
TLR9 Monotherapy in Immune-Competent Mice Suppresses Orthotopic Prostate Tumor Development
 , , , , ,
, , , , ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Orthotopic Murine Model of Prostate Cancer
2.2. Flow Cytometric Analysis of Immune Populations
2.3. Cell Culture
2.4. Cell Proliferation
2.5. Cell Survival
2.6. Gene Expression by Real-Time qPCR
2.7. Protein Quantification
2.8. Statistical Analysis
3. Results
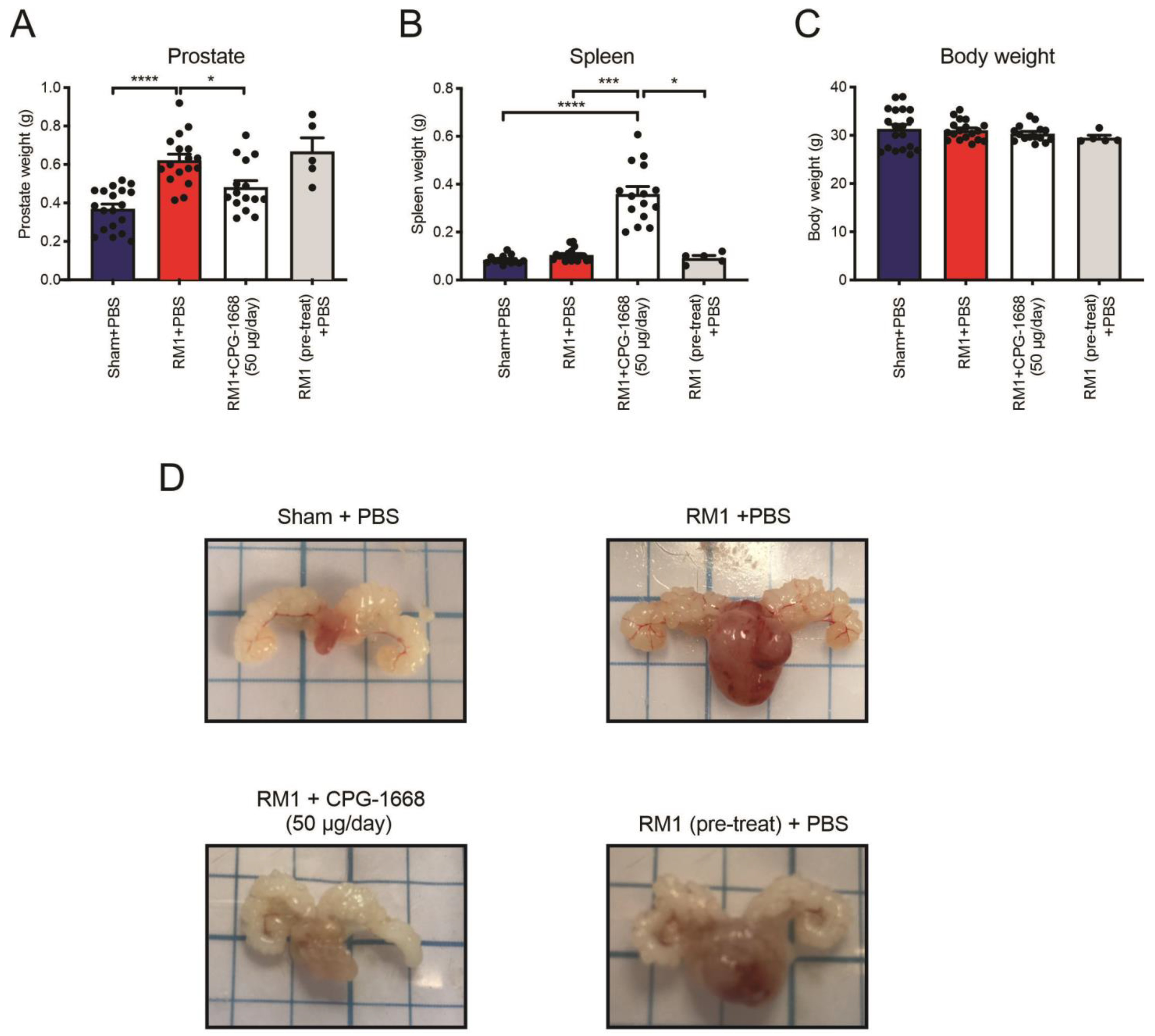
3.1. Systemic CPG-1668 Administration Murine Prostate Cancer Tumorigenesis
3.2. CPG-1668 Treatment Reduces T Cell and M1 Macrophage Populations in Prostate Tumors
3.3. CPG-1668 Treatment Boosts Systemic Macrophage, Neutrophil, and Cytotoxic CD8+ T-Cell Numbers
3.4. In Vitro Stimulation of TLR9 in Immune Cells but Not RM1 Cells Limit Cancer Cell Proliferation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Miller, K.D.; Nogueira, L.; Mariotto, A.B.; Rowland, J.H.; Yabroff, K.R.; Alfano, C.M.; Jemal, A.; Kramer, J.L.; Siegel, R.L. Cancer treatment and survivorship statistics, 2019. CA Cancer J. Clin. 2019, 69, 363–385. [Google Scholar] [CrossRef] [PubMed]
- Rawla, P. Epidemiology of Prostate Cancer. World J. Oncol. 2019, 10, 63–89. [Google Scholar] [CrossRef] [PubMed]
- Sfanos, K.S.; De Marzo, A.M. Prostate cancer and inflammation: The evidence. Histopathology 2012, 60, 199–215. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int. Immunol. 2009, 21, 317–337. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef]
- Takeda, K.; Akira, S. Toll-like receptors. Curr. Protoc. Immunol. 2015, 109. [Google Scholar] [CrossRef]
- Urban-Wojciuk, Z.; Khan, M.M.; Oyler, B.L.; Fåhraeus, R.; Marek-Trzonkowska, N.; Nita-Lazar, A.; Hupp, T.R.; Goodlett, D.R. The Role of TLRs in Anti-cancer Immunity and Tumor Rejection. Front. Immunol. 2019, 10, 2388. [Google Scholar] [CrossRef]
- Razack, A.H. Bacillus Calmette-Guerin and bladder cancer. Asian J. Surg. 2007, 30, 302–309. [Google Scholar] [CrossRef]
- Kikkawa, F.; Kawai, M.; Oguchi, H.; Kojima, M.; Ishikawa, H.; Iwata, M.; Maeda, O.; Tomoda, Y.; Arii, Y.; Kuzuya, K.; et al. Randomised study of immunotherapy with OK-432 in uterine cervical carcinoma. Eur. J. Cancer 1993, 29, 1542–1546. [Google Scholar] [CrossRef]
- Maehara, Y.; Okuyama, T.; Kakeji, Y.; Baba, H.; Furusawa, M.; Sugimachi, K. Postoperative immunochemotherapy including streptococcal lysate OK-432 is effective for patients with gastric cancer and serosal invasion. Am. J. Surg. 1994, 168, 36–40. [Google Scholar] [CrossRef]
- Sato, M.; Harada, K.; Yoshida, H.; Yura, Y.; Azuma, M.; Iga, H.; Bando, T.; Kawamata, H.; Takegawa, Y. Therapy for oral squamous cell carcinoma by tegafur and streptococcal agent OK-432 in combination with radiotherapy: Association of the therapeutic effect with differentiation and apoptosis in the cancer cells. Apoptosis 1997, 2, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, M.; Oshikawa, T.; Tano, T.; Ahmed, S.U.; Kan, S.; Sasai, A.; Akashi, S.; Miyake, K.; Moriya, Y.; Ryoma, Y.; et al. Mechanism of anticancer host response induced by OK-432, a streptococcal preparation, mediated by phagocytosis and Toll-like receptor 4 signaling. J. Immunother. 2006, 29, 78–86. [Google Scholar] [CrossRef]
- Pei, Z.; Lin, D.; Song, X.; Li, H.; Yao, H. TLR4 signaling promotes the expression of VEGF and TGFβ1 in human prostate epithelial PC3 cells induced by lipopolysaccharide. Cell. Immunol. 2008, 254, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Hua, D.; Liu, M.Y.; Cheng, Z.D.; Qin, X.J.; Zhang, H.M.; Chen, Y.; Qin, G.J.; Liang, G.; Li, J.N.; Han, X.F.; et al. Small interfering RNA-directed targeting of Toll-like receptor 4 inhibits human prostate cancer cell invasion, survival, and tumorigenicity. Mol. Immunol. 2009, 46, 2876–2884. [Google Scholar] [CrossRef] [PubMed]
- Paone, A.; Starace, D.; Galli, R.; Padula, F.; De Cesaris, P.; Filippini, A.; Ziparo, E.; Riccioli, A. Toll-like receptor 3 triggers apoptosis of human prostate cancer cells through a PKC-alpha-dependent mechanism. Carcinogenesis 2008, 29, 1334–1342. [Google Scholar] [CrossRef] [PubMed]
- Salaun, B.; Coste, I.; Rissoan, M.C.; Lebecque, S.J.; Renno, T. TLR3 can directly trigger apoptosis in human cancer cells. J. Immunol. 2006, 176, 4894–4901. [Google Scholar] [CrossRef] [PubMed]
- Hemmi, H.; Takeuchi, O.; Kawai, T.; Kaisho, T.; Sato, S.; Sanjo, H.; Matsumoto, M.; Hoshino, K.; Wagner, H.; Takeda, K.; et al. A Toll-like receptor recognizes bacterial DNA. Nature 2000, 408, 740–745. [Google Scholar] [CrossRef]
- Ohto, U.; Shibata, T.; Tanji, H.; Ishida, H.; Krayukhina, E.; Uchiyama, S.; Miyake, K.; Shimizu, T. Structural basis of CpG and inhibitory DNA recognition by Toll-like receptor 9. Nature 2015, 520, 702–705. [Google Scholar] [CrossRef]
- Berger, R.; Fiegl, H.; Goebel, G.; Obexer, P.; Ausserlechner, M.; Doppler, W.; Hauser-Kronberger, C.; Reitsamer, R.; Egle, D.; Reimer, D.; et al. Toll-like receptor 9 expression in breast and ovarian cancer is associated with poorly differentiated tumors. Cancer Sci. 2010, 101, 1059–1066. [Google Scholar] [CrossRef]
- Väisänen, M.R.; Väisänen, T.; Jukkola-Vuorinen, A.; Vuopala, K.S.; Desmond, R.; Selander, K.S.; Vaarala, M.H. Expression of toll-like receptor-9 is increased in poorly differentiated prostate tumors. Prostate 2010, 70, 817–824. [Google Scholar] [CrossRef]
- Di, J.M.; Pang, J.; Pu, X.Y.; Zhang, Y.; Liu, X.P.; Fang, Y.Q.; Ruan, X.X.; Gao, X. Toll-like receptor 9 agonists promote IL-8 and TGF-beta1 production via activation of nuclear factor kappaB in PC-3 cells. Cancer Genet. Cytogenet. 2009, 192, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Slaton, J.W.; Eve, B.Y.; Kim, S.J.; Perrotte, P.; Balbay, M.D.; Yano, S.; Bar-Eli, M.; Radinsky, R.; Pettaway, C.A.; et al. Interleukin 8 expression regulates tumorigenicity and metastases in androgen-independent prostate cancer. Clin. Cancer Res. 2000, 6, 2104–2119. [Google Scholar] [PubMed]
- Ilvesaro, J.M.; Merrell, M.A.; Swain, T.M.; Davidson, J.; Zayzafoon, M.; Harris, K.W.; Selander, K.S. Toll like receptor-9 agonists stimulate prostate cancer invasion in vitro. Prostate 2007, 67, 774–781. [Google Scholar] [CrossRef] [PubMed]
- Kudo, Y.; Iizuka, S.; Yoshida, M.; Tsunematsu, T.; Kondo, T.; Subarnbhesaj, A.; Deraz, E.M.; Siriwardena, S.B.; Tahara, H.; Ishimaru, N.; et al. Matrix metalloproteinase-13 (MMP-13) directly and indirectly promotes tumor angiogenesis. J. Biol. Chem. 2012, 287, 38716–38728. [Google Scholar] [CrossRef] [PubMed]
- Won, H.; Moreira, D.; Gao, C.; Duttagupta, P.; Zhao, X.; Manuel, E.; Diamond, D.; Yuan, Y.C.; Liu, Z.; Jones, J.; et al. TLR9 expression and secretion of LIF by prostate cancer cells stimulates accumulation and activity of polymorphonuclear MDSCs. J. Leukoc. Biol. 2017, 102, 423–436. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Hossain, D.M.S.; Nechaev, S.; Kozlowska, A.; Zhang, W.; Liu, Y.; Kowolik, C.M.; Swiderski, P.; Rossi, J.J.; Forman, S.; et al. TLR9-mediated siRNA delivery for targeting of normal and malignant human hematopoietic cells in vivo. Blood 2013, 121, 1304–1315. [Google Scholar] [CrossRef]
- Moreira, D.; Zhang, Q.; Hossain, D.M.; Nechaev, S.; Li, H.; Kowolik, C.M.; D’Apuzzo, M.; Forman, S.; Jones, J.; Pal, S.K.; et al. TLR9 signaling through NF-κB/RELA and STAT3 promotes tumor-propagating potential of prostate cancer cells. Oncotarget 2015, 6, 17302–17313. [Google Scholar] [CrossRef]
- Tokunaga, T.; Yano, O.; Kuramoto, E.; Kimura, Y.; Yamamoto, T.; Kataoka, T.; Yamamoto, S. Synthetic oligonucleotides with particular base sequences from the cDNA encoding proteins of Mycobacterium bovis BCG induce interferons and activate natural killer cells. Microbiol. Immunol. 1992, 36, 55–66. [Google Scholar] [CrossRef]
- Tokunaga, T.; Yamamoto, T.; Yamamoto, S. How BCG led to the discovery of immunostimulatory DNA. Jpn. J. Infect. Dis. 1999, 52, 1–11. [Google Scholar] [CrossRef]
- Krieg, A.M. Development of TLR9 agonists for cancer therapy. J. Clin. Investig. 2007, 117, 1184–1194. [Google Scholar] [CrossRef]
- Lonsdorf, A.S.; Kuekrek, H.; Stern, B.V.; Boehm, B.O.; Lehmann, P.V.; Tary-Lehmann, M. Intratumor CpG-oligodeoxynucleotide injection induces protective antitumor T cell immunity. J. Immunol. 2003, 171, 3941–3946. [Google Scholar] [CrossRef] [PubMed]
- Baines, J.; Celis, E. Immune-mediated tumor regression induced by CpG-containing oligodeoxynucleotides. Clin. Cancer Res. 2003, 9, 2693–2700. [Google Scholar] [PubMed]
- Sagiv-Barfi, I.; Czerwinski, D.K.; Levy, S.; Alam, I.S.; Mayer, A.T.; Gambhir, S.S.; Levy, R. Eradication of spontaneous malignancy by local immunotherapy. Sci. Transl. Med. 2018, 10, eaan4488. [Google Scholar] [CrossRef] [PubMed]
- Krieg, A.M. CpG motifs in bacterial DNA and their immune effects. Annu. Rev. Immunol. 2002, 20, 709–760. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, T.; Yamamoto, S.; Yamamoto, T.; Kuramoto, E.; Kimura, Y.; Yano, O.; Tokunaga, T. Antitumor activity of synthetic oligonucleotides with sequences from cDNA encoding proteins of Mycobacterium bovis BCG. Jpn. J. Cancer Res. 1992, 83, 244–247. [Google Scholar] [CrossRef] [PubMed]
- Moreira, D.; Adamus, T.; Zhao, X.; Su, Y.-L.; Zhang, Z.; White, S.V.; Swiderski, P.; Lu, X.; DePinho, R.A.; Pal, S.K.; et al. STAT3 Inhibition Combined with CpG Immunostimulation Activates Antitumor Immunity to Eradicate Genetically Distinct Castration-Resistant Prostate Cancers. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 5948–5962. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Waldschmidt, M.; Zhao, X.; Ratliff, T.; Krieg, A.M. Antitumor mechanisms of oligodeoxynucleotides with CpG and polyG motifs in murine prostate cancer cells: Decrease of NF-kappaB and AP-1 binding activities and induction of apoptosis. Antisense Nucleic Acid. Drug Dev. 2002, 12, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Mueller, M.; Reichardt, W.; Koerner, J.; Groettrup, M. Coencapsulation of tumor lysate and CpG-ODN in PLGA-microspheres enables successful immunotherapy of prostate carcinoma in TRAMP mice. J. Control Release 2012, 162, 159–166. [Google Scholar] [CrossRef]
- Mutwiri, G.K.; Nichani, A.K.; Babiuk, S.; Babiuk, L.A. Strategies for enhancing the immunostimulatory effects of CpG oligodeoxynucleotides. J. Control Release 2004, 97, 1–17. [Google Scholar] [CrossRef]
- Scheiermann, J.; Klinman, D.M. Clinical evaluation of CpG oligonucleotides as adjuvants for vaccines targeting infectious diseases and cancer. Vaccine 2014, 32, 6377–6389. [Google Scholar] [CrossRef]
- Harrison, I.P.; Vinh, A.; Johnson, I.R.D.; Luong, R.; Drummond, G.R.; Sobey, C.G.; Tiganis, T.; Williams, E.D.; JJ, O.L.; Brooks, D.A.; et al. NOX2 oxidase expressed in endosomes promotes cell proliferation and prostate tumour development. Oncotarget 2018, 9, 35378–35393. [Google Scholar] [CrossRef] [PubMed]
- Showalter, A.; Limaye, A.; Oyer, J.L.; Igarashi, R.; Kittipatarin, C.; Copik, A.J.; Khaled, A.R. Cytokines in immunogenic cell death: Applications for cancer immunotherapy. Cytokine 2017, 97, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Krieg, A.M. Antitumor applications of stimulating toll-like receptor 9 with CpG oligodeoxynucleotides. Curr. Oncol. Rep. 2004, 6, 88–95. [Google Scholar] [CrossRef]
- Brennan, T.V.; Lin, L.; Brandstadter, J.D.; Rendell, V.R.; Dredge, K.; Huang, X.; Yang, Y. Heparan sulfate mimetic PG545-mediated antilymphoma effects require TLR9-dependent NK cell activation. J. Clin. Investig. 2016, 126, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Zarling, J.M.; Tevethia, S.S. Transplantation immunity to simian virus 40-transformed cells in tumor-bearing mice. II. Evidence for macrophage participation at the effector level of tumor cell rejection. J. Natl. Cancer Inst. 1973, 50, 149–157. [Google Scholar] [CrossRef]
- Gallimore, A.M.; Simon, A.K. Positive and negative influences of regulatory T cells on tumour immunity. Oncogene 2008, 27, 5886–5893. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Saxena, S.; Awaji, M.; Singh, R.K. Tumor-Associated Neutrophils in Cancer: Going Pro. Cancers 2019, 11, 564. [Google Scholar] [CrossRef]
- Barron, D.A.; Rowley, D.R. The reactive stroma microenvironment and prostate cancer progression. Endocr. Relat. Cancer 2012, 19, R187–R204. [Google Scholar] [CrossRef]
- De Marzo, A.M.; Meeker, A.K.; Zha, S.; Luo, J.; Nakayama, M.; Platz, E.A.; Isaacs, W.B.; Nelson, W.G. Human prostate cancer precursors and pathobiology. Urology 2003, 62, 55–62. [Google Scholar] [CrossRef]
- Stultz, J.; Fong, L. How to turn up the heat on the cold immune microenvironment of metastatic prostate cancer. Prostate Cancer Prostatic Dis. 2021, 24, 697–717. [Google Scholar] [CrossRef]
- Li, D.; Zhou, X.; Xu, W.; Chen, Y.; Mu, C.; Zhao, X.; Yang, T.; Wang, G.; Wei, L.; Ma, B. Prostate cancer cells synergistically defend against CD8(+) T cells by secreting exosomal PD-L1. Cancer Med. 2023, 12, 16405–16415. [Google Scholar] [CrossRef] [PubMed]
- Hirz, T.; Mei, S.; Sarkar, H.; Kfoury, Y.; Wu, S.; Verhoeven, B.M.; Subtelny, A.O.; Zlatev, D.V.; Wszolek, M.W.; Salari, K.; et al. Dissecting the immune suppressive human prostate tumor microenvironment via integrated single-cell and spatial transcriptomic analyses. Nat. Commun. 2023, 14, 663. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, H.; Li, G.; Zhou, X.; Shi, Y.; Zou, F.; Chen, Y.; Gao, J.; Yang, S.; Wu, S.; et al. Increased Tim-3 expression on TILs during treatment with the Anchored GM-CSF vaccine and anti-PD-1 antibodies is inversely correlated with response in prostate cancer. J. Cancer 2020, 11, 648–656. [Google Scholar] [CrossRef] [PubMed]
- Czernin, J.; Current, K.; Mona, C.E.; Nyiranshuti, L.; Hikmat, F.; Radu, C.G.; Lückerath, K. Immune-Checkpoint Blockade Enhances (225)Ac-PSMA617 Efficacy in a Mouse Model of Prostate Cancer. J. Nucl. Med. 2021, 62, 228–231. [Google Scholar] [CrossRef] [PubMed]
- Mita, Y.; Kimura, M.Y.; Hayashizaki, K.; Koyama-Nasu, R.; Ito, T.; Motohashi, S.; Okamoto, Y.; Nakayama, T. Crucial role of CD69 in anti-tumor immunity through regulating the exhaustion of tumor-infiltrating T cells. Int. Immunol. 2018, 30, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Esplugues, E.; Sancho, D.; Vega-Ramos, J.; Martínez, C.; Syrbe, U.; Hamann, A.; Engel, P.; Sánchez-Madrid, F.; Lauzurica, P. Enhanced antitumor immunity in mice deficient in CD69. J. Exp. Med. 2003, 197, 1093–1106. [Google Scholar] [CrossRef] [PubMed]
- Hossain, D.M.; Pal, S.K.; Moreira, D.; Duttagupta, P.; Zhang, Q.; Won, H.; Jones, J.; D’Apuzzo, M.; Forman, S.; Kortylewski, M. TLR9-Targeted STAT3 Silencing Abrogates Immunosuppressive Activity of Myeloid-Derived Suppressor Cells from Prostate Cancer Patients. Clin. Cancer Res. 2015, 21, 3771–3782. [Google Scholar] [CrossRef]
- Wong, R.M.; Smith, K.A.; Tam, V.L.; Pagarigan, R.R.; Meisenburg, B.L.; Quach, A.M.; Carrillo, M.A.; Qiu, Z.; Bot, A.I. TLR-9 signaling and TCR stimulation co-regulate CD8(+) T cell-associated PD-1 expression. Immunol. Lett. 2009, 127, 60–67. [Google Scholar] [CrossRef]
- Yin, P.; Liu, X.; Mansfield, A.S.; Harrington, S.M.; Li, Y.; Yan, Y.; Dong, H. CpG-induced antitumor immunity requires IL-12 in expansion of effector cells and down-regulation of PD-1. Oncotarget 2016, 7, 70223–70231. [Google Scholar] [CrossRef]
- Ren, T.; Wen, Z.K.; Liu, Z.M.; Qian, C.; Liang, Y.J.; Jin, M.L.; Cai, Y.Y.; Xu, L. Targeting toll-like receptor 9 with CpG oligodeoxynucleotides enhances anti-tumor responses of peripheral blood mononuclear cells from human lung cancer patients. Cancer Investig. 2008, 26, 448–455. [Google Scholar] [CrossRef]
- Najafi, M.; Hashemi Goradel, N.; Farhood, B.; Salehi, E.; Nashtaei, M.S.; Khanlarkhani, N.; Khezri, Z.; Majidpoor, J.; Abouzaripour, M.; Habibi, M.; et al. Macrophage polarity in cancer: A review. J. Cell Biochem. 2019, 120, 2756–2765. [Google Scholar] [CrossRef]
- Tuong, Z.K.; Loudon, K.W.; Berry, B.; Richoz, N.; Jones, J.; Tan, X.; Nguyen, Q.; George, A.; Hori, S.; Field, S.; et al. Resolving the immune landscape of human prostate at a single-cell level in health and cancer. Cell Rep. 2021, 37, 110132. [Google Scholar] [CrossRef]
- Guiducci, C.; Vicari, A.P.; Sangaletti, S.; Trinchieri, G.; Colombo, M.P. Redirecting in vivo elicited tumor infiltrating macrophages and dendritic cells towards tumor rejection. Cancer Res. 2005, 65, 3437–3446. [Google Scholar] [CrossRef]
- Lu, C.-H.; Lai, C.-Y.; Yeh, D.-W.; Liu, Y.-L.; Su, Y.-W.; Hsu, L.-C.; Chang, C.-H.; Catherine Jin, S.-L.; Chuang, T.-H. Involvement of M1 macrophage polarization in endosomal toll-like receptors activated psoriatic inflammation. Mediat. Inflamm. 2018, 2018, 3523642. [Google Scholar] [CrossRef]
- Lindau, D.; Mussard, J.; Wagner, B.J.; Ribon, M.; Rönnefarth, V.M.; Quettier, M.; Jelcic, I.; Boissier, M.C.; Rammensee, H.G.; Decker, P. Primary blood neutrophils express a functional cell surface Toll-like receptor 9. Eur. J. Immunol. 2013, 43, 2101–2113. [Google Scholar] [CrossRef]
- El Kebir, D.; Damlaj, A.; Filep, J.G. Toll-like receptor 9 signaling delays neutrophil apoptosis by increasing transcription of Mcl-1. PLoS ONE 2014, 9, e87006. [Google Scholar] [CrossRef]
- Knuefermann, P.; Baumgarten, G.; Koch, A.; Schwederski, M.; Velten, M.; Ehrentraut, H.; Mersmann, J.; Meyer, R.; Hoeft, A.; Zacharowski, K.; et al. CpG oligonucleotide activates Toll-like receptor 9 and causes lung inflammation in vivo. Respir. Res. 2007, 8, 72. [Google Scholar] [CrossRef]
- Granot, Z. Neutrophils as a Therapeutic Target in Cancer. Front. Immunol. 2019, 10, 1710. [Google Scholar] [CrossRef]
- Coso, S.; Harrison, I.; Harrison, C.B.; Vinh, A.; Sobey, C.G.; Drummond, G.R.; Williams, E.D.; Selemidis, S. NADPH oxidases as regulators of tumor angiogenesis: Current and emerging concepts. Antioxid. Redox Signal 2012, 16, 1229–1247. [Google Scholar] [CrossRef]
- Bulbul, M.A.; Huben, R.P.; Murphy, G.P. Interferon-beta treatment of metastatic prostate cancer. J. Surg. Oncol. 1986, 33, 231–233. [Google Scholar] [CrossRef] [PubMed]
- Ren, C.; Kumar, S.; Chanda, D.; Kallman, L.; Chen, J.; Mountz, J.D.; Ponnazhagan, S. Cancer gene therapy using mesenchymal stem cells expressing interferon-beta in a mouse prostate cancer lung metastasis model. Gene Ther. 2008, 15, 1446–1453. [Google Scholar] [CrossRef]
- Simonsson, B.; Gedde-Dahl, T.; Markevärn, B.; Remes, K.; Stentoft, J.; Almqvist, A.; Björeman, M.; Flogegård, M.; Koskenvesa, P.; Lindblom, A.; et al. Combination of pegylated IFN-α2b with imatinib increases molecular response rates in patients with low- or intermediate-risk chronic myeloid leukemia. Blood 2011, 118, 3228–3235. [Google Scholar] [CrossRef]
- Owen, K.L.; Gearing, L.J.; Zanker, D.J.; Brockwell, N.K.; Khoo, W.H.; Roden, D.L.; Cmero, M.; Mangiola, S.; Hong, M.K.; Spurling, A.J.; et al. Prostate cancer cell-intrinsic interferon signaling regulates dormancy and metastatic outgrowth in bone. EMBO Rep. 2020, 21, e50162. [Google Scholar] [CrossRef]
- Luo, Y.; Jiang, Q.W.; Wu, J.Y.; Qiu, J.G.; Zhang, W.J.; Mei, X.L.; Shi, Z.; Di, J.M. Regulation of migration and invasion by Toll-like receptor-9 signaling network in prostate cancer. Oncotarget 2015, 6, 22564–22574. [Google Scholar] [CrossRef]
- Bauer, M.; Heeg, K.; Wagner, H.; Lipford, G.B. DNA activates human immune cells through a CpG sequence-dependent manner. Immunology 1999, 97, 699–705. [Google Scholar] [CrossRef]
- Honda, K.; Ohba, Y.; Yanai, H.; Negishi, H.; Mizutani, T.; Takaoka, A.; Taya, C.; Taniguchi, T. Spatiotemporal regulation of MyD88-IRF-7 signalling for robust type-I interferon induction. Nature 2005, 434, 1035–1040. [Google Scholar] [CrossRef]
- Zhang, Z.; Kuo, J.C.; Yao, S.; Zhang, C.; Khan, H.; Lee, R.J. CpG Oligodeoxynucleotides for Anticancer Monotherapy from Preclinical Stages to Clinical Trials. Pharmaceutics 2021, 14, 73. [Google Scholar] [CrossRef] [PubMed]
- Zent, C.S.; Smith, B.J.; Ballas, Z.K.; Wooldridge, J.E.; Link, B.K.; Call, T.G.; Shanafelt, T.D.; Bowen, D.A.; Kay, N.E.; Witzig, T.E.; et al. Phase I clinical trial of CpG oligonucleotide 7909 (PF-03512676) in patients with previously treated chronic lymphocytic leukemia. Leuk Lymphoma 2012, 53, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Kayraklioglu, N.; Horuluoglu, B.; Klinman, D.M. CpG Oligonucleotides as Vaccine Adjuvants. Methods Mol. Biol. 2021, 2197, 51–85. [Google Scholar] [CrossRef] [PubMed]
- Johnson, I.R.; Parkinson-Lawrence, E.J.; Shandala, T.; Weigert, R.; Butler, L.M.; Brooks, D.A. Altered endosome biogenesis in prostate cancer has biomarker potential. Mol. Cancer Res. 2014, 12, 1851–1862. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miles, M.A.; Luong, R.; To, E.E.; Erlich, J.R.; Liong, S.; Liong, F.; Logan, J.M.; O’Leary, J.; Brooks, D.A.; Selemidis, S. TLR9 Monotherapy in Immune-Competent Mice Suppresses Orthotopic Prostate Tumor Development. Cells 2024, 13, 97. https://doi.org/10.3390/cells13010097
Miles MA, Luong R, To EE, Erlich JR, Liong S, Liong F, Logan JM, O’Leary J, Brooks DA, Selemidis S. TLR9 Monotherapy in Immune-Competent Mice Suppresses Orthotopic Prostate Tumor Development. Cells. 2024; 13(1):97. https://doi.org/10.3390/cells13010097
Chicago/Turabian StyleMiles, Mark A., Raymond Luong, Eunice E. To, Jonathan R. Erlich, Stella Liong, Felicia Liong, Jessica M. Logan, John O’Leary, Doug A. Brooks, and Stavros Selemidis. 2024. "TLR9 Monotherapy in Immune-Competent Mice Suppresses Orthotopic Prostate Tumor Development" Cells 13, no. 1: 97. https://doi.org/10.3390/cells13010097
APA StyleMiles, M. A., Luong, R., To, E. E., Erlich, J. R., Liong, S., Liong, F., Logan, J. M., O’Leary, J., Brooks, D. A., & Selemidis, S. (2024). TLR9 Monotherapy in Immune-Competent Mice Suppresses Orthotopic Prostate Tumor Development. Cells, 13(1), 97. https://doi.org/10.3390/cells13010097