
Regulation of m6A Methylome in Cancer: Mechanisms, Implications, and Therapeutic Strategies

Abstract
:1. Introduction
1.1. Writers
1.2. Readers
1.3. Erasers
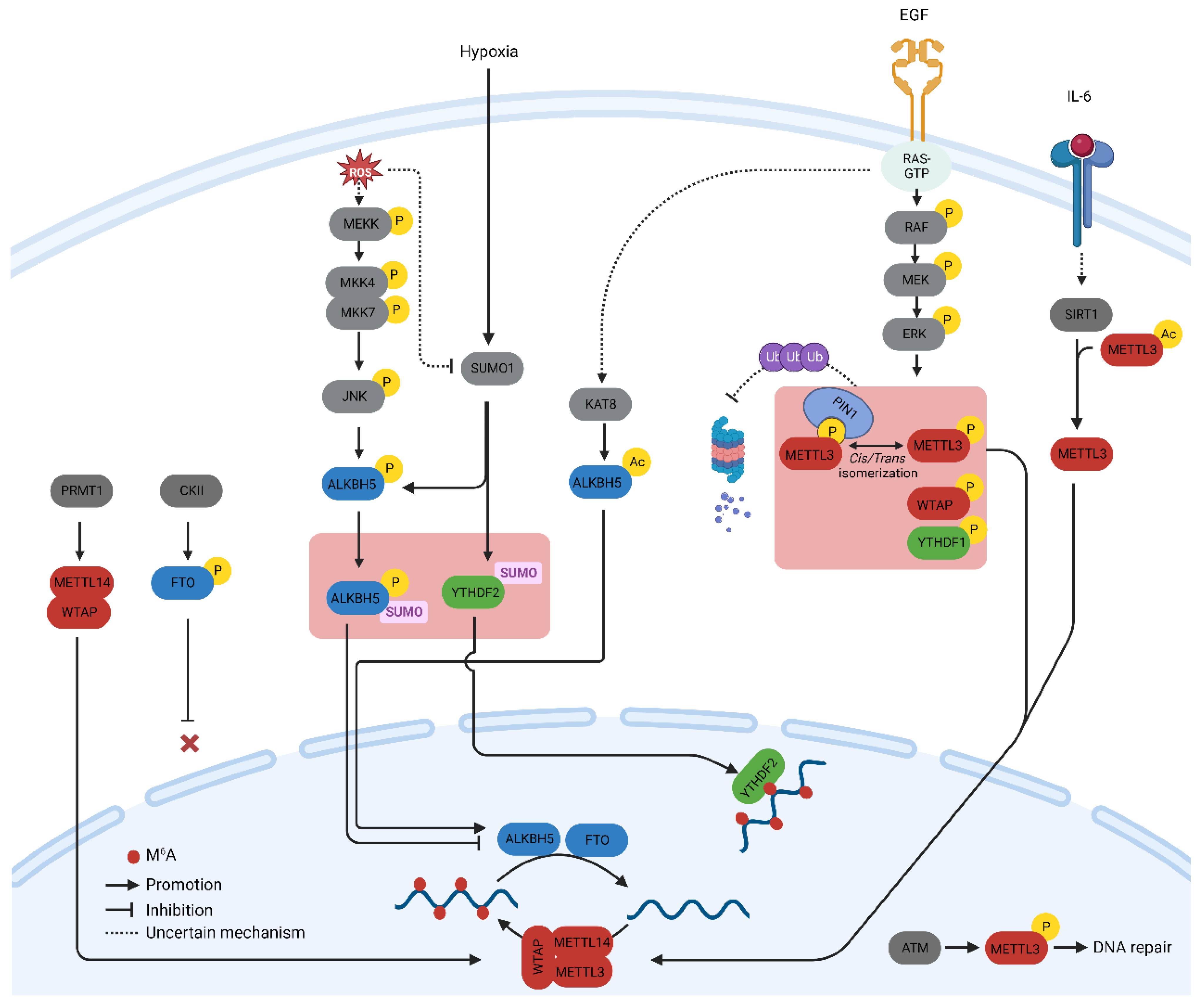
2. Post-Translational Modification of m6A-Related Proteins
2.1. Phosphorylation
2.2. Methylation
2.3. Acetylation
2.4. SUMOylation
2.5. O-GlcNAcylation
3. Transcriptional Activation of m6A-Related Genes
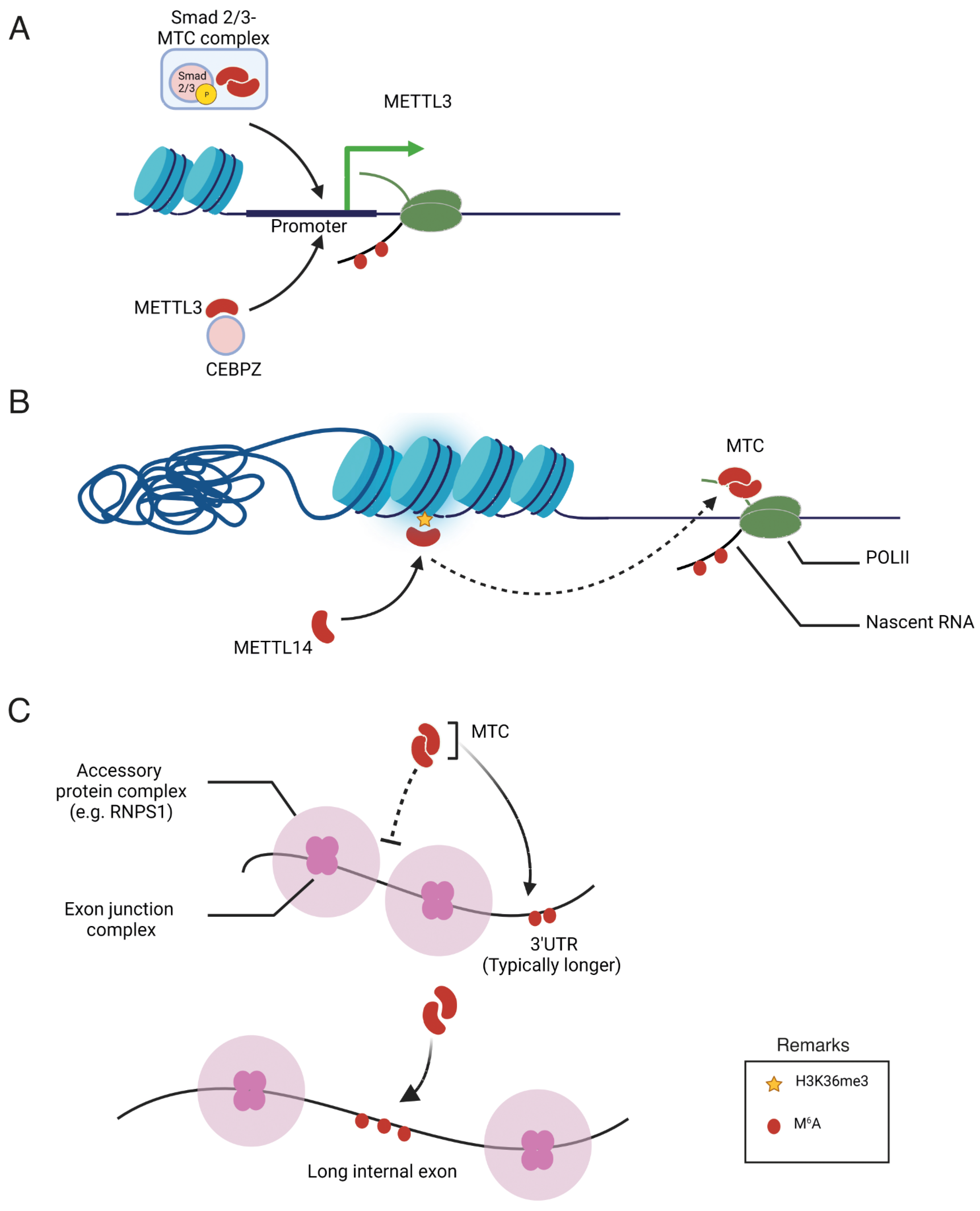
4. Regulation of m6A Specificity
4.1. Transcription Factors and Co-Activators
4.2. Epigenetic Modification
4.3. Exon Architecture
5. Dysregulation of m6A in Cancer and Chemoresistance
6. Future Perspective
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kumar, S.; Mohapatra, T. Deciphering Epitranscriptome: Modification of mRNA Bases Provides a New Perspective for Post-transcriptional Regulation of Gene Expression. Front. Cell Dev. Biol. 2021, 9, 628415. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yue, Y.; Han, D.; Wang, X.; Fu, Y.; Zhang, L.; Jia, G.; Yu, M.; Lu, Z.; Deng, X.; et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 2014, 10, 93–95. [Google Scholar] [CrossRef] [PubMed]
- Zaccara, S.; Ries, R.J.; Jaffrey, S.R. Reading, writing and erasing mRNA methylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 608–624. [Google Scholar] [CrossRef] [PubMed]
- Ping, X.-L.; Sun, B.-F.; Wang, L.; Xiao, W.; Yang, X.; Wang, W.-J.; Adhikari, S.; Shi, Y.; Lv, Y.; Chen, Y.-S.; et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014, 24, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Doxtader, K.A.; Nam, Y. Structural Basis for Cooperative Function of Mettl3 and Mettl14 Methyltransferases. Mol. Cell 2016, 63, 306–317. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Feng, J.; Xue, Y.; Guan, Z.; Zhang, D.; Liu, Z.; Gong, Z.; Wang, Q.; Huang, J.; Tang, C.; et al. Structural basis of N6-adenosine methylation by the METTL3–METTL14 complex. Nature 2016, 534, 575–578. [Google Scholar] [CrossRef] [PubMed]
- Bokar, J.A.; Shambaugh, M.E.; Polayes, D.; Matera, A.G.; Rottman, F.M. Purification and cDNA cloning of the AdoMet-binding subunit of the human mRNA (N6-adenosine)-methyltransferase. Rna 1997, 3, 1233–1247. [Google Scholar]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; Salmon-Divon, M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Kupiec, M.; et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012, 485, 201–206. [Google Scholar] [CrossRef]
- Roost, C.; Lynch, S.R.; Batista, P.J.; Qu, K.; Chang, H.Y.; Kool, E.T. Structure and thermodynamics of N6-methyladenosine in RNA: A spring-loaded base modification. J. Am. Chem. Soc. 2015, 137, 2107–2115. [Google Scholar] [CrossRef]
- Liu, N.; Dai, Q.; Zheng, G.; He, C.; Parisien, M.; Pan, T. N6-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature 2015, 518, 560–564. [Google Scholar] [CrossRef]
- Gerken, T.; Girard, C.A.; Tung, Y.C.; Webby, C.J.; Saudek, V.; Hewitson, K.S.; Yeo, G.S.; McDonough, M.A.; Cunliffe, S.; McNeill, L.A.; et al. The obesity-associated FTO gene encodes a 2-oxoglutarate-dependent nucleic acid demethylase. Science 2007, 318, 1469–1472. [Google Scholar] [CrossRef] [PubMed]
- Fedeles, B.I.; Singh, V.; Delaney, J.C.; Li, D.; Essigmann, J.M. The AlkB Family of Fe(II)/α-Ketoglutarate-dependent Dioxygenases: Repairing Nucleic Acid Alkylation Damage and Beyond. J. Biol. Chem. 2015, 290, 20734–20742. [Google Scholar] [CrossRef] [PubMed]
- Mauer, J.; Luo, X.; Blanjoie, A.; Jiao, X.; Grozhik, A.V.; Patil, D.P.; Linder, B.; Pickering, B.F.; Vasseur, J.J.; Chen, Q.; et al. Reversible methylation of m6Am in the 5’ cap controls mRNA stability. Nature 2017, 541, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Ardito, F.; Giuliani, M.; Perrone, D.; Troiano, G.; Lo Muzio, L. The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy (Review). Int. J. Mol. Med. 2017, 40, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.L.; Zhu, A.C.; Gao, Y.; Terajima, H.; Fei, Q.; Liu, S.; Zhang, L.; Zhang, Z.; Harada, B.T.; He, Y.Y.; et al. Stabilization of ERK-Phosphorylated METTL3 by USP5 Increases m6A Methylation. Mol. Cell 2020, 80, 633–647.e7. [Google Scholar] [CrossRef] [PubMed]
- Perez-Pepe, M.; Desotell, A.W.; Li, H.; Li, W.; Han, B.; Lin, Q.; Klein, D.E.; Liu, Y.; Goodarzi, H.; Alarcón, C.R. 7SK methylation by METTL3 promotes transcriptional activity. Sci. Adv. 2023, 9, eade7500. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Chen, L.; Peng, D.; Jiang, A.; He, Y.; Zeng, Y.; Xie, C.; Zhou, H.; Luo, X.; Liu, H.; et al. METTL3 and N6-Methyladenosine Promote Homologous Recombination-Mediated Repair of DSBs by Modulating DNA-RNA Hybrid Accumulation. Mol. Cell 2020, 79, 425–442.e7. [Google Scholar] [CrossRef]
- Bhattarai, P.Y.; Kim, G.; Lim, S.-C.; Mariappan, R.; Ohn, T.; Choi, H.S. METTL3 stabilization by PIN1 promotes breast tumorigenesis via enhanced m6A-dependent translation. Oncogene 2023, 42, 1010–1023. [Google Scholar] [CrossRef]
- Schöller, E.; Weichmann, F.; Treiber, T.; Ringle, S.; Treiber, N.; Flatley, A.; Feederle, R.; Bruckmann, A.; Meister, G. Interactions, localization, and phosphorylation of the m6A generating METTL3-METTL14-WTAP complex. Rna 2018, 24, 499–512. [Google Scholar] [CrossRef]
- Patil, D.P.; Pickering, B.F.; Jaffrey, S.R. Reading m6A in the Transcriptome: m6A-Binding Proteins. Trends Cell Biol. 2018, 28, 113–127. [Google Scholar] [CrossRef]
- Fang, R.; Chen, X.; Zhang, S.; Shi, H.; Ye, Y.; Shi, H.; Zou, Z.; Li, P.; Guo, Q.; Ma, L.; et al. EGFR/SRC/ERK-stabilized YTHDF2 promotes cholesterol dysregulation and invasive growth of glioblastoma. Nat. Commun. 2021, 12, 177. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, M.; Wei, F.Y.; Chujo, T.; Oki, S.; Yakita, M.; Kobayashi, D.; Araki, N.; Takahashi, N.; Yoshida, R.; Nakayama, H.; et al. FTO Demethylates Cyclin D1 mRNA and Controls Cell-Cycle Progression. Cell Rep. 2020, 31, 107464. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Wei, J.; Cui, X.; Yu, C.; Ni, W.; Bungert, J.; Wu, L.; He, C.; Qian, Z. Post-translational modification of RNA m6A demethylase ALKBH5 regulates ROS-induced DNA damage response. Nucleic Acids Res. 2021, 49, 5779–5797. [Google Scholar] [CrossRef] [PubMed]
- Biggar, K.K.; Li, S.S.C. Non-histone protein methylation as a regulator of cellular signalling and function. Nat. Rev. Mol. Cell Biol. 2015, 16, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, H.; Zhao, X.; Luo, Q.; Wang, Q.; Tan, K.; Wang, Z.; Jiang, J.; Cui, J.; Du, E.; et al. Arginine methylation of METTL14 promotes RNA N6-methyladenosine modification and endoderm differentiation of mouse embryonic stem cells. Nat. Commun. 2021, 12, 3780. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, Z.; Inuzuka, H.; Wei, W.; Liu, J. PRMT1 methylates METTL14 to modulate its oncogenic function. Neoplasia 2023, 42, 100912. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Yu, X.; Liu, R.; Shi, L.; Jin, H.; Yang, D.; Zhang, X.; Shen, Y.; Feng, Y.; Zhang, P.; et al. PRMT1 methylation of WTAP promotes multiple myeloma tumorigenesis by activating oxidative phosphorylation via m6A modification of NDUFS6. Cell Death Dis. 2023, 14, 512. [Google Scholar] [CrossRef]
- Shang, S.; Liu, J.; Hua, F. Protein acylation: Mechanisms, biological functions and therapeutic targets. Signal Transduct. Target. Ther. 2022, 7, 396. [Google Scholar] [CrossRef]
- Li, Y.; He, X.; Lu, X.; Gong, Z.; Li, Q.; Zhang, L.; Yang, R.; Wu, C.; Huang, J.; Ding, J.; et al. METTL3 acetylation impedes cancer metastasis via fine-tuning its nuclear and cytosolic functions. Nat. Commun. 2022, 13, 6350. [Google Scholar] [CrossRef]
- Zhang, G.; Huang, R.; Zhao, H.; Xia, Y.; Huang, H.; Qian, M.; Fu, Y.; Cui, Y. ACAT1-mediated METTL3 acetylation inhibits cell migration and invasion in triple negative breast cancer. Genes Immun. 2023, 24, 99–107. [Google Scholar] [CrossRef]
- Zhang, X.-L.; Chen, X.-H.; Xu, B.; Chen, M.; Zhu, S.; Meng, N.; Wang, J.-Z.; Zhu, H.; Chen, D.; Liu, J.-B.; et al. K235 acetylation couples with PSPC1 to regulate the m6A demethylation activity of ALKBH5 and tumorigenesis. Nat. Commun. 2023, 14, 3815. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Hou, G.; Zhang, H.; Dou, J.; He, J.; Guo, Y.; Li, L.; Chen, R.; Wang, Y.; Deng, R.; et al. SUMOylation of the m6A-RNA methyltransferase METTL3 modulates its function. Nucleic Acids Res. 2018, 46, 5195–5208. [Google Scholar] [CrossRef] [PubMed]
- Hou, G.; Zhao, X.; Li, L.; Yang, Q.; Liu, X.; Huang, C.; Lu, R.; Chen, R.; Wang, Y.; Jiang, B.; et al. SUMOylation of YTHDF2 promotes mRNA degradation and cancer progression by increasing its binding affinity with m6A-modified mRNAs. Nucleic Acids Res. 2021, 49, 2859–2877. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ahmad, M.; Sang, L.; Zhan, Y.; Wang, Y.; Yan, Y.; Liu, Y.; Mi, W.; Lu, M.; Dai, Y.; et al. O-GlcNAcylation promotes the cytosolic localization of the m6A reader YTHDF1 and colorectal cancer tumorigenesis. J. Biol. Chem. 2023, 299, 104738. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Chen, C.; Ding, Q.; Zhao, Y.; Wang, Z.; Chen, J.; Jiang, Z.; Zhang, Y.; Xu, G.; Zhang, J.; et al. METTL3-mediated m6A modification of HDGF mRNA promotes gastric cancer progression and has prognostic significance. Gut 2020, 69, 1193–1205. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Bai, R.; Li, M.; Ye, H.; Wu, C.; Wang, C.; Li, S.; Tan, L.; Mai, D.; Li, G.; et al. Excessive miR-25-3p maturation via N6-methyladenosine stimulated by cigarette smoke promotes pancreatic cancer progression. Nat. Commun. 2019, 10, 1858. [Google Scholar] [CrossRef]
- Du, M.; Peng, Y.; Li, Y.; Sun, W.; Zhu, H.; Wu, J.; Zong, D.; Wu, L.; He, X. MYC-activated RNA N6-methyladenosine reader IGF2BP3 promotes cell proliferation and metastasis in nasopharyngeal carcinoma. Cell Death Discov. 2022, 8, 53. [Google Scholar] [CrossRef]
- Meyer, K.D.; Saletore, Y.; Zumbo, P.; Elemento, O.; Mason, C.E.; Jaffrey, S.R. Comprehensive Analysis of mRNA Methylation Reveals Enrichment in 3′ UTRs and near Stop Codons. Cell 2012, 149, 1635–1646. [Google Scholar] [CrossRef]
- Jang, K.H.; Heras, C.R.; Lee, G. m6A in the Signal Transduction Network. Mol. Cells 2022, 45, 435–443. [Google Scholar] [CrossRef]
- Barbieri, I.; Tzelepis, K.; Pandolfini, L.; Shi, J.; Millán-Zambrano, G.; Robson, S.C.; Aspris, D.; Migliori, V.; Bannister, A.J.; Han, N.; et al. Promoter-bound METTL3 maintains myeloid leukaemia by m6A-dependent translation control. Nature 2017, 552, 126–131. [Google Scholar] [CrossRef]
- Bertero, A.; Brown, S.; Madrigal, P.; Osnato, A.; Ortmann, D.; Yiangou, L.; Kadiwala, J.; Hubner, N.C.; de Los Mozos, I.R.; Sadée, C.; et al. The SMAD2/3 interactome reveals that TGFβ controls m6A mRNA methylation in pluripotency. Nature 2018, 555, 256–259. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Weng, H.; Zhou, K.; Wu, T.; Zhao, B.S.; Sun, M.; Chen, Z.; Deng, X.; Xiao, G.; Auer, F.; et al. Histone H3 trimethylation at lysine 36 guides m6A RNA modification co-transcriptionally. Nature 2019, 567, 414–419. [Google Scholar] [CrossRef] [PubMed]
- He, P.C.; Wei, J.; Dou, X.; Harada, B.T.; Zhang, Z.; Ge, R.; Liu, C.; Zhang, L.-S.; Yu, X.; Wang, S.; et al. Exon architecture controls mRNA m6A suppression and gene expression. Science 2023, 379, 677–682. [Google Scholar] [CrossRef] [PubMed]
- Uzonyi, A.; Dierks, D.; Nir, R.; Kwon, O.S.; Toth, U.; Barbosa, I.; Burel, C.; Brandis, A.; Rossmanith, W.; Le Hir, H.; et al. Exclusion of m6A from splice-site proximal regions by the exon junction complex dictates m6A topologies and mRNA stability. Mol. Cell 2023, 83, 237–251.e7. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Triboulet, R.; Liu, Q.; Sendinc, E.; Gregory, R.I. Exon junction complex shapes the m6A epitranscriptome. Nat. Commun. 2022, 13, 7904. [Google Scholar] [CrossRef] [PubMed]
- Covelo-Molares, H.; Obrdlik, A.; Poštulková, I.; Dohnálková, M.; Gregorová, P.; Ganji, R.; Potěšil, D.; Gawriyski, L.; Varjosalo, M.; Vaňáčová, Š. The comprehensive interactomes of human adenosine RNA methyltransferases and demethylases reveal distinct functional and regulatory features. Nucleic Acids Res. 2021, 49, 10895–10910. [Google Scholar] [CrossRef] [PubMed]
- Choe, J.; Lin, S.; Zhang, W.; Liu, Q.; Wang, L.; Ramirez-Moya, J.; Du, P.; Kim, W.; Tang, S.; Sliz, P.; et al. mRNA circularization by METTL3–eIF3h enhances translation and promotes oncogenesis. Nature 2018, 561, 556–560. [Google Scholar] [CrossRef]
- Visvanathan, A.; Patil, V.; Arora, A.; Hegde, A.S.; Arivazhagan, A.; Santosh, V.; Somasundaram, K. Essential role of METTL3-mediated m6A modification in glioma stem-like cells maintenance and radioresistance. Oncogene 2018, 37, 522–533. [Google Scholar] [CrossRef]
- Vu, L.P.; Pickering, B.F.; Cheng, Y.; Zaccara, S.; Nguyen, D.; Minuesa, G.; Chou, T.; Chow, A.; Saletore, Y.; MacKay, M.; et al. The N6-methyladenosine (m6A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat. Med. 2017, 23, 1369–1376. [Google Scholar] [CrossRef]
- Li, T.; Hu, P.-S.; Zuo, Z.; Lin, J.-F.; Li, X.; Wu, Q.-N.; Chen, Z.-H.; Zeng, Z.-L.; Wang, F.; Zheng, J.; et al. METTL3 facilitates tumor progression via an m6A-IGF2BP2-dependent mechanism in colorectal carcinoma. Mol. Cancer 2019, 18, 112. [Google Scholar] [CrossRef]
- Hua, W.; Zhao, Y.; Jin, X.; Yu, D.; He, J.; Xie, D.; Duan, P. METTL3 promotes ovarian carcinoma growth and invasion through the regulation of AXL translation and epithelial to mesenchymal transition. Gynecol. Oncol. 2018, 151, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Dahal, U.; Le, K.; Gupta, M. RNA m6A methyltransferase METTL3 regulates invasiveness of melanoma cells by matrix metallopeptidase 2. Melanoma Res. 2019, 29, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Chen, Q.; Tian, K.; Liang, R.; Chen, T.; Gong, A.; Mathy, N.W.; Yu, T.; Chen, X. m6A methyltransferase METTL3 maintains colon cancer tumorigenicity by suppressing SOCS2 to promote cell proliferation. Oncol. Rep. 2020, 44, 973–986. [Google Scholar] [CrossRef]
- Wang, H.; Xu, B.; Shi, J. N6-methyladenosine METTL3 promotes the breast cancer progression via targeting Bcl-2. Gene 2020, 722, 144076. [Google Scholar] [CrossRef]
- Hu, S.; Song, Y.; Zhou, Y.; Jiao, Y.; Li, G. METTL3 Accelerates Breast Cancer Progression via Regulating EZH2 m6A Modification. J. Heal. Eng. 2022, 2022, 5794422. [Google Scholar] [CrossRef] [PubMed]
- Yankova, E.; Blackaby, W.; Albertella, M.; Rak, J.; De Braekeleer, E.; Tsagkogeorga, G.; Pilka, E.S.; Aspris, D.; Leggate, D.; Hendrick, A.G.; et al. Small-molecule inhibition of METTL3 as a strategy against myeloid leukaemia. Nature 2021, 593, 597–601. [Google Scholar] [CrossRef] [PubMed]
- Ofir-Rosenfeld, Y.; Vasiliauskaitė, L.; Saunders, C.; Sapetschnig, A.; Tsagkogeorga, G.; Albertella, M.; Carkill, M.; Self-Fordham, J.; Holz, J.B.; Rausch, O. STC-15, an oral small molecule inhibitor of the RNA methyltransferase METTL3, inhibits tumour growth through activation of anti-cancer immune responses associated with increased interferon signalling, and synergises with T cell checkpoint blockade. Eur. J. Cancer 2022, 174, S123. [Google Scholar] [CrossRef]
- Micaelli, M.; Dalle Vedove, A.; Cerofolini, L.; Vigna, J.; Sighel, D.; Zaccara, S.; Bonomo, I.; Poulentzas, G.; Rosatti, E.F.; Cazzanelli, G.; et al. Small-Molecule Ebselen Binds to YTHDF Proteins Interfering with the Recognition of N6-Methyladenosine-Modified RNAs. ACS Pharmacol. Transl. Sci. 2022, 5, 872–891. [Google Scholar] [CrossRef]
- Moroz-Omori, E.V.; Huang, D.; Kumar Bedi, R.; Cheriyamkunnel, S.J.; Bochenkova, E.; Dolbois, A.; Rzeczkowski, M.D.; Li, Y.; Wiedmer, L.; Caflisch, A. METTL3 Inhibitors for Epitranscriptomic Modulation of Cellular Processes. ChemMedChem 2021, 16, 3035–3043. [Google Scholar] [CrossRef]
- Lee, J.H.; Choi, N.; Kim, S.; Jin, M.S.; Shen, H.; Kim, Y.C. Eltrombopag as an Allosteric Inhibitor of the METTL3-14 Complex Affecting the m6A Methylation of RNA in Acute Myeloid Leukemia Cells. Pharmaceuticals 2022, 15, 440. [Google Scholar] [CrossRef]
- Huang, Y.; Su, R.; Sheng, Y.; Dong, L.; Dong, Z.; Xu, H.; Ni, T.; Zhang, Z.S.; Zhang, T.; Li, C.; et al. Small-Molecule Targeting of Oncogenic FTO Demethylase in Acute Myeloid Leukemia. Cancer Cell 2019, 35, 677–691.e10. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liang, G.; Xu, H.; Dong, W.; Dong, Z.; Qiu, Z.; Zhang, Z.; Li, F.; Huang, Y.; Li, Y.; et al. Tumors exploit FTO-mediated regulation of glycolytic metabolism to evade immune surveillance. Cell Metab. 2021, 33, 1221–1233.e11. [Google Scholar] [CrossRef] [PubMed]
- Su, R.; Dong, L.; Li, Y.; Gao, M.; Han, L.; Wunderlich, M.; Deng, X.; Li, H.; Huang, Y.; Gao, L.; et al. Targeting FTO Suppresses Cancer Stem Cell Maintenance and Immune Evasion. Cancer Cell 2020, 38, 79–96.e11. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yan, J.; Li, Q.; Li, J.; Gong, S.; Zhou, H.; Gan, J.; Jiang, H.; Jia, G.F.; Luo, C.; et al. Meclofenamic acid selectively inhibits FTO demethylation of m6A over ALKBH5. Nucleic Acids Res. 2015, 43, 373–384. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Cancer Type | Expression | mRNA Substrate | Biological Function | Phenotype | Ref. |
|---|---|---|---|---|---|
| Lung cancer | METTL3, Increased | EGFR, BRD4, MGMT, TIMP1 | Translation ↑ | Tumorigenesis | [47] |
| Glioma | METTL3, Increased | SOX2 | mRNA stability ↑ | Stem-like cell maintenance and radioresistance | [48] |
| Leukemia | METTL3, Increased | c-My cBCL2 PTEN | Translation ↑ | Inhibits differentiation and increases cell growth | [49] |
| Leukemia | METTL3, Increased | SP1, SP2 | Translation ↑ | Cell proliferation and differentiation | [40] |
| Colorectal cancer | METTL3, Increased | SOX2 | mRNA stability ↑ | Self-renewal, stem cell frequency, and migration | [50] |
| Ovarian cancer | METTL3, Increased | AXL | Translation ↑ | Promotes the proliferation, invasion, and tumor formation | [51] |
| Melanoma | METTL3, Increased | MMP2 | Translation ↑ | Promotes invasion | [52] |
| HCC | METTL3, Increased | SOCS2 | mRNA stability ↑ | Cell proliferation, migration, and colony formation | [53] |
| Breast | METTL3, Increased | BCL2 EZH2 | mRNA stability ↑ Translation ↑ | Promotes proliferation and inhibits apoptosis | [54,55] |
| Compound | Mechanism of Action | IC50 | Ref. |
|---|---|---|---|
| STM2457 | METTL3 inhibitor | 16.9 nM | [56] |
| Ebselen | YTHDF1/DF2 | 1.63/1.66 µM | [58] |
| UZh1a | METTL3 inhibitor | 280 nM | [59] |
| Eltrombopag | Allosteric inhibitor of METTL3-14 complex | 4.55 µM | [60] |
| FB23 | FTO inhibitor | 60 nM | [61] |
| FB23-2 | FTO inhibitor | 2.6 µM | [61] |
| Dac51 | FTO inhibitor | 0.4 µM | [62] |
| Bisantrene | FTO inhibitor | 712.8 nM | [63] |
| Meclofenamate sodium | FTO inhibitor | 7 µM | [64] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhattarai, P.Y.; Kim, G.; Bhandari, D.; Shrestha, P.; Choi, H.S. Regulation of m6A Methylome in Cancer: Mechanisms, Implications, and Therapeutic Strategies. Cells 2024, 13, 66. https://doi.org/10.3390/cells13010066
Bhattarai PY, Kim G, Bhandari D, Shrestha P, Choi HS. Regulation of m6A Methylome in Cancer: Mechanisms, Implications, and Therapeutic Strategies. Cells. 2024; 13(1):66. https://doi.org/10.3390/cells13010066
Chicago/Turabian StyleBhattarai, Poshan Yugal, Garam Kim, Dibikshya Bhandari, Pratikshya Shrestha, and Hong Seok Choi. 2024. "Regulation of m6A Methylome in Cancer: Mechanisms, Implications, and Therapeutic Strategies" Cells 13, no. 1: 66. https://doi.org/10.3390/cells13010066