 2014, 1, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Shi, Y.; Du, Q.; Zhang, R.R.; Wu, H.; Qiao, S.; Liu, X. Clinical Review and Prognostic Analysis of alpha-Amino-3-Hydroxy-5-Methyl-4-Isoxazole Propionate Receptor-Associated Encephalitis. Front. Neurol. 2021, 12, 665229. [Google Scholar] [CrossRef] [PubMed]
- Petit-Pedrol, M.; Armangue, T.; Peng, X.Y.; Bataller, L.; Cellucci, T.; Davis, R.; McCracken, L.; Martinez-Hernandez, E.; Mason, W.P.; Kruer, M.C.; et al. Encephalitis with refractory seizures, status epilepticus, and antibodies to the GABA(A) receptor: A case series, characterisation of the antigen, and analysis of the effects of antibodies. Lancet Neurol. 2014, 13, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Ohkawa, T.; Satake, S.; Yokoi, N.; Miyazaki, Y.; Ohshita, T.; Sobue, G.; Takashima, H.; Watanabe, O.; Fukata, Y.; Fukata, M. Identification and Characterization of GABA(A) Receptor Autoantibodies in Autoimmune Encephalitis. J. Neurosci. 2014, 34, 8151–8163. [Google Scholar] [CrossRef] [PubMed]
- Pettingill, P.; Kramer, H.B.; Coebergh, J.A.; Pettingill, R.; Maxwell, S.; Nibber, A.; Malaspina, A.; Jacob, A.; Irani, S.R.; Buckley, C.; et al. Antibodies to GABA(A) receptor alpha 1 and gamma 2 subunits Clinical and serologic characterization. Neurology 2015, 84, 1233–1241. [Google Scholar] [CrossRef] [PubMed]
- Noviello, C.M.; Kreye, J.; Teng, J.; Pruss, H.; Hibbs, R.E. Structural mechanisms of GABA(A) receptor autoimmune encephalitis. Cell 2022, 185, 2469–2477.e2413. [Google Scholar] [CrossRef]
- Brandle, S.M.; Cerina, M.; Weber, S.; Held, K.; Menke, A.F.; Alcala, C.; Gebert, D.; Herrmann, A.M.; Pellkofer, H.; Gerdes, L.A.; et al. Cross-reactivity of a pathogenic autoantibody to a tumor antigen in GABA(A) receptor encephalitis. Proc. Natl. Acad. Sci. USA 2021, 118, e1916337118. [Google Scholar] [CrossRef] [PubMed]
- Kreye, J.; Wright, S.K.; van Casteren, A.; Stoffler, L.; Machule, M.L.; Reincke, S.M.; Nikolaus, M.; van Hoof, S.; Sanchez-Sendin, E.; Homeyer, M.A.; et al. Encephalitis patient-derived monoclonal GABAA receptor antibodies cause epileptic seizures. J. Exp. Med. 2021, 218, e20210012. [Google Scholar] [CrossRef]
- Hara, M.; Arino, H.; Petit-Pedrol, M.; Sabater, L.; Titulaer, M.J.; Martinez-Hernandez, E.; Schreurs, M.W.J.; Rosenfeld, M.R.; Graus, F.; Dalmau, J. DPPX antibody-associated encephalitis Main syndrome and antibody effects. Neurology 2017, 88, 1340–1348. [Google Scholar] [CrossRef]
- Piepgras, J.; Holtje, M.; Michel, K.; Li, Q.; Otto, C.; Drenckhahn, C.; Probst, C.; Schemann, M.; Jarius, S.; Stocker, W.; et al. Anti-DPPX encephalitis: Pathogenic effects of antibodies on gut and brain neurons. Neurology 2015, 85, 890–897. [Google Scholar] [CrossRef]
- Tobin, W.O.; Lennon, V.A.; Komorowski, L.; Probst, C.; Clardy, S.L.; Aksamit, A.J.; Appendino, J.P.; Lucchinetti, C.F.; Matsumoto, J.Y.; Pittock, S.J.; et al. DPPX potassium channel antibody: Frequency, clinical accompaniments, and outcomes in 20 patients. Neurology 2014, 83, 1797–1803. [Google Scholar] [CrossRef]
- Sabater, L.; Planaguma, J.; Dalmau, J.; Graus, F. Cellular investigations with human antibodies associated with the anti-IgLON5 syndrome. J. Neuroinflamm. 2016, 13, 226. [Google Scholar] [CrossRef]
- Ryding, M.; Gamre, M.; Nissen, M.S.; Nilsson, A.C.; Okarmus, J.; Poulsen, A.A.E.; Meyer, M.; Blaabjerg, M. Neurodegeneration Induced by Anti-IgLON5 Antibodies Studied in Induced Pluripotent Stem Cell-Derived Human Neurons. Cells 2021, 10, 837. [Google Scholar] [CrossRef]
- Ni, Y.; Feng, Y.; Shen, D.; Chen, M.; Zhu, X.; Zhou, Q.; Gao, Y.; Liu, J.; Zhang, Q.; Shen, Y.; et al. Anti-IgLON5 antibodies cause progressive behavioral and neuropathological changes in mice. J. Neuroinflamm. 2022, 19, 140. [Google Scholar] [CrossRef]
- Landa, J.; Gaig, C.; Plaguma, J.; Saiz, A.; Antonell, A.; Sanchez-Valle, R.; Dalmau, J.; Graus, F.; Sabater, L. Effects of IgLON5 Antibodies on Neuronal Cytoskeleton: A Link between Autoimmunity and Neurodegeneration. Ann. Neurol. 2020, 88, 1023–1027. [Google Scholar] [CrossRef]
- Alvente, S.; Matteoli, G.; Molina-Porcel, L.; Landa, J.; Alba, M.; Bastianini, S.; Berteotti, C.; Graus, F.; Lo Martire, V.; Sabater, L.; et al. Pilot Study of the Effects of Chronic Intracerebroventricular Infusion of Human Anti-IgLON5 Disease Antibodies in Mice. Cells 2022, 11, 1024. [Google Scholar] [CrossRef]
- Sabater, L.; Gaig, C.; Gelpi, E.; Bataller, L.; Lewerenz, J.; Torres-Vega, E.; Contreras, A.; Giometto, B.; Compta, Y.; Embid, C.; et al. A novel non-rapid-eye movement and rapid-eye-movement parasomnia with sleep breathing disorder associated with antibodies to IgLON5: A case series, characterisation of the antigen, and post-mortem study. Lancet Neurol. 2014, 13, 575–586. [Google Scholar] [CrossRef]
- Carvajal-Gonzalez, A.; Leite, M.I.; Waters, P.; Woodhall, M.; Coutinho, E.; Balint, B.; Lang, B.; Pettingill, P.; Carr, A.; Sheerin, U.M.; et al. Glycine receptor antibodies in PERM and related syndromes: Characteristics, clinical features and outcomes (vol 137, pg 2178, 2014). Brain 2014, 137, e315. [Google Scholar] [CrossRef]
- Crisp, S.J.; Dixon, C.L.; Jacobson, L.; Chabrol, E.; Irani, S.R.; Leite, M.I.; Leschziner, G.; Slaght, S.J.; Vincent, A.; Kullmann, D.M. Glycine receptor autoantibodies disrupt inhibitory neurotransmission. Brain 2019, 142, 3398–3410. [Google Scholar] [CrossRef]
- Carvajal, A.; Jacobsen, L.; Clover, L.; Lang, B.; Vincent, A. Passive Transfer of Glycine Receptor-Antibody IgG Induces Motor Dysfunction in Mice. Ann. Neurol. 2014, 76, S76–S77. [Google Scholar]
- Nibber, A.; Pettingill, P.; Waters, P.; Mann, E.; Vincent, A.; Lang, B. Pathogenic Potential of Antibodies Directed against Ampar and Gaba(B)R in Autoimmune Encephalitis. Epilepsia 2014, 55, 9. [Google Scholar]
- Lancaster, E.; Lai, M.; Peng, X.; Hughes, E.; Constantinescu, R.; Raizer, J.; Friedman, D.; Skeen, M.B.; Grisold, W.; Kimura, A.; et al. Antibodies to the GABA(B) receptor in limbic encephalitis with seizures: Case series and characterisation of the antigen. Lancet Neurol. 2010, 9, 67–76. [Google Scholar] [CrossRef]
- Spatola, M.; Petit Pedrol, M.; Maudes, E.; Simabukuro, M.; Muniz-Castrillo, S.; Pinto, A.L.; Wandinger, K.P.; Spiegler, J.; Schramm, P.; Dutra, L.A.; et al. Clinical features, prognostic factors, and antibody effects in anti-mGluR1 encephalitis. Neurology 2020, 95, e3012–e3025. [Google Scholar] [CrossRef]
- Coesmans, M.; Smitt, P.A.; Linden, D.J.; Shigemoto, R.; Hirano, T.; Yamakawa, Y.; van Alphen, A.M.; Luo, C.; van der Geest, J.N.; Kros, J.M.; et al. Mechanisms underlying cerebellar motor deficits due to mGluR1-autoantibodies. Ann. Neurol. 2003, 53, 325–336. [Google Scholar] [CrossRef]
- Sillevis Smitt, P.; Kinoshita, A.; De Leeuw, B.; Moll, W.; Coesmans, M.; Jaarsma, D.; Henzen-Logmans, S.; Vecht, C.; De Zeeuw, C.; Sekiyama, N.; et al. Paraneoplastic cerebellar ataxia due to autoantibodies against a glutamate receptor. N. Engl. J. Med. 2000, 342, 21–27. [Google Scholar] [CrossRef]
- Spatola, M.; Sabater, L.; Planaguma, J.; Martinez-Hernandez, E.; Armangue, T.; Pruss, H.; Iizuka, T.; Caparo Oblitas, R.L.; Antoine, J.C.; Li, R.; et al. Encephalitis with mGluR5 antibodies: Symptoms and antibody effects. Neurology 2018, 90, e1964–e1972. [Google Scholar] [CrossRef]
- Gresa-Arribas, N.; Planaguma, J.; Petit-Pedrol, M.; Kawachi, I.; Katada, S.; Glaser, C.A.; Simabukuro, M.M.; Armangue, T.; Martinez-Hernandez, E.; Graus, F.; et al. Human neurexin-3alpha antibodies associate with encephalitis and alter synapse development. Neurology 2016, 86, 2235–2242. [Google Scholar] [CrossRef]
- Landa, J.; Guasp, M.; Miguez-Cabello, F.; Guimaraes, J.; Mishima, T.; Oda, F.; Zipp, F.; Krajinovic, V.; Fuhr, P.; Honnorat, J.; et al. Encephalitis with Autoantibodies against the Glutamate Kainate Receptors GluK2. Ann. Neurol. 2021, 90, 101–117. [Google Scholar] [CrossRef]
- Lee, S.T.; Lee, B.J.; Bae, J.Y.; Kim, Y.S.; Han, D.H.; Shin, H.S.; Kim, S.; Park, D.K.; Seo, S.W.; Chu, K.; et al. CaV alpha2delta Autoimmune Encephalitis: A Novel Antibody and its Characteristics. Ann. Neurol. 2021, 89, 740–752. [Google Scholar] [CrossRef]
- Dale, R.C.; Merheb, V.; Pillai, S.; Wang, D.; Cantrill, L.; Murphy, T.K.; Ben-Pazi, H.; Varadkar, S.; Aumann, T.D.; Horne, M.K.; et al. Antibodies to surface dopamine-2 receptor in autoimmune movement and psychiatric disorders. Brain 2012, 135, 3453–3468. [Google Scholar] [CrossRef]
- Ruiz-Garcia, R.; Martinez-Hernandez, E.; Joubert, B.; Petit-Pedrol, M.; Pajaron-Boix, E.; Fernandez, V.; Salais, L.; Del Pozo, M.; Armangue, T.; Sabater, L.; et al. Paraneoplastic cerebellar ataxia and antibodies to metabotropic glutamate receptor 2. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7, e658. [Google Scholar] [CrossRef]
- Landa, J.; Guasp, M.; Petit-Pedrol, M.; Martinez-Hernandez, E.; Planaguma, J.; Saiz, A.; Ruiz-Garcia, R.; Garcia-Fernandez, L.; Verschuuren, J.; Saunders-Pullman, R.; et al. Seizure-related 6 homolog like 2 autoimmunity: Neurologic syndrome and antibody effects. Neurol. Neuroimmunol. Neuroinflamm. 2021, 8, e916. [Google Scholar] [CrossRef]
- Jarius, S.; Wildemann, B. ‘Medusa head ataxia’: The expanding spectrum of Purkinje cell antibodies in autoimmune cerebellar ataxia. Part 2: Anti-PKC-gamma, anti-GluR-delta2, anti-Ca/ARHGAP26 and anti-VGCC. J. Neuroinflamm. 2015, 12, 167. [Google Scholar] [CrossRef]
- Dalmau, J.; Tuzun, E.; Wu, H.Y.; Masjuan, J.; Rossi, J.E.; Voloschin, A.; Baehring, J.M.; Shimazaki, H.; Koide, R.; King, D.; et al. Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann. Neurol. 2007, 61, 25–36. [Google Scholar] [CrossRef]
- Saraya, A.W.; Worachotsueptrakun, K.; Vutipongsatorn, K.; Sonpee, C.; Hemachudha, T. Differences and diversity of autoimmune encephalitis in 77 cases from a single tertiary care center. BMC Neurol. 2019, 19, 273. [Google Scholar] [CrossRef]
- Tanaka, K.; Kawamura, M.; Sakimura, K.; Kato, N. Significance of Autoantibodies in Autoimmune Encephalitis in Relation to Antigen Localization: An Outline of Frequently Reported Autoantibodies with a Non-Systematic Review. Int. J. Mol. Sci. 2020, 21, 4941. [Google Scholar] [CrossRef]
- Hunt, D.L.; Castillo, P.E. Synaptic plasticity of NMDA receptors: Mechanisms and functional implications. Curr. Opin. Neurobiol. 2012, 22, 496–508. [Google Scholar] [CrossRef]
- Paoletti, P.; Bellone, C.; Zhou, Q. NMDA receptor subunit diversity: Impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 2013, 14, 383–400. [Google Scholar] [CrossRef]
- Luscher, C.; Malenka, R.C. NMDA receptor-dependent long-term potentiation and long-term depression (LTP/LTD). Cold Spring Harb. Perspect. Biol. 2012, 4, a005710. [Google Scholar] [CrossRef]
- Nakazawa, K.; McHugh, T.J.; Wilson, M.A.; Tonegawa, S. NMDA receptors, place cells and hippocampal spatial memory. Nat. Rev. Neurosci. 2004, 5, 361–372. [Google Scholar] [CrossRef]
- Willard, S.S.; Koochekpour, S. Glutamate, glutamate receptors, and downstream signaling pathways. Int. J. Biol. Sci. 2013, 9, 948–959. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Hansen, K.B.; Vance, K.M.; Ogden, K.K.; Traynelis, S.F. Control of NMDA receptor function by the NR2 subunit amino-terminal domain. J. Neurosci. 2009, 29, 12045–12058. [Google Scholar] [CrossRef] [PubMed]
- Ulbrich, M.H.; Isacoff, E.Y. Rules of engagement for NMDA receptor subunits. Proc. Natl. Acad. Sci. USA 2008, 105, 14163–14168. [Google Scholar] [CrossRef] [PubMed]
- Stroebel, D.; Casado, M.; Paoletti, P. Triheteromeric NMDA receptors: From structure to synaptic physiology. Curr. Opin. Physiol. 2018, 2, 1–12. [Google Scholar] [CrossRef]
- Kumar, A. NMDA Receptor Function During Senescence: Implication on Cognitive Performance. Front. Neurosci. 2015, 9, 473. [Google Scholar] [CrossRef]
- Hansen, K.B.; Yi, F.; Perszyk, R.E.; Menniti, F.S.; Traynelis, S.F. NMDA Receptors in the Central Nervous System. Methods Mol. Biol. 2017, 1677, 1–80. [Google Scholar] [CrossRef]
- Young, D. The NMDA Receptor Antibody Paradox: A Possible Approach to Developing Immunotherapies Targeting the NMDA Receptor. Front. Neurol. 2020, 11, 635. [Google Scholar] [CrossRef]
- Tuzun, E.; Zhou, L.; Baehring, J.M.; Bannykh, S.; Rosenfeld, M.R.; Dalmau, J. Evidence for antibody-mediated pathogenesis in anti-NMDAR encephalitis associated with ovarian teratoma. Acta Neuropathol. 2009, 118, 737–743. [Google Scholar] [CrossRef]
- Kreye, J.; Wenke, N.K.; Chayka, M.; Leubner, J.; Murugan, R.; Maier, N.; Jurek, B.; Ly, L.T.; Brandl, D.; Rost, B.R.; et al. Human cerebrospinal fluid monoclonal N-methyl-D-aspartate receptor autoantibodies are sufficient for encephalitis pathogenesis. Brain 2016, 139, 2641–2652. [Google Scholar] [CrossRef]
- Goi, L.D.S.; Altenhofen, S.; Nabinger, D.D.; Bonan, C.D.; Sato, D.K. Decreased convulsive threshold and memory loss after anti-NMDAR positive CSF injection in zebrafish. J. Neuroimmunol. 2021, 359, 577689. [Google Scholar] [CrossRef]
- Andrzejak, E.; Rabinovitch, E.; Kreye, J.; Pruss, H.; Rosenmund, C.; Ziv, N.E.; Garner, C.C.; Ackermann, F. Patient-Derived Anti-NMDAR Antibody Disinhibits Cortical Neuronal Networks through Dysfunction of Inhibitory Neuron Output. J. Neurosci. 2022, 42, 3253–3270. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, K.; Zsiros, V.; Jiang, Z.; Nakao, K.; Kolata, S.; Zhang, S.; Belforte, J.E. GABAergic interneuron origin of schizophrenia pathophysiology. Neuropharmacology 2012, 62, 1574–1583. [Google Scholar] [CrossRef] [PubMed]
- Saab, A.S.; Tzvetavona, I.D.; Trevisiol, A.; Baltan, S.; Dibaj, P.; Kusch, K.; Mobius, W.; Goetze, B.; Jahn, H.M.; Huang, W.; et al. Oligodendroglial NMDA Receptors Regulate Glucose Import and Axonal Energy Metabolism. Neuron 2016, 91, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Mannara, F.; Radosevic, M.; Planaguma, J.; Soto, D.; Aguilar, E.; Garcia-Serra, A.; Maudes, E.; Pedreno, M.; Paul, S.; Doherty, J.; et al. Allosteric modulation of NMDA receptors prevents the antibody effects of patients with anti-NMDAR encephalitis. Brain 2020, 143, 2709–2720. [Google Scholar] [CrossRef] [PubMed]
- Wright, S.K.; Rosch, R.E.; Wilson, M.A.; Upadhya, M.A.; Dhangar, D.R.; Clarke-Bland, C.; Wahid, T.T.; Barman, S.; Goebels, N.; Kreye, J.; et al. Multimodal electrophysiological analyses reveal that reduced synaptic excitatory neurotransmission underlies seizures in a model of NMDAR antibody-mediated encephalitis. Commun. Biol. 2021, 4, 1106. [Google Scholar] [CrossRef] [PubMed]
- Buckley, C.; Oger, J.; Clover, L.; Tuzun, E.; Carpenter, K.; Jackson, M.; Vincent, A. Potassium channel antibodies in two patients with reversible limbic encephalitis. Ann. Neurol. 2001, 50, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Lai, M.; Huijbers, M.G.; Lancaster, E.; Graus, F.; Bataller, L.; Balice-Gordon, R.; Cowell, J.K.; Dalmau, J. Investigation of LGI1 as the antigen in limbic encephalitis previously attributed to potassium channels: A case series. Lancet Neurol. 2010, 9, 776–785. [Google Scholar] [CrossRef]
- Lang, B.; Makuch, M.; Moloney, T.; Dettmann, I.; Mindorf, S.; Probst, C.; Stoecker, W.; Buckley, C.; Newton, C.R.; Leite, M.I.; et al. Intracellular and non-neuronal targets of voltage-gated potassium channel complex antibodies. J. Neurol. Neurosurg. Psychiatry 2017, 88, 353–361. [Google Scholar] [CrossRef]
- Irani, S.R.; Michell, A.W.; Lang, B.; Pettingill, P.; Waters, P.; Johnson, M.R.; Schott, J.M.; Armstrong, R.J.; Zagami, A.S.; Bleasel, A.; et al. Faciobrachial dystonic seizures precede Lgi1 antibody limbic encephalitis. Ann. Neurol. 2011, 69, 892–900. [Google Scholar] [CrossRef]
- Chater, T.E.; Goda, Y. The role of AMPA receptors in postsynaptic mechanisms of synaptic plasticity. Front. Cell Neurosci. 2014, 8, 401. [Google Scholar] [CrossRef]
- van Sonderen, A.; Petit-Pedrol, M.; Dalmau, J.; Titulaer, M.J. The value of LGI1, Caspr2 and voltage-gated potassium channel antibodies in encephalitis. Nat. Rev. Neurol. 2017, 13, 290–301. [Google Scholar] [CrossRef] [PubMed]
- Grizel, A.V.; Glukhov, G.S.; Sokolova, O.S. Mechanisms of activation of voltage-gated potassium channels. Acta Nat. 2014, 6, 10–26. [Google Scholar] [CrossRef]
- Fels, E.; Muniz-Castrillo, S.; Vogrig, A.; Joubert, B.; Honnorat, J.; Pascual, O. Role of LGI1 protein in synaptic transmission: From physiology to pathology. Neurobiol. Dis. 2021, 160, 105537. [Google Scholar] [CrossRef] [PubMed]
- Lugara, E.; Kaushik, R.; Leite, M.; Chabrol, E.; Dityatev, A.; Lignani, G.; Walker, M.C. LGI1 downregulation increases neuronal circuit excitability. Epilepsia 2020, 61, 2836–2846. [Google Scholar] [CrossRef] [PubMed]
- Extremet, J.; El Far, O.; Ankri, N.; Irani, S.R.; Debanne, D.; Russier, M. An Epitope-Specific LGI1-Autoantibody Enhances Neuronal Excitability by Modulating Kv1.1 Channel. Cells 2022, 11, 2713. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Franco, J.; Debreux, K.; Extremet, J.; Maulet, Y.; Belghazi, M.; Villard, C.; Sangiardi, M.; Youssouf, F.; El Far, L.; Leveque, C.; et al. Patient-derived antibodies reveal the subcellular distribution and heterogeneous interactome of LGI1. Brain 2022, 145, 3843–3858. [Google Scholar] [CrossRef] [PubMed]
- Olsen, A.L.; Lai, Y.; Dalmau, J.; Scherer, S.S.; Lancaster, E. Caspr2 autoantibodies target multiple epitopes. Neurol. Neuroimmunol. Neuroinflamm. 2015, 2, e127. [Google Scholar] [CrossRef]
- van Sonderen, A.; Arino, H.; Petit-Pedrol, M.; Leypoldt, F.; Kortvelyessy, P.; Wandinger, K.P.; Lancaster, E.; Wirtz, P.W.; Schreurs, M.W.; Sillevis Smitt, P.A.; et al. The clinical spectrum of Caspr2 antibody-associated disease. Neurology 2016, 87, 521–528. [Google Scholar] [CrossRef]
- Saint-Martin, M.; Joubert, B.; Pellier-Monnin, V.; Pascual, O.; Noraz, N.; Honnorat, J. Contactin-associated protein-like 2, a protein of the neurexin family involved in several human diseases. Eur. J. Neurosci. 2018, 48, 1906–1923. [Google Scholar] [CrossRef]
- Fernandes, D.; Santos, S.D.; Coutinho, E.; Whitt, J.L.; Beltrao, N.; Rondao, T.; Leite, M.I.; Buckley, C.; Lee, H.K.; Carvalho, A.L. Disrupted AMPA Receptor Function upon Genetic- or Antibody-Mediated Loss of Autism-Associated CASPR2. Cereb Cortex 2019, 29, 4919–4931. [Google Scholar] [CrossRef]
- Corti, E.; Duarte, C.B. The role of post-translational modifications in synaptic AMPA receptor activity. Biochem. Soc. Trans. 2023, 51, 315–330. [Google Scholar] [CrossRef] [PubMed]
- Dalmau, J.; Geis, C.; Graus, F. Autoantibodies to Synaptic Receptors and Neuronal Cell Surface Proteins in Autoimmune Diseases of the Central Nervous System. Physiol. Rev. 2017, 97, 839–887. [Google Scholar] [CrossRef]
- Scheggia, D.; Stanic, J.; Italia, M.; La Greca, F.; Zianni, E.; Benussi, A.; Borroni, B.; Di Luca, M.; Gardoni, F. GluA3 autoantibodies induce alterations in dendritic spine and behavior in mice. Brain Behav. Immun. 2021, 97, 89–101. [Google Scholar] [CrossRef]
- Borroni, B.; Stanic, J.; Verpelli, C.; Mellone, M.; Bonomi, E.; Alberici, A.; Bernasconi, P.; Culotta, L.; Zianni, E.; Archetti, S.; et al. Anti-AMPA GluA3 antibodies in Frontotemporal dementia: A new molecular target. Sci. Rep. 2017, 7, 6723. [Google Scholar] [CrossRef] [PubMed]
- Khayenko, V.; Maric, H.M. Targeting GABA(A)R-Associated Proteins: New Modulators, Labels and Concepts. Front. Mol. Neurosci. 2019, 12, 162. [Google Scholar] [CrossRef] [PubMed]
- Olsen, R.W.; Sieghart, W. GABA A receptors: Subtypes provide diversity of function and pharmacology. Neuropharmacology 2009, 56, 141–148. [Google Scholar] [CrossRef]
- Spatola, M.; Petit-Pedrol, M.; Simabukuro, M.M.; Armangue, T.; Castro, F.J.; Barcelo Artigues, M.I.; Julia Benique, M.R.; Benson, L.; Gorman, M.; Felipe, A.; et al. Investigations in GABA(A) receptor antibody-associated encephalitis. Neurology 2017, 88, 1012–1020. [Google Scholar] [CrossRef]
- Menke, A.F.; Ismail, F.S.; Dornmair, K.; Cerina, M.; Meuth, S.G.; Melzer, N. GABAA Receptor Autoantibodies Decrease GABAergic Synaptic Transmission in the Hippocampal CA3 Network. Int. J. Mol. Sci. 2022, 23, 3707. [Google Scholar] [CrossRef]
- Boronat, A.; Gelfand, J.M.; Gresa-Arribas, N.; Jeong, H.Y.; Walsh, M.; Roberts, K.; Martinez-Hernandez, E.; Rosenfeld, M.R.; Balice-Gordon, R.; Graus, F.; et al. Encephalitis and antibodies to dipeptidyl-peptidase-like protein-6, a subunit of Kv4.2 potassium channels. Ann. Neurol. 2013, 73, 120–128. [Google Scholar] [CrossRef]
- Jerng, H.H.; Pfaffinger, P.J.; Covarrubias, M. Molecular physiology and modulation of somatodendritic A-type potassium channels. Mol. Cell Neurosci. 2004, 27, 343–369. [Google Scholar] [CrossRef]
- Kim, J.; Nadal, M.S.; Clemens, A.M.; Baron, M.; Jung, S.C.; Misumi, Y.; Rudy, B.; Hoffman, D.A. Kv4 accessory protein DPPX (DPP6) is a critical regulator of membrane excitability in hippocampal CA1 pyramidal neurons. J. Neurophysiol. 2008, 100, 1835–1847. [Google Scholar] [CrossRef] [PubMed]
- Nadal, M.S.; Ozaita, A.; Amarillo, Y.; Vega-Saenz de Miera, E.; Ma, Y.; Mo, W.; Goldberg, E.M.; Misumi, Y.; Ikehara, Y.; Neubert, T.A.; et al. The CD26-related dipeptidyl aminopeptidase-like protein DPPX is a critical component of neuronal A-type K+ channels. Neuron 2003, 37, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Maffie, J.K.; Lin, L.; Petralia, R.S.; Rudy, B.; Hoffman, D.A. DPP6 establishes the A-type K(+) current gradient critical for the regulation of dendritic excitability in CA1 hippocampal neurons. Neuron 2011, 71, 1102–1115. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Maekawa, S.; Miyata, S. IgLON cell adhesion molecules regulate synaptogenesis in hippocampal neurons. Cell Biochem. Funct. 2009, 27, 496–498. [Google Scholar] [CrossRef] [PubMed]
- Gaig, C.; Graus, F.; Compta, Y.; Hogl, B.; Bataller, L.; Bruggemann, N.; Giordana, C.; Heidbreder, A.; Kotschet, K.; Lewerenz, J.; et al. Clinical manifestations of the anti-IgLON5 disease. Neurology 2017, 88, 1736–1743. [Google Scholar] [CrossRef] [PubMed]
- Gelpi, E.; Hoftberger, R.; Graus, F.; Ling, H.; Holton, J.L.; Dawson, T.; Popovic, M.; Pretnar-Oblak, J.; Hogl, B.; Schmutzhard, E.; et al. Neuropathological criteria of anti-IgLON5-related tauopathy. Acta Neuropathol. 2016, 132, 531–543. [Google Scholar] [CrossRef] [PubMed]
- Lynch, J.W. Molecular structure and function of the glycine receptor chloride channel. Physiol. Rev. 2004, 84, 1051–1095. [Google Scholar] [CrossRef]
- Avila, A.; Nguyen, L.; Rigo, J.M. Glycine receptors and brain development. Front. Cell Neurosci. 2013, 7, 184. [Google Scholar] [CrossRef]
- Hutchinson, M.; Waters, P.; McHugh, J.; Gorman, G.; O’Riordan, S.; Connolly, S.; Hager, H.; Yu, P.; Becker, C.M.; Vincent, A. Progressive encephalomyelitis, rigidity, and myoclonus: A novel glycine receptor antibody. Neurology 2008, 71, 1291–1292. [Google Scholar] [CrossRef]
- Rauschenberger, V.; von Wardenburg, N.; Schaefer, N.; Ogino, K.; Hirata, H.; Lillesaar, C.; Kluck, C.J.; Meinck, H.M.; Borrmann, M.; Weishaupt, A.; et al. Glycine Receptor Autoantibodies Impair Receptor Function and Induce Motor Dysfunction. Ann. Neurol. 2020, 88, 544–561. [Google Scholar] [CrossRef]
- Bettler, B.; Kaupmann, K.; Mosbacher, J.; Gassmann, M. Molecular structure and physiological functions of GABA(B) receptors. Physiol. Rev. 2004, 84, 835–867. [Google Scholar] [CrossRef] [PubMed]
- Terunuma, M. Diversity of structure and function of GABA(B) receptors: A complexity of GABA(B)-mediated signaling. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2018, 94, 390–411. [Google Scholar] [CrossRef] [PubMed]
- Nibber, A.; Mann, E.O.; Pettingill, P.; Waters, P.; Irani, S.R.; Kullmann, D.M.; Vincent, A.; Lang, B. Pathogenic potential of antibodies to the GABAB receptor. Epilepsia Open 2017, 2, 355–359. [Google Scholar] [CrossRef] [PubMed]
- Kano, M.; Watanabe, T. Type-1 metabotropic glutamate receptor signaling in cerebellar Purkinje cells in health and disease. F1000Research 2017, 6, 416. [Google Scholar] [CrossRef] [PubMed]
- Khojah, O.; Makkawi, S.; Alghamdi, S. Anti-mGluR1 encephalitis: Case illustration and systematic review. Front. Neurol. 2023, 14, 1142160. [Google Scholar] [CrossRef]
- Rodrigues, S.M.; Bauer, E.P.; Farb, C.R.; Schafe, G.E.; LeDoux, J.E. The group I metabotropic glutamate receptor mGluR5 is required for fear memory formation and long-term potentiation in the lateral amygdala. J. Neurosci. 2002, 22, 5219–5229. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S. Inside story of Group I Metabotropic Glutamate Receptors (mGluRs). Int. J. Biochem. Cell Biol. 2016, 77, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Benquet, P.; Gee, C.E.; Gerber, U. Two distinct signaling pathways upregulate NMDA receptor responses via two distinct metabotropic glutamate receptor subtypes. J. Neurosci. 2002, 22, 9679–9686. [Google Scholar] [CrossRef]
- De Blasi, A.; Conn, P.J.; Pin, J.; Nicoletti, F. Molecular determinants of metabotropic glutamate receptor signaling. Trends Pharmacol. Sci. 2001, 22, 114–120. [Google Scholar] [CrossRef]
- Riedel, G.; Casabona, G.; Platt, B.; Macphail, E.M.; Nicoletti, F. Fear conditioning-induced time- and subregion-specific increase in expression of mGlu5 receptor protein in rat hippocampus. Neuropharmacology 2000, 39, 1943–1951. [Google Scholar] [CrossRef]
- Schulz, B.; Fendt, M.; Gasparini, F.; Lingenhohl, K.; Kuhn, R.; Koch, M. The metabotropic glutamate receptor antagonist 2-methyl-6-(phenylethynyl)-pyridine (MPEP) blocks fear conditioning in rats. Neuropharmacology 2001, 41, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.M.; Jia, Z.; Janus, C.; Henderson, J.T.; Gerlai, R.; Wojtowicz, J.M.; Roder, J.C. Mice lacking metabotropic glutamate receptor 5 show impaired learning and reduced CA1 long-term potentiation (LTP) but normal CA3 LTP. J. Neurosci. 1997, 17, 5196–5205. [Google Scholar] [CrossRef] [PubMed]
- Niswender, C.M.; Conn, P.J. Metabotropic glutamate receptors: Physiology, pharmacology, and disease. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 295–322. [Google Scholar] [CrossRef]
- Lancaster, E.; Martinez-Hernandez, E.; Titulaer, M.J.; Boulos, M.; Weaver, S.; Antoine, J.C.; Liebers, E.; Kornblum, C.; Bien, C.G.; Honnorat, J.; et al. Antibodies to metabotropic glutamate receptor 5 in the Ophelia syndrome. Neurology 2011, 77, 1698–1701. [Google Scholar] [CrossRef] [PubMed]
- Maudes, E.; Mannara, F.; Garcia-Serra, A.; Radosevic, M.; Mellado, A.; Serafim, A.B.; Planaguma, J.; Sabater, L.; Dalmau, J.; Spatola, M. Human Metabotropic Glutamate Receptor 5 Antibodies Alter Receptor Levels and Behavior in Mice. Ann. Neurol. 2022, 92, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Aoto, J.; Foldy, C.; Ilcus, S.M.; Tabuchi, K.; Sudhof, T.C. Distinct circuit-dependent functions of presynaptic neurexin-3 at GABAergic and glutamatergic synapses. Nat. Neurosci. 2015, 18, 997–1007. [Google Scholar] [CrossRef] [PubMed]
- Missler, M.; Zhang, W.; Rohlmann, A.; Kattenstroth, G.; Hammer, R.E.; Gottmann, K.; Sudhof, T.C. Alpha-neurexins couple Ca2+ channels to synaptic vesicle exocytosis. Nature 2003, 423, 939–948. [Google Scholar] [CrossRef] [PubMed]
- Contractor, A.; Mulle, C.; Swanson, G.T. Kainate receptors coming of age: Milestones of two decades of research. Trends Neurosci. 2011, 34, 154–163. [Google Scholar] [CrossRef]
- Lerma, J.; Marques, J.M. Kainate receptors in health and disease. Neuron 2013, 80, 292–311. [Google Scholar] [CrossRef]
- Sihra, T.S.; Rodriguez-Moreno, A. Metabotropic actions of kainate receptors in the control of GABA release. Adv. Exp. Med. Biol. 2011, 717, 1–10. [Google Scholar] [CrossRef]
- Lerma, J. Kainate receptor physiology. Curr. Opin. Pharmacol. 2006, 6, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, P.S.; Mulle, C. Presynaptic glutamate receptors: Physiological functions and mechanisms of action. Nat. Rev. Neurosci. 2008, 9, 423–436. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Moreno, A.; Sihra, T.S. Metabotropic actions of kainate receptors in the CNS. J. Neurochem. 2007, 103, 2121–2135. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.W.; Tang, Z.Q. Focusing on the Emerging Role of Kainate Receptors in the Dorsal Cochlear Nucleus (DCN) and Cerebellum. Int. J. Mol. Sci. 2023, 24, 1718. [Google Scholar] [CrossRef] [PubMed]
- Contractor, A.; Swanson, G.; Heinemann, S.F. Kainate receptors are involved in short- and long-term plasticity at mossy fiber synapses in the hippocampus. Neuron 2001, 29, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Jane, D.E.; Lodge, D.; Collingridge, G.L. Kainate receptors: Pharmacology, function and therapeutic potential. Neuropharmacology 2009, 56, 90–113. [Google Scholar] [CrossRef] [PubMed]
- Petrovic, M.M.; Viana da Silva, S.; Clement, J.P.; Vyklicky, L.; Mulle, C.; Gonzalez-Gonzalez, I.M.; Henley, J.M. Metabotropic action of postsynaptic kainate receptors triggers hippocampal long-term potentiation. Nat. Neurosci. 2017, 20, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Simms, B.A.; Zamponi, G.W. Neuronal voltage-gated calcium channels: Structure, function, and dysfunction. Neuron 2014, 82, 24–45. [Google Scholar] [CrossRef]
- Hoppa, M.B.; Lana, B.; Margas, W.; Dolphin, A.C.; Ryan, T.A. alpha2delta expression sets presynaptic calcium channel abundance and release probability. Nature 2012, 486, 122–125. [Google Scholar] [CrossRef]
- Lai, M.; Li, Y.; Luo, D.; Xu, J.; Li, J. Dopamine-2 receptor antibody encephalitis presenting as pure tongue-biting in a tourette syndrome patient: A case report. BMC Psychiatry 2022, 22, 47. [Google Scholar] [CrossRef]
- Yamakawa, M.; Mukaino, A.; Kimura, A.; Nagasako, Y.; Kitazaki, Y.; Maeda, Y.; Higuchi, O.; Takamatsu, K.; Watari, M.; Yoshikura, N.; et al. Antibodies to the alpha3 subunit of the ganglionic-type nicotinic acetylcholine receptors in patients with autoimmune encephalitis. J. Neuroimmunol. 2020, 349, 577399. [Google Scholar] [CrossRef] [PubMed]
- Irani, S.R. ‘Moonlighting’ surface antigens: A paradigm for autoantibody pathogenicity in neurology? Brain 2016, 139, 304–306. [Google Scholar] [CrossRef] [PubMed]
- Dade, M.; Berzero, G.; Izquierdo, C.; Giry, M.; Benazra, M.; Delattre, J.Y.; Psimaras, D.; Alentorn, A. Neurological Syndromes Associated with Anti-GAD Antibodies. Int. J. Mol. Sci. 2020, 21, 3701. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.L.; Rimvall, K. Regulation of gamma-aminobutyric acid synthesis in the brain. J. Neurochem. 1993, 60, 395–407. [Google Scholar] [CrossRef]
- Kanaani, J.; Kolibachuk, J.; Martinez, H.; Baekkeskov, S. Two distinct mechanisms target GAD67 to vesicular pathways and presynaptic clusters. J. Cell Biol. 2010, 190, 911–925. [Google Scholar] [CrossRef]
- Mitoma, H.; Manto, M.; Hampe, C.S. Pathogenic Roles of Glutamic Acid Decarboxylase 65 Autoantibodies in Cerebellar Ataxias. J. Immunol. Res. 2017, 2017, 2913297. [Google Scholar] [CrossRef]
- Jin, H.; Wu, H.; Osterhaus, G.; Wei, J.; Davis, K.; Sha, D.; Floor, E.; Hsu, C.C.; Kopke, R.D.; Wu, J.Y. Demonstration of functional coupling between gamma -aminobutyric acid (GABA) synthesis and vesicular GABA transport into synaptic vesicles. Proc. Natl. Acad. Sci. USA 2003, 100, 4293–4298. [Google Scholar] [CrossRef]
- Saiz, A.; Blanco, Y.; Sabater, L.; Gonzalez, F.; Bataller, L.; Casamitjana, R.; Ramio-Torrenta, L.; Graus, F. Spectrum of neurological syndromes associated with glutamic acid decarboxylase antibodies: Diagnostic clues for this association. Brain 2008, 131, 2553–2563. [Google Scholar] [CrossRef]
- Bjork, E.; Velloso, L.A.; Kampe, O.; Karlsson, F.A. GAD autoantibodies in IDDM, stiff-man syndrome, and autoimmune polyendocrine syndrome type I recognize different epitopes. Diabetes 1994, 43, 161–165. [Google Scholar] [CrossRef]
- Honnorat, J.; Saiz, A.; Giometto, B.; Vincent, A.; Brieva, L.; de Andres, C.; Maestre, J.; Fabien, N.; Vighetto, A.; Casamitjana, R.; et al. Cerebellar ataxia with anti-glutamic acid decarboxylase antibodies: Study of 14 patients. Arch. Neurol. 2001, 58, 225–230. [Google Scholar] [CrossRef]
- Mata, S.; Muscas, G.C.; Cincotta, M.; Bartolozzi, M.L.; Ambrosini, S.; Sorbi, S. GAD antibodies associated neurological disorders: Incidence and phenotype distribution among neurological inflammatory diseases. J. Neuroimmunol. 2010, 227, 175–177. [Google Scholar] [CrossRef] [PubMed]
- Rakocevic, G.; Raju, R.; Semino-Mora, C.; Dalakas, M.C. Stiff person syndrome with cerebellar disease and high-titer anti-GAD antibodies. Neurology 2006, 67, 1068–1070. [Google Scholar] [CrossRef] [PubMed]
- Gresa-Arribas, N.; Arino, H.; Martinez-Hernandez, E.; Petit-Pedrol, M.; Sabater, L.; Saiz, A.; Dalmau, J.; Graus, F. Antibodies to inhibitory synaptic proteins in neurological syndromes associated with glutamic acid decarboxylase autoimmunity. PLoS ONE 2015, 10, e0121364. [Google Scholar] [CrossRef] [PubMed]
- Biljecki, M.; Eisenhut, K.; Beltran, E.; Winklmeier, S.; Mader, S.; Thaller, A.; Eichhorn, P.; Steininger, P.; Flierl-Hecht, A.; Lewerenz, J.; et al. Antibodies Against Glutamic Acid Decarboxylase 65 Are Locally Produced in the CSF and Arise During Affinity Maturation. Neurol. Neuroimmunol. Neuroinflamm. 2023, 10, e200090. [Google Scholar] [CrossRef] [PubMed]
- Towns, R.; Pietropaolo, M. GAD65 autoantibodies and its role as biomarker of Type 1 diabetes and Latent Autoimmune Diabetes in Adults (LADA). Drugs Future 2011, 36, 847. [Google Scholar] [CrossRef] [PubMed]
- Meinck, H.M.; Faber, L.; Morgenthaler, N.; Seissler, J.; Maile, S.; Butler, M.; Solimena, M.; DeCamilli, P.; Scherbaum, W.A. Antibodies against glutamic acid decarboxylase: Prevalence in neurological diseases. J. Neurol. Neurosurg. Psychiatry 2001, 71, 100–103. [Google Scholar] [CrossRef]
- Hampe, C.S.; Petrosini, L.; De Bartolo, P.; Caporali, P.; Cutuli, D.; Laricchiuta, D.; Foti, F.; Radtke, J.R.; Vidova, V.; Honnorat, J.; et al. Monoclonal antibodies to 65kDa glutamate decarboxylase induce epitope specific effects on motor and cognitive functions in rats. Orphanet. J. Rare Dis. 2013, 8, 82. [Google Scholar] [CrossRef]
- Vega-Flores, G.; Rubio, S.E.; Jurado-Parras, M.T.; Gomez-Climent, M.A.; Hampe, C.S.; Manto, M.; Soriano, E.; Pascual, M.; Gruart, A.; Delgado-Garcia, J.M. The GABAergic septohippocampal pathway is directly involved in internal processes related to operant reward learning. Cereb Cortex 2014, 24, 2093–2107. [Google Scholar] [CrossRef]
- Reetz, A.; Solimena, M.; Matteoli, M.; Folli, F.; Takei, K.; De Camilli, P. GABA and pancreatic beta-cells: Colocalization of glutamic acid decarboxylase (GAD) and GABA with synaptic-like microvesicles suggests their role in GABA storage and secretion. EMBO J. 1991, 10, 1275–1284. [Google Scholar] [CrossRef]
- Christgau, S.; Aanstoot, H.J.; Schierbeck, H.; Begley, K.; Tullin, S.; Hejnaes, K.; Baekkeskov, S. Membrane anchoring of the autoantigen GAD65 to microvesicles in pancreatic beta-cells by palmitoylation in the NH2-terminal domain. J. Cell Biol. 1992, 118, 309–320. [Google Scholar] [CrossRef]
- Dinkel, K.; Meinck, H.M.; Jury, K.M.; Karges, W.; Richter, W. Inhibition of gamma-aminobutyric acid synthesis by glutamic acid decarboxylase autoantibodies in stiff-man syndrome. Ann. Neurol. 1998, 44, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Raju, R.; Foote, J.; Banga, J.P.; Hall, T.R.; Padoa, C.J.; Dalakas, M.C.; Ortqvist, E.; Hampe, C.S. Analysis of GAD65 autoantibodies in Stiff-Person syndrome patients. J. Immunol. 2005, 175, 7755–7762. [Google Scholar] [CrossRef] [PubMed]
- Daw, K.; Ujihara, N.; Atkinson, M.; Powers, A.C. Glutamic acid decarboxylase autoantibodies in stiff-man syndrome and insulin-dependent diabetes mellitus exhibit similarities and differences in epitope recognition. J. Immunol. 1996, 156, 818–825. [Google Scholar] [CrossRef] [PubMed]
- Baekkeskov, S.; Aanstoot, H.J.; Christgau, S.; Reetz, A.; Solimena, M.; Cascalho, M.; Folli, F.; Richter-Olesen, H.; De Camilli, P. Identification of the 64K autoantigen in insulin-dependent diabetes as the GABA-synthesizing enzyme glutamic acid decarboxylase. Nature 1990, 347, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Namchuk, M.; Bugawan, T.; Fu, Q.; Jaffe, M.; Shi, Y.; Aanstoot, H.J.; Turck, C.W.; Erlich, H.; Lennon, V.; et al. Higher autoantibody levels and recognition of a linear NH2-terminal epitope in the autoantigen GAD65, distinguish stiff-man syndrome from insulin-dependent diabetes mellitus. J. Exp. Med. 1994, 180, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.H.; Solimena, M.; Dirkx, R., Jr.; Hayday, A.; De Camilli, P. Identification of a dominant epitope of glutamic acid decarboxylase (GAD-65) recognized by autoantibodies in stiff-man syndrome. J. Exp. Med. 1993, 178, 2097–2106. [Google Scholar] [CrossRef] [PubMed]
- Manto, M.; Honnorat, J.; Hampe, C.S.; Guerra-Narbona, R.; Lopez-Ramos, J.C.; Delgado-Garcia, J.M.; Saitow, F.; Suzuki, H.; Yanagawa, Y.; Mizusawa, H.; et al. Disease-specific monoclonal antibodies targeting glutamate decarboxylase impair GABAergic neurotransmission and affect motor learning and behavioral functions. Front. Behav. Neurosci. 2015, 9, 78. [Google Scholar] [CrossRef] [PubMed]
- Manto, M.U.; Hampe, C.S.; Rogemond, V.; Honnorat, J. Respective implications of glutamate decarboxylase antibodies in stiff person syndrome and cerebellar ataxia. Orphanet. J. Rare Dis. 2011, 6, 3. [Google Scholar] [CrossRef]
- Fouka, P.; Alexopoulos, H.; Akrivou, S.; Trohatou, O.; Politis, P.K.; Dalakas, M.C. GAD65 epitope mapping and search for novel autoantibodies in GAD-associated neurological disorders. J. Neuroimmunol. 2015, 281, 73–77. [Google Scholar] [CrossRef]
- Manto, M.U.; Laute, M.A.; Aguera, M.; Rogemond, V.; Pandolfo, M.; Honnorat, J. Effects of anti-glutamic acid decarboxylase antibodies associated with neurological diseases. Ann. Neurol. 2007, 61, 544–551. [Google Scholar] [CrossRef]
- Ishida, K.; Mitoma, H.; Song, S.Y.; Uchihara, T.; Inaba, A.; Eguchi, S.; Kobayashi, T.; Mizusawa, H. Selective suppression of cerebellar GABAergic transmission by an autoantibody to glutamic acid decarboxylase. Ann. Neurol. 1999, 46, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Mitoma, H.; Ishida, K.; Shizuka-Ikeda, M.; Mizusawa, H. Dual impairment of GABAA- and GABAB-receptor-mediated synaptic responses by autoantibodies to glutamic acid decarboxylase. J. Neurol. Sci. 2003, 208, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Vianello, M.; Bisson, G.; Dal Maschio, M.; Vassanelli, S.; Girardi, S.; Mucignat, C.; Fountzoulas, K.; Giometto, B. Increased spontaneous activity of a network of hippocampal neurons in culture caused by suppression of inhibitory potentials mediated by anti-gad antibodies. Autoimmunity 2008, 41, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Takenoshita, H.; Shizuka-Ikeda, M.; Mitoma, H.; Song, S.; Harigaya, Y.; Igeta, Y.; Yaguchi, M.; Ishida, K.; Shoji, M.; Tanaka, M.; et al. Presynaptic inhibition of cerebellar GABAergic transmission by glutamate decarboxylase autoantibodies in progressive cerebellar ataxia. J. Neurol. Neurosurg. Psychiatry 2001, 70, 386–389. [Google Scholar] [CrossRef] [PubMed]
- Mitoma, H.; Song, S.Y.; Ishida, K.; Yamakuni, T.; Kobayashi, T.; Mizusawa, H. Presynaptic impairment of cerebellar inhibitory synapses by an autoantibody to glutamate decarboxylase. J. Neurol. Sci. 2000, 175, 40–44. [Google Scholar] [CrossRef]
- Stemmler, N.; Rohleder, K.; Malter, M.P.; Widman, G.; Elger, C.E.; Beck, H.; Surges, R. Serum from a Patient with GAD65 Antibody-Associated Limbic Encephalitis Did Not Alter GABAergic Neurotransmission in Cultured Hippocampal Networks. Front. Neurol. 2015, 6, 189. [Google Scholar] [CrossRef]
- Hackert, J.K.; Muller, L.; Rohde, M.; Bien, C.G.; Kohling, R.; Kirschstein, T. Anti-GAD65 Containing Cerebrospinal Fluid Does not Alter GABAergic Transmission. Front. Cell Neurosci. 2016, 10, 130. [Google Scholar] [CrossRef]
- Hansen, N.; Grunewald, B.; Weishaupt, A.; Colaco, M.N.; Toyka, K.V.; Sommer, C.; Geis, C. Human Stiff person syndrome IgG-containing high-titer anti-GAD65 autoantibodies induce motor dysfunction in rats. Exp. Neurol. 2013, 239, 202–209. [Google Scholar] [CrossRef]
- Geis, C.; Weishaupt, A.; Grunewald, B.; Wultsch, T.; Reif, A.; Gerlach, M.; Dirkx, R.; Solimena, M.; Perani, D.; Heckmann, M.; et al. Human stiff-person syndrome IgG induces anxious behavior in rats. PLoS ONE 2011, 6, e16775. [Google Scholar] [CrossRef]
- Han, G.; Li, Y.; Wang, J.; Wang, R.; Chen, G.; Song, L.; Xu, R.; Yu, M.; Wu, X.; Qian, J.; et al. Active tolerance induction and prevention of autoimmune diabetes by immunogene therapy using recombinant adenoassociated virus expressing glutamic acid decarboxylase 65 peptide GAD(500-585). J. Immunol. 2005, 174, 4516–4524. [Google Scholar] [CrossRef]
- Chang, T.; Alexopoulos, H.; Pettingill, P.; McMenamin, M.; Deacon, R.; Erdelyi, F.; Szabo, G.; Buckley, C.J.; Vincent, A. Immunization against GAD induces antibody binding to GAD-independent antigens and brainstem GABAergic neuronal loss. PLoS ONE 2013, 8, e72921. [Google Scholar] [CrossRef]
- De Camilli, P.; Thomas, A.; Cofiell, R.; Folli, F.; Lichte, B.; Piccolo, G.; Meinck, H.M.; Austoni, M.; Fassetta, G.; Bottazzo, G.; et al. The synaptic vesicle-associated protein amphiphysin is the 128-kD autoantigen of Stiff-Man syndrome with breast cancer. J. Exp. Med. 1993, 178, 2219–2223. [Google Scholar] [CrossRef] [PubMed]
- Lichte, B.; Veh, R.W.; Meyer, H.E.; Kilimann, M.W. Amphiphysin, a novel protein associated with synaptic vesicles. EMBO J. 1992, 11, 2521–2530. [Google Scholar] [CrossRef] [PubMed]
- Folli, F.; Solimena, M.; Cofiell, R.; Austoni, M.; Tallini, G.; Fassetta, G.; Bates, D.; Cartlidge, N.; Bottazzo, G.F.; Piccolo, G.; et al. Autoantibodies to a 128-kd synaptic protein in three women with the stiff-man syndrome and breast cancer. N. Engl. J. Med. 1993, 328, 546–551. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Zelhof, A.C. Amphiphysins: Raising the BAR for synaptic vesicle recycling and membrane dynamics. Bin-Amphiphysin-Rvsp. Traffic 2002, 3, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Prokic, I.; Cowling, B.S.; Laporte, J. Amphiphysin 2 (BIN1) in physiology and diseases. J. Mol. Med. 2014, 92, 453–463. [Google Scholar] [CrossRef]
- Wu, Y.; Matsui, H.; Tomizawa, K. Amphiphysin I and regulation of synaptic vesicle endocytosis. Acta. Med. Okayama 2009, 63, 305–323. [Google Scholar] [CrossRef]
- Sommer, C.; Weishaupt, A.; Brinkhoff, J.; Biko, L.; Wessig, C.; Gold, R.; Toyka, K.V. Paraneoplastic stiff-person syndrome: Passive transfer to rats by means of IgG antibodies to amphiphysin. Lancet 2005, 365, 1406–1411. [Google Scholar] [CrossRef]
- Geis, C.; Beck, M.; Jablonka, S.; Weishaupt, A.; Toyka, K.V.; Sendtner, M.; Sommer, C. Stiff person syndrome associated anti-amphiphysin antibodies reduce GABA associated [Ca2+]i rise in embryonic motoneurons. Neurobiol. Dis. 2009, 36, 191–199. [Google Scholar] [CrossRef]
- Geis, C.; Weishaupt, A.; Hallermann, S.; Grunewald, B.; Wessig, C.; Wultsch, T.; Reif, A.; Byts, N.; Beck, M.; Jablonka, S.; et al. Stiff person syndrome-associated autoantibodies to amphiphysin mediate reduced GABAergic inhibition. Brain 2010, 133, 3166–3180. [Google Scholar] [CrossRef]
- Werner, C.; Pauli, M.; Doose, S.; Weishaupt, A.; Haselmann, H.; Grunewald, B.; Sauer, M.; Heckmann, M.; Toyka, K.V.; Asan, E.; et al. Human autoantibodies to amphiphysin induce defective presynaptic vesicle dynamics and composition. Brain 2016, 139, 365–379. [Google Scholar] [CrossRef] [PubMed]
- Geis, C.; Grunewald, B.; Weishaupt, A.; Wultsch, T.; Toyka, K.V.; Reif, A.; Sommer, C. Human IgG directed against amphiphysin induces anxiety behavior in a rat model after intrathecal passive transfer. J. Neural. Transm. 2012, 119, 981–985. [Google Scholar] [CrossRef] [PubMed]
- Pitsch, J.; Kamalizade, D.; Braun, A.; Kuehn, J.C.; Gulakova, P.E.; Ruber, T.; Lubec, G.; Dietrich, D.; von Wrede, R.; Helmstaedter, C.; et al. Drebrin Autoantibodies in Patients with Seizures and Suspected Encephalitis. Ann. Neurol. 2020, 87, 869–884. [Google Scholar] [CrossRef] [PubMed]
- Shirao, T.; Sekino, Y. General Introduction to Drebrin. Adv. Exp. Med. Biol. 2017, 1006, 3–22. [Google Scholar] [CrossRef] [PubMed]
- Koganezawa, N.; Hanamura, K.; Sekino, Y.; Shirao, T. The role of drebrin in dendritic spines. Mol. Cell Neurosci. 2017, 84, 85–92. [Google Scholar] [CrossRef]
- Dzamba, D.; Honsa, P.; Anderova, M. NMDA Receptors in Glial Cells: Pending Questions. Curr. Neuropharmacol. 2013, 11, 250–262. [Google Scholar] [CrossRef]
- Kim, J.E.; Choi, H.C.; Song, H.K.; Kang, T.C. Blockade of AMPA Receptor Regulates Mitochondrial Dynamics by Modulating ERK1/2 and PP1/PP2A-Mediated DRP1-S616 Phosphorylations in the Normal Rat Hippocampus. Front. Cell Neurosci. 2019, 13, 179. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Classification of AE Autoantibodies |
|---|
| Class I—Confirmed pathogenicity |
| NMDAR, LGI1, CASPR2, AMPAR, GABAAR, DPPX, IgLON5, GlyR, GAD65 |
| Class II—Highly suspected pathogenicity |
| GABABR, mGluR1, mGluR5, Neurexin-3α, GluK2, CaVα2δ, Amphiphysin, Drebrin |
| Class III—Pathogenicity not established |
| D2R, mGluR2, SEZ6L2, GluRδ2 |
| Antigen/ IgG Subtype | Effect on the Antigen | Cellular Consequences | In Vivo Manifestations | Clinical Manifestations |
|---|---|---|---|---|
| Class I: Autoantibodies with confirmed pathogenicity | ||||
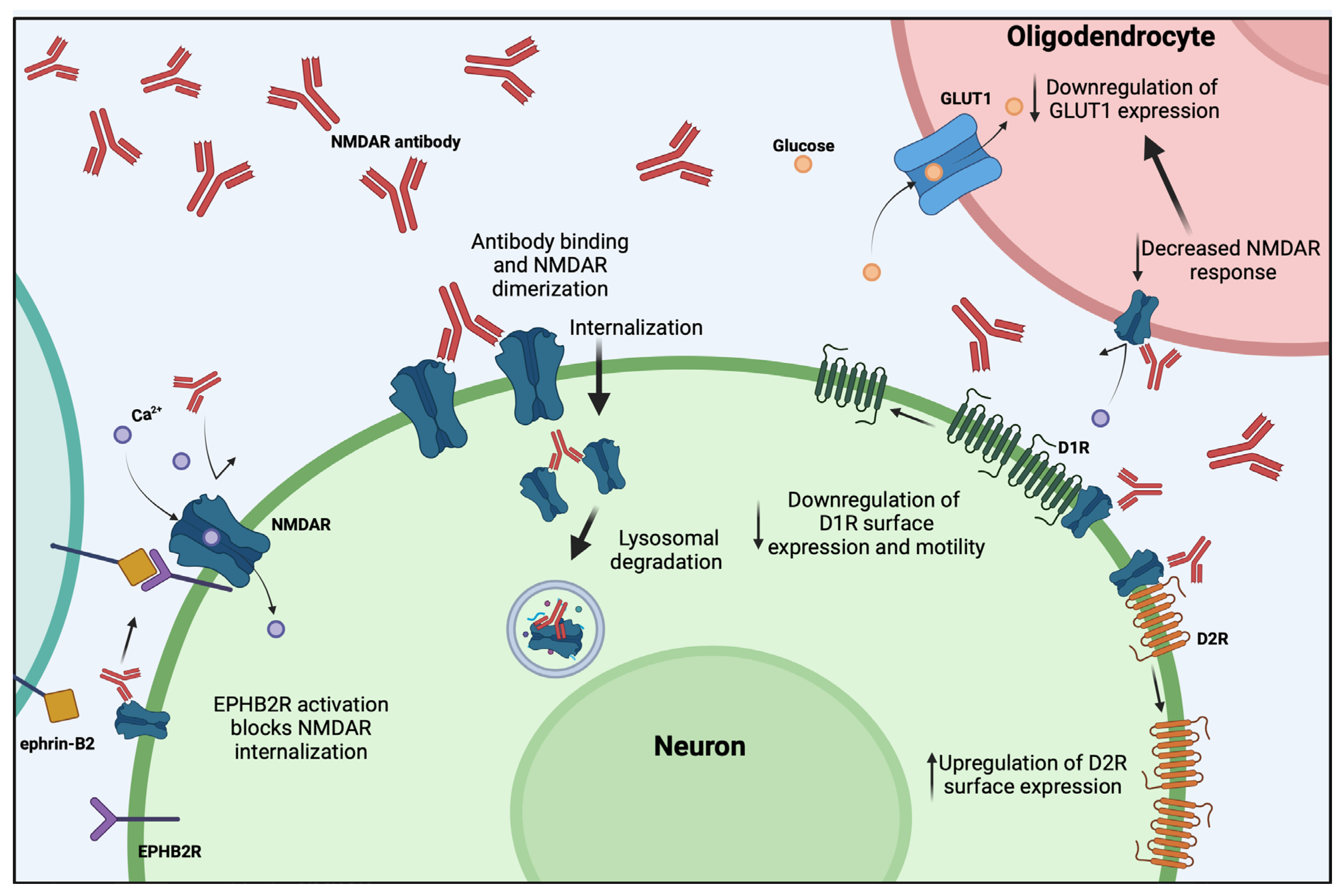
| NMDAR/ Primarily IgG1 | Cross-linking, internalization and lysosomal degradation of NMDA receptors (reversible) [31,32,33,34]. Prevented by activation of EPHB2R [34,35]. | Decreased neuronal excitability [36] and suppressed NMDAR dependent long term plasticity but not short term [35,37]. Decreased GLUT1 expression in oligodendrocytes [38]. Altered D1R and D2R surface expression and dynamics [39,40]. | Memory impairment and depression [41,42,43]. One study reports infiltration of immune cells [44] while one reports no infiltration [43]. Seizures [45]. | Psychiatric and cognitive dysfunction including anxiety and memory impairment. Dyskinesias, seizures, reduced verbal output, insomnia, autonomic dysfunction [31]. |
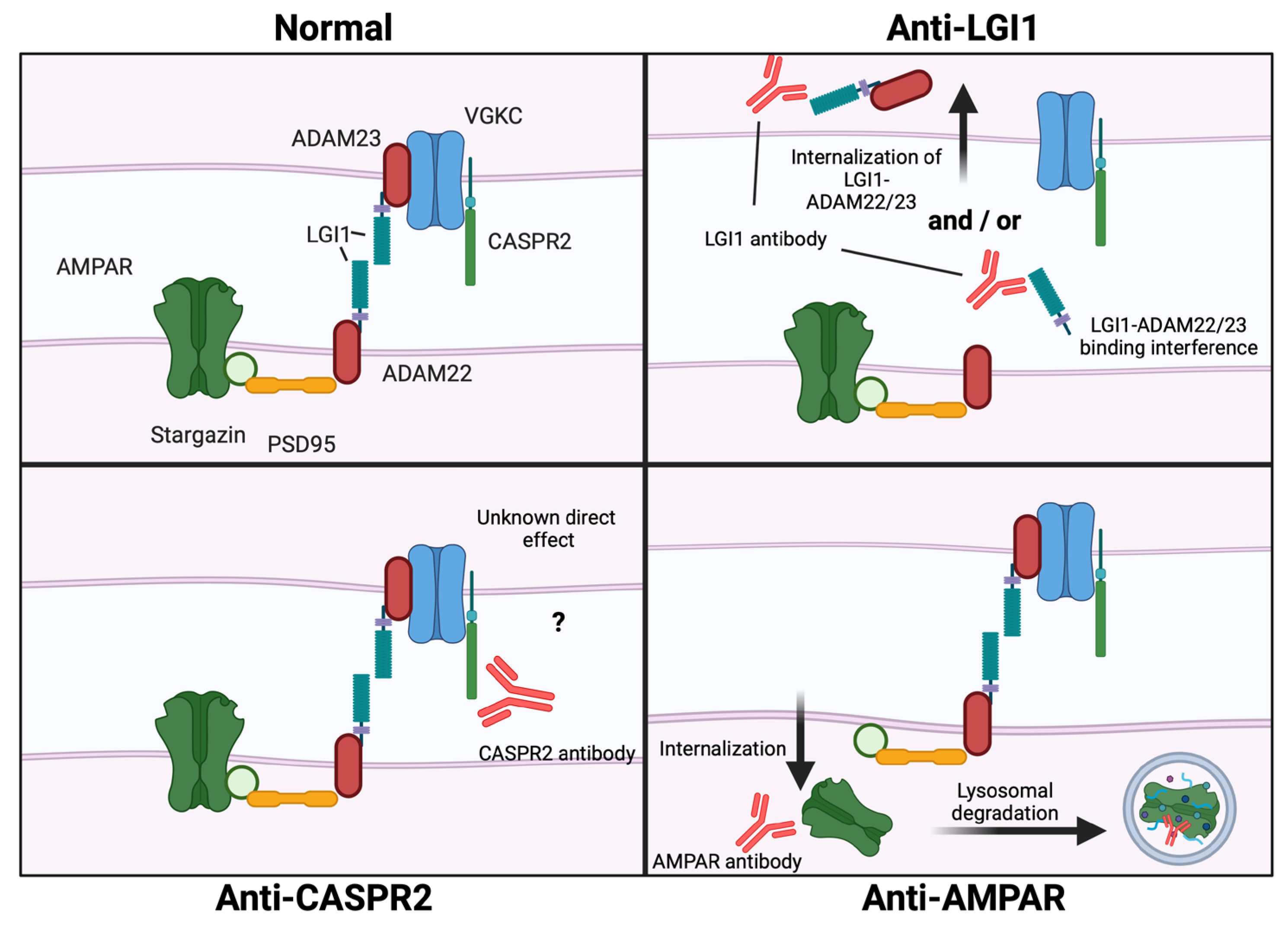
| LGI1/ Primarily IgG4 | Interference of LGI1—ADAM22/ADAM23 interaction [46,47,48]. One study reports LGI1—ADAM22/ADAM23 complex internalization dependent on autoantibody domain binding characteristics [49]. | Increased neuronal excitability (including increased spike frequency and amplitude) [47,48,50]. Reduced synaptic AMPARs and Kv1.1 clusters [46,47]. Impaired LTP [47,49]. | Memory impairments [47,49] and tight junction breakdown in BBB [51]. | Limbic encephalitis, Facio-brachial dystonic seizures (FBDS) [52]. |
| Caspr2/ Primarily IgG4 | Interference of Caspr2—contactin-2 interaction [53], Caspr2 cluster formation [54], and potentially increased Caspr2 expression on the cell surface [55]. One publication reports no changes in Caspr2 surface expression [53]. | Increased Kv1_2 channel expression [54,56] and Increased neuronal excitability [50,56]. Increased microglial density, altered glial morphology and raised complement C3 expression [55]. | Increased sensitivity towards pain [56], memory impairments and behavioral changes [55,57]. | Peripheral neuropathy, neuropathic pain, neuromyotonia, Morvan syndrome, limbic encephalitis [52]. |
| AMPAR/ Unknown | Decrease in GluA1 and GluA2 containing AMPARS by internalization and degradation [58,59,60]. | Decreased AMPAR mediated currents and impaired LTP [59,60,61]. | Memory deficits and increased anxiety [60]. | Limbic encephalitis, encephalopathy, seizures, prominent psychiatric symptoms [62]. |
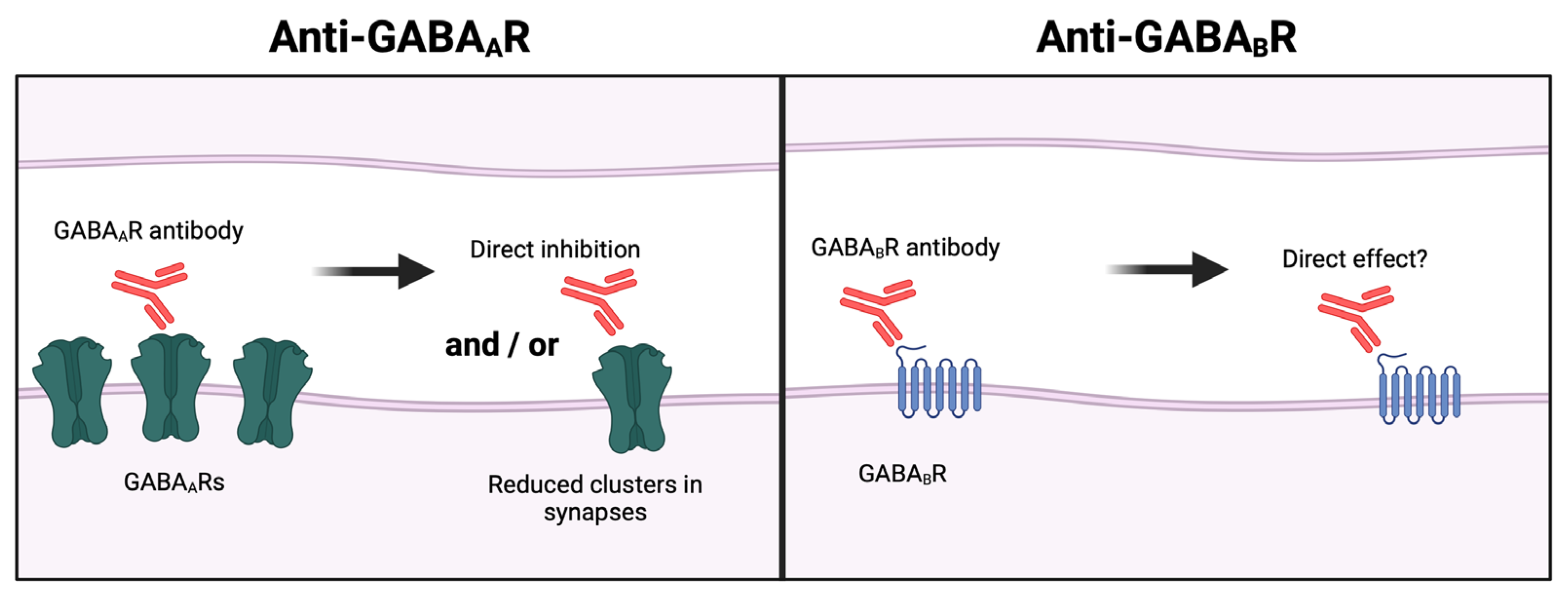
| GABAAR/ Primarily IgG1 | Reduction in GABAaR clusters in synapses [63,64,65] and/or direct inhibition [66]. | Decreased mean amplitude of inhibitory postsynaptic currents [64,67,68]. | Seizures [68]. | Subacute encephalopathy, seizures, status epilepticus [63]. |
| DPPX/ IgG1 and IgG 4 | Reduction in DPPX clusters and Kv4.2 subtype potassium channels [69,70]. | Increased excitability and action potential rate of enteric neurons [70]. | Encephalopathy with diarrhea and weight loss, myoclonus, seizures [71]. | |
| IgLON5/ IgG4 > IgG1 | Irreversible internalization of IgLON5 clusters [72,73,74]. | Decreased number of synapses, and spike rate [73,74]. Axonal degenerative changes [75] Accumulation of phosphorylated Tau and increased cell death [73]. | Cognitive abnormalities, astrocyte and microglia recruitment [74]. Tau deposition [76]. | Non-REM parasomnia, obstructive sleep apnea, bulbar symptoms, cognitive decline, gait instability, dystonia, chorea. May resemble neurodegenerative diseases or motor neuron disease [77]. |
| GlycineR IgG1 and IgG 3 | Internalization and lysosomal degradation of glycine receptors, and possible complement activation through binding of C3 [78]. Or, direct inhibition of GlycineR [79]. | Reduced glycinergic transmission [79]. | Increased anxiety [80]. | Progressive Encephalopathy with Rigidity and Myoclonus (PERM), limb paralysis, cognitive impairment, seizures [78]. |
| Class II: Autoantibodies with highly suspected pathogenicity | ||||
| GABABR/ Primarily IgG1 | No change in GABAbR expression, but direct inhibition of receptor function [81]. | One study reports decreased excitability of neurons [81] while another reports no changes [50]. | Limbic encephalitis, seizures [82]. | |
| mGluR1/ Primarily IgG1 | Decreased number of mGluR1 clusters [83]. | Reduced LTD [84]. | Reversible ataxia [85]. Decreased compensatory eye movements [83]. | Subacute cerebellar ataxia syndrome [83]. |
| mGluR5/ Primarily IgG1 | Decrease in mGluR5 clusters, reversible upon removal [86]. | Encephalopathy, Ophelia syndrome (Hodgkins lymphoma) [86]. | ||
| Neurexin-3α/ Primarily IgG1 | Reduction in Neurexin-3α clusters [87]. | Decreased number of synapses [87]. | Confusion, seizures and decreased level of consciousness [87]. | |
| GluK-2/ Primarily IgG1 | Reversible internalization [88]. | Reduced GluK-2 mediated currents [88]. | Headache, nausea, vomiting, decreased level of consciousness. Or acute cerebellitis with hydrocephalus [88]. | |
| CaVα2δ/ Unknown | Reduced excitatory and inhibitory signaling [89]. | Memory loss, psychosis and seizures [89]. | ||
| Class III: Autoantibodies without established pathogenicity | ||||
| D2R/ Unknown | Lethargy, dystonia, agitation, confusion parkinsonism [90]. | |||
| mGluR2/ Primarily IgG1 | Cerebellar ataxia [91]. | |||
| SEZ6L2/ Primarily IgG4 | Dysarthria, ataxia, bradykinesia [92]. | |||
| GluRδ2/ Unknown | Cerebellar ataxia [93]. | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ryding, M.; Mikkelsen, A.W.; Nissen, M.S.; Nilsson, A.C.; Blaabjerg, M. Pathophysiological Effects of Autoantibodies in Autoimmune Encephalitides. Cells 2024, 13, 15. https://doi.org/10.3390/cells13010015
Ryding M, Mikkelsen AW, Nissen MS, Nilsson AC, Blaabjerg M. Pathophysiological Effects of Autoantibodies in Autoimmune Encephalitides. Cells. 2024; 13(1):15. https://doi.org/10.3390/cells13010015
Chicago/Turabian StyleRyding, Matias, Anne With Mikkelsen, Mette Scheller Nissen, Anna Christine Nilsson, and Morten Blaabjerg. 2024. "Pathophysiological Effects of Autoantibodies in Autoimmune Encephalitides" Cells 13, no. 1: 15. https://doi.org/10.3390/cells13010015