
Current and Future Therapeutics for Treating Patients with Sickle Cell Disease
{kind=link}
{kind=link}
{kind=link}
Abstract
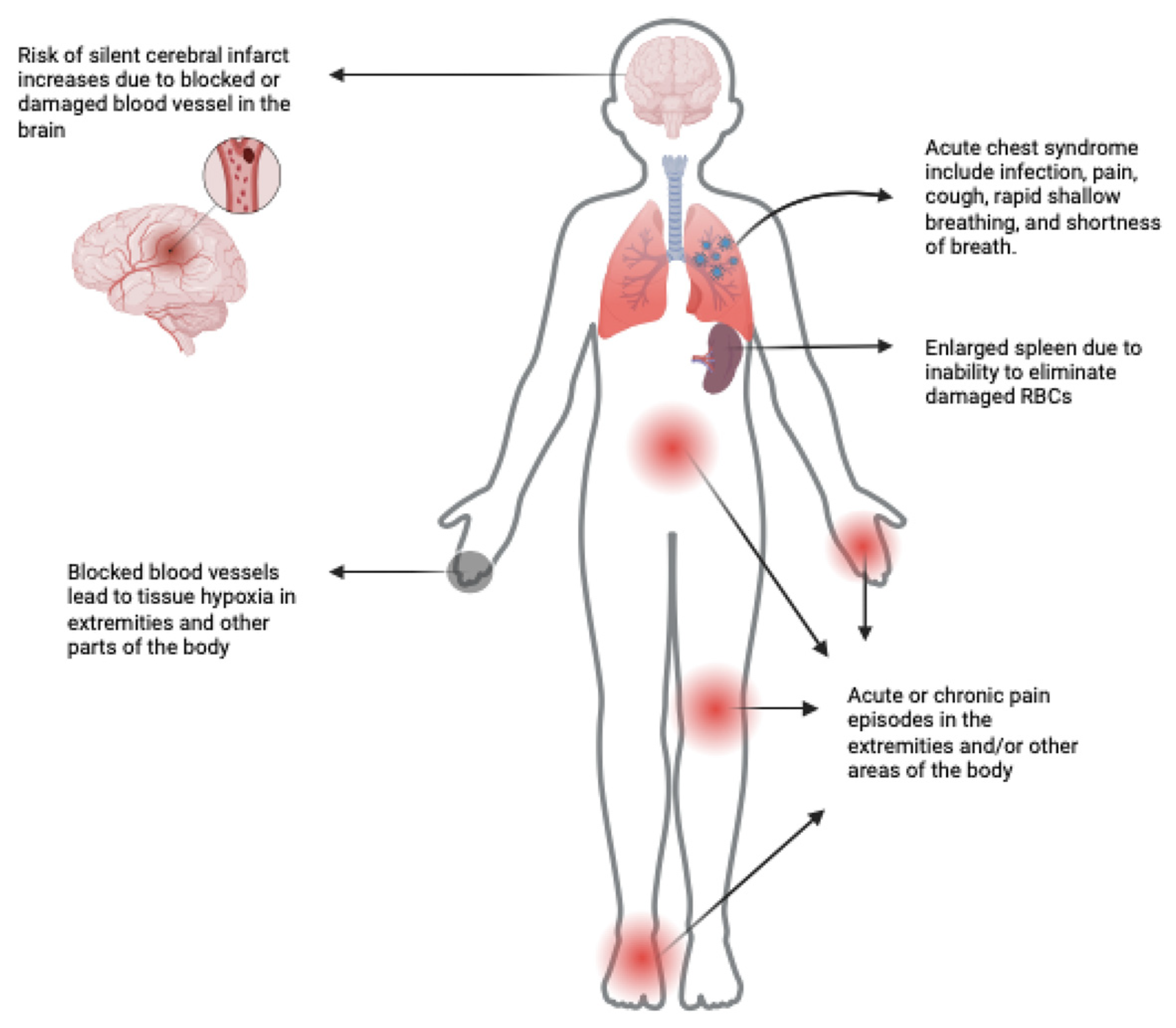
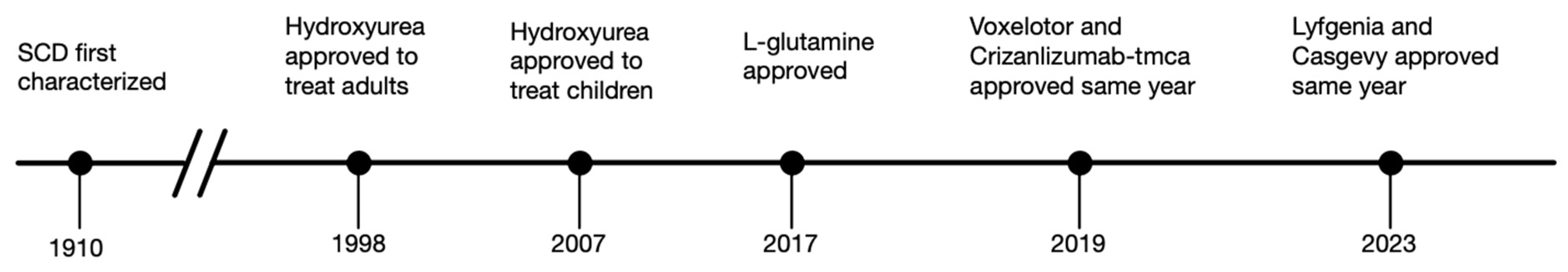
:1. Introduction
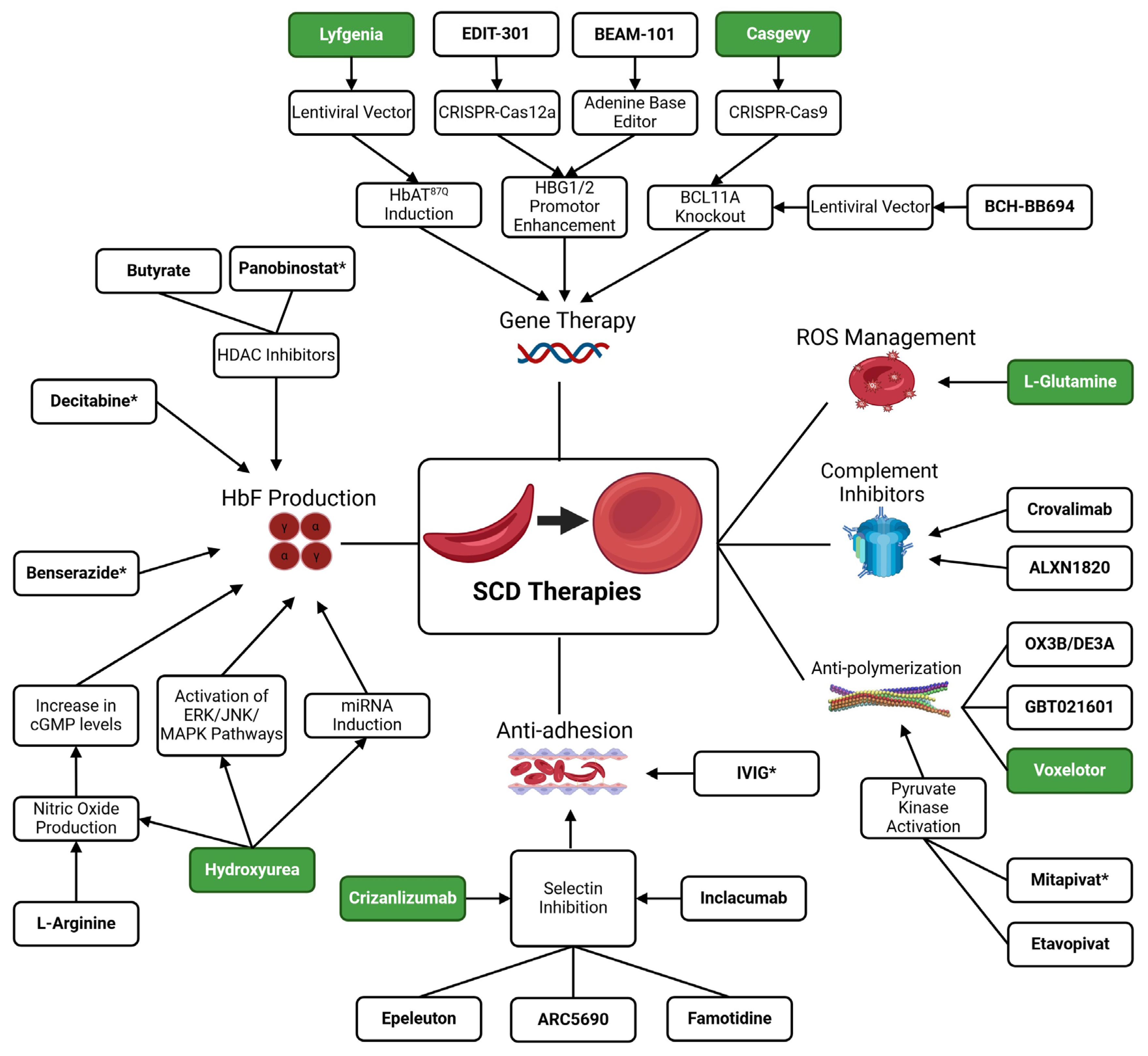
2. Hemoglobin F Induction Therapies
2.1. Hydroxyurea (HU)
2.2. Decitabine
2.3. HDAC Inhibitors
2.4. Benserazide
3. Anti-Polymerization Therapies
3.1. Voxelotor (GBT440)
3.2. GBT021601
4. Pyruvate Kinase Activators
4.1. Mitapivat
4.2. Etavopivat
5. Anti-Adhesion Therapies
5.1. Crizanlizumab-tmca (ADAKVEO)
5.2. Epeleuton
5.3. Famotidine
5.4. Inclacumab
5.5. Intravenous Gamma Globulin (IVIG)
6. Gene Therapy
6.1. BCH-BB694
6.2. Casgevy/Exa-Cel (CTX001)
6.3. Lyfgenia/Lov-Cel (BB305)
6.4. EDIT-301
6.5. BEAM-101
7. Other Novel Therapeutics to Treat SCD Complications
7.1. L-Glutamine (Endari)
7.2. Defibrotide
7.3. ALXN1820
7.4. Crovalimab
7.5. RNA Aptamers
7.6. Nitric Oxide (NO) Modulation
7.7. Alternatives to Drug Therapies
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Onimoe, G.; Rotz, S. Sickle Cell Disease: A Primary Care Update. Cleve Clin. J. Med. 2020, 87, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Ware, R.E.; de Montalembert, M.; Tshilolo, L.; Abboud, M.R. Sickle Cell Disease. Lancet 2017, 390, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Piel, F.B.; Steinberg, M.H.; Rees, D.C. Sickle Cell Disease. New Engl. J. Med. 2017, 376, 1561–1573. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.T.; Smith-Whitley, K.; Banks, S.; Puckrein, G. Reducing Health Care Disparities in Sickle Cell Disease: A Review. Public Health Rep. 2019, 134, 599–607. [Google Scholar] [CrossRef] [PubMed]
- Ashorobi, D.; Ramsey, A.; Yarrarapu, S.N.S.; Bhatt, R. Sickle Cell Trait. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Williams, T.N.; Thein, S.L. Sickle Cell Anemia and Its Phenotypes. Annu. Rev. Genom. Hum. Genet. 2018, 19, 113–147. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Bao, G. CRISPR/Cas9 Gene Editing for Curing Sickle Cell Disease. Transfus. Apher. Sci. 2021, 60, 103060. [Google Scholar] [CrossRef] [PubMed]
- Sadaf, A.; Quinn, C.T. L-Glutamine for Sickle Cell Disease: Knight or Pawn? Exp. Biol. Med. 2020, 245, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Alayash, A.I. Oxidative Pathways in the Sickle Cell and Beyond. Blood Cells Mol. Dis. 2018, 70, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Ilesanmi, O.O. Pathological Basis of Symptoms and Crises in Sickle Cell Disorder: Implications for Counseling and Psychotherapy. Hematol. Rev. 2010, 2, e2. [Google Scholar] [CrossRef]
- Ballas, S.K.; Gupta, K.; Adams-Graves, P. Sickle Cell Pain: A Critical Reappraisal. Blood 2012, 120, 3647–3656. [Google Scholar] [CrossRef]
- Darbari, D.S.; Sheehan, V.A.; Ballas, S.K. The Vaso-Occlusive Pain Crisis in Sickle Cell Disease: Definition, Pathophysiology, and Management. Eur. J. Haematol. 2020, 105, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Wun, T.; Cordoba, M.; Rangaswami, A.; Cheung, A.W.; Paglieroni, T. Activated Monocytes and Platelet-Monocyte Aggregates in Patients with Sickle Cell Disease. Clin. Lab. Haematol. 2002, 24, 81–88. [Google Scholar] [CrossRef]
- Lum, A.F.H.; Wun, T.; Staunton, D.; Simon, S.I. Inflammatory Potential of Neutrophils Detected in Sickle Cell Disease. Am. J. Hematol. 2004, 76, 126–133. [Google Scholar] [CrossRef]
- Okpala, I.; Daniel, Y.; Haynes, R.; Odoemene, D.; Goldman, J. Relationship between the Clinical Manifestations of Sickle Cell Disease and the Expression of Adhesion Molecules on White Blood Cells. Eur. J. Haematol. 2002, 69, 135–144. [Google Scholar] [CrossRef]
- Afrin, L.B. Mast Cell Activation Syndrome as a Significant Comorbidity in Sickle Cell Disease. Am. J. Med. Sci. 2014, 348, 460–464. [Google Scholar] [CrossRef] [PubMed]
- Rees, D.C.; Kilinc, Y.; Unal, S.; Dampier, C.; Pace, B.S.; Kaya, B.; Trompeter, S.; Odame, I.; Mahlangu, J.; Unal, S.; et al. A Randomized, Placebo-Controlled, Double-Blind Trial of Canakinumab in Children and Young Adults with Sickle Cell Anemia. Blood 2022, 139, 2642–2652. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, F.A.; Mahdi, N.; Sater, M.A.; Al-Ola, K.; Almawi, W.Y. The Relation of C-Reactive Protein to Vasoocclusive Crisis in Children with Sickle Cell Disease. Blood Cells Mol. Dis. 2010, 45, 293–296. [Google Scholar] [CrossRef]
- Rees, D.C.; Williams, T.N.; Gladwin, M.T. Sickle-Cell Disease. Lancet 2010, 376, 2018–2031. [Google Scholar] [CrossRef]
- Gragert, L.; Eapen, M.; Williams, E.; Freeman, J.; Spellman, S.; Baitty, R.; Hartzman, R.; Rizzo, J.D.; Horowitz, M.; Confer, D.; et al. HLA Match Likelihoods for Hematopoietic Stem-Cell Grafts in the U.S. Registry. New Engl. J. Med. 2014, 371, 339–348. [Google Scholar] [CrossRef]
- Parikh, S.; Brochstein, J.A.; Galamidi, E.; Schwarzbach, A.; Kurtzberg, J. Allogeneic Stem Cell Transplantation with Omidubicel in Sickle Cell Disease. Blood Adv. 2021, 5, 843–852. [Google Scholar] [CrossRef]
- Charache, S.; Terrin, M.L.; Moore, R.D.; Dover, G.J.; Barton, F.B.; Eckert, S.V.; McMahon, R.P.; Bonds, D.R. Effect of Hydroxyurea on the Frequency of Painful Crises in Sickle Cell Anemia. New Engl. J. Med. 1995, 332, 1317–1322. [Google Scholar] [CrossRef]
- Steinberg, M.H.; McCarthy, W.F.; Castro, O.; Ballas, S.K.; Armstrong, F.D.; Smith, W.; Ataga, K.; Swerdlow, P.; Kutlar, A.; DeCastro, L.; et al. The Risks and Benefits of Long-Term Use of Hydroxyurea in Sickle Cell Anemia: A 17.5 Year Follow-Up. Am. J. Hematol. 2010, 85, 403–408. [Google Scholar] [CrossRef]
- Kinney, T.R.; Helms, R.W.; O’Branski, E.E.; Ohene-Frempong, K.; Wang, W.; Daeschner, C.; Vichinsky, E.; Redding-Lallinger, R.; Gee, B.; Platt, O.S.; et al. Safety of Hydroxyurea in Children with Sickle Cell Anemia: Results of the HUG-KIDS Study, a Phase I/II Trial. Blood 1999, 94, 1550–1554. [Google Scholar]
- Wang, W.C.; Helms, R.W.; Lynn, H.S.; Redding-Lallinger, R.; Gee, B.E.; Ohene-Frempong, K.; Smith-Whitley, K.; Waclawiw, M.A.; Vichinsky, E.P.; Styles, L.A.; et al. Effect of Hydroxyurea on Growth in Children with Sickle Cell Anemia: Results of the HUG-KIDS Study. J. Pediatr. 2002, 140, 225–229. [Google Scholar] [CrossRef]
- Lanzkron, S.; Strouse, J.J.; Wilson, R.; Beach, M.C.; Haywood, C.; Park, H.; Witkop, C.; Bass, E.B.; Segal, J.B. Systematic Review: Hydroxyurea for the Treatment of Adults with Sickle Cell Disease. Ann. Intern. Med. 2008, 148, 939–955. [Google Scholar] [CrossRef]
- Jeste, N.D.; Sánchez, L.M.; Urcuyo, G.S.; Bergés, M.E.; Luden, J.P.; Stuber, S.E.; Latham, T.S.; Mena, R.; Nieves, R.M.; Ware, R.E. Stroke Avoidance for Children in República Dominicana (SACRED): Protocol for a Prospective Study of Stroke Risk and Hydroxyurea Treatment in Sickle Cell Anemia. JMIR Res. Protoc. 2017, 6, e107. [Google Scholar] [CrossRef]
- Molokie, R.; Lavelle, D.; Gowhari, M.; Pacini, M.; Krauz, L.; Hassan, J.; Ibanez, V.; Ruiz, M.A.; Ng, K.P.; Woost, P.; et al. Oral Tetrahydrouridine and Decitabine for Non-Cytotoxic Epigenetic Gene Regulation in Sickle Cell Disease: A Randomized Phase 1 Study. PLoS Med. 2017, 14, e1002382. [Google Scholar] [CrossRef]
- Esrick, E.B.; Mcconkey, M.; Lin, K.; Frisbee, A.; Ebert, B.L. Inactivation of HDAC1 or HDAC2 Induces Gamma Globin Expression without Altering Cell Cycle or Proliferation. Am. J. Hematol. 2015, 90, 624–628. [Google Scholar] [CrossRef]
- Paikari, A.; Sheehan, V.A. Fetal Haemoglobin Induction in Sickle Cell Disease. Br. J. Haematol. 2018, 180, 189–200. [Google Scholar] [CrossRef]
- Weinberg, R.S.; Ji, X.; Sutton, M.; Perrine, S.; Galperin, Y.; Li, Q.; Liebhaber, S.A.; Stamatoyannopoulos, G.; Atweh, G.F. Butyrate Increases the Efficiency of Translation of γ-Globin MRNA. Blood 2005, 105, 1807–1809. [Google Scholar] [CrossRef]
- Kutlar, A.; Reid, M.E.; Inati, A.; Taher, A.T.; Abboud, M.R.; El-Beshlawy, A.; Buchanan, G.R.; Smith, H.; Ataga, K.I.; Perrine, S.P.; et al. A Dose-Escalation Phase IIa Study of 2,2-Dimethylbutyrate (HQK-1001), an Oral Fetal Globin Inducer, in Sickle Cell Disease. Am. J. Hematol. 2013, 88, E255–E260. [Google Scholar] [CrossRef]
- Pace, B.S.; Perrine, S.; Li, B.; Makala, L.; Xu, H.; Takezaki, M.; Wolf, R.F.; Wang, A.; Xu, X.; Huang, J.; et al. Benserazide Racemate and Enantiomers Induce Fetal Globin Gene Expression in Vivo: Studies to Guide Clinical Development for Beta Thalassemia and Sickle Cell Disease. Blood Cells Mol. Dis. 2021, 89, 102561. [Google Scholar] [CrossRef]
- Santos, M.E.H.P.; Olops, L.; Vendrame, F.; Tavares, A.H.J.; Leonardo, D.P.; de Azevedo, P.C.; Piovesana, L.G.; Costa, F.F.; Fertrin, K.Y. Benserazide as a Potential Novel Fetal Hemoglobin Inducer: An Observational Study in Non-Carriers of Hemoglobin Disorders. Blood Cells Mol. Dis. 2021, 87, 102511. [Google Scholar] [CrossRef]
- Herity, L.B.; Vaughan, D.M.M.; Rodriguez, L.R.; Lowe, D.K. Voxelotor: A Novel Treatment for Sickle Cell Disease. Ann. Pharmacother. 2021, 55, 240–245. [Google Scholar] [CrossRef]
- Oksenberg, D.; Dufu, K.; Patel, M.P.; Chuang, C.; Li, Z.; Xu, Q.; Silva-Garcia, A.; Zhou, C.; Hutchaleelaha, A.; Patskovska, L.; et al. GBT440 Increases Haemoglobin Oxygen Affinity, Reduces Sickling and Prolongs RBC Half-Life in a Murine Model of Sickle Cell Disease. Br. J. Haematol. 2016, 175, 141–153. [Google Scholar] [CrossRef]
- Brown, C.; Idowu, M.; Drachtman, R.; Beaubrun, A.; Agodoa, I.; Nguyen, A.; Lipman, K.; Moshkovich, O.; Murphy, R.; Bellenger, M.A.; et al. Patient-Reported Experiences in Voxelotor-Treated Children and Adults with Sickle Cell Disease: A Semistructured Interview Study. Biomed. Res. Int. 2023, 2023, 7533111. [Google Scholar] [CrossRef]
- Blyden, G.; Bridges, K.R.; Bronte, L. Case Series of Patients with Severe Sickle Cell Disease Treated with Voxelotor (GBT440) by Compassionate Access. Am. J. Hematol. 2018, 93, E188–E190. [Google Scholar] [CrossRef]
- Telfer, P.; Agodoa, I.; Fox, K.M.; Burke, L.; Mant, T.; Jurek, M.; Tonda, M.; Lehrer-Graiwer, J. Impact of Voxelotor (GBT440) on Unconjugated Bilirubin and Jaundice in Sickle Cell Disease: A Patient Case Report. Hematol. Rep. 2018, 10, 50–52. [Google Scholar] [CrossRef]
- Han, J.; Molokie, R.E.; Hussain, F.; Njoku, F.; Gordeuk, V.R.; Saraf, S.L. Voxelotor and Albuminuria in Adults with Sickle Cell Anaemia. Br. J. Haematol. 2022, 197, E63–E64. [Google Scholar] [CrossRef] [PubMed]
- Vichinsky, E.; Hoppe, C.C.; Ataga, K.I.; Ware, R.E.; Nduba, V.; El-Beshlawy, A.; Hassab, H.; Achebe, M.M.; Alkindi, S.; Brown, R.C.; et al. A Phase 3 Randomized Trial of Voxelotor in Sickle Cell Disease. New Engl. J. Med. 2019, 381, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Howard, J.; Ataga, K.I.; Brown, R.C.; Achebe, M.; Nduba, V.; El-Beshlawy, A.; Hassab, H.; Agodoa, I.; Tonda, M.; Gray, S.; et al. Voxelotor in Adolescents and Adults with Sickle Cell Disease (HOPE): Long-Term Follow-up Results of an International, Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet Haematol. 2021, 8, e323–e333. [Google Scholar] [CrossRef] [PubMed]
- Estepp, J.H.; Kalpatthi, R.; Woods, G.; Trompeter, S.; Liem, R.I.; Sims, K.; Inati, A.; Inusa, B.P.D.; Campbell, A.; Piccone, C.; et al. Safety and Efficacy of Voxelotor in Pediatric Patients with Sickle Cell Disease Aged 4 to 11 Years. Pediatr. Blood Cancer 2022, 69, e29716. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.; Key, C.; Agodoa, I.; Olbertz, J.; Duchin, K.; Barth, A.; Lisbon, E. S268: Safety, Tolerability, and Pharmacokinetic/Pharmacodynamic Results from Phase 1 Studies of GBT021601, a Next-Generation HBS Polymerization Inhibitor for Treatment of Sickle Cell Disease. Hemasphere 2022, 6, 169–170. [Google Scholar] [CrossRef]
- Pochron, M.P.; Sun, J.; Potdar, A.A.; Alt, C.; DeGuzman, F.; Connor, M.W.; Cathers, B.; Dufu, K.; Archer, D.R. Predictive Biomarker Analysis from the GBT021601 Survival Study in Townes Sickle Mice. Blood 2023, 142, 14. [Google Scholar] [CrossRef]
- van Dijk, M.J.; Rab, M.A.E.; van Oirschot, B.A.; Bos, J.; Derichs, C.; Rijneveld, A.W.; Cnossen, M.H.; Nur, E.; Biemond, B.J.; Bartels, M.; et al. One-Year Safety and Efficacy of Mitapivat in Sickle Cell Disease: Follow-up Results of a Phase 2, Open-Label Study. Blood Adv. 2023, 7, 7539–7550. [Google Scholar] [CrossRef] [PubMed]
- Grace, R.F.; Mark Layton, D.; Barcellini, W. How We Manage Patients with Pyruvate Kinase Deficiency. Br. J. Haematol. 2019, 184, 721–734. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, P.; Fulzele, K.; Forsyth, S.; Ribadeneira, M.D.; Guichard, S.; Wilker, E.; Marshall, C.G.; Drake, A.; Fessler, R.; Konstantinidis, D.G.; et al. Etavopivat, a Pyruvate Kinase Activator in Red Blood Cells, for the Treatment of Sickle Cell Disease. J. Pharmacol. Exp. Ther. 2022, 380, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Blair, H.A. Crizanlizumab: First Approval. Drugs 2020, 80, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Cheplowitz, H.; Block, S.; Groesbeck, J.; Sacknoff, S.; Nguyen, A.L.; Gopal, S. Real-World Data of Crizanlizumab in Sickle Cell Disease: A Single-Center Analysis. J. Hematol. 2023, 12, 105–108. [Google Scholar] [CrossRef]
- Liles, D.; Shah, N.; Scullin, B.; Gordeuk, V.; Smith, W.; Kanter, J.; Achebe, M.; Boccia, R.; Crary, S.; Kraft, W.; et al. S853 Successor: A Multicenter Retrospective Noninterventional Follow-Up Study in Patients with Sickle Cell Pain Crises Who Previously Participated in the Sustain Trial in the United States. Hemasphere 2019, 3, 380–381. [Google Scholar] [CrossRef]
- Jacobs, J.W.; Stephens, L.D.; Chooljian, D.M.; Sharma, D.; Adkins, B.D.; Booth, G.S. Crizanlizumab and Sickle Cell Disease: When Should Medications Have Their Approval Status Revoked? Am. J. Hematol. 2024. Online ahead of print. [Google Scholar] [CrossRef]
- Hines, P.C.; Zen, Q.; Burney, S.N.; Shea, D.A.; Ataga, K.I.; Orringer, E.P.; Telen, M.J.; Parise, L.V. Novel Epinephrine and Cyclic AMP-Mediated Activation of BCAM/Lu-Dependent Sickle (SS) RBC Adhesion. Blood 2003, 101, 3281–3287. [Google Scholar] [CrossRef] [PubMed]
- Brittain, J.E.; Mlinar, K.J.; Anderson, C.S.; Orringer, E.P.; Parise, L.V. Activation of Sickle Red Blood Cell Adhesion via Integrin-Associated Protein/CD47-Induced Signal Transduction. J. Clin. Investig. 2001, 107, 1555–1562. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Xu, C.; Manwani, D.; Frenette, P.S. Neutrophils, Platelets, and Inflammatory Pathways at the Nexus of Sickle Cell Disease Pathophysiology. Blood 2016, 127, 801–809. [Google Scholar] [CrossRef]
- Matsui, N.M.; Borsig, L.; Rosen, S.D.; Yaghmai, M.; Varki, A.; Embury, S.H. P-Selectin Mediates the Adhesion of Sickle Erythrocytes to the Endothelium. Blood 2001, 98, 1955–1962. [Google Scholar] [CrossRef] [PubMed]
- Kato, G.J.; Steinberg, M.H.; Gladwin, M.T. Intravascular Hemolysis and the Pathophysiology of Sickle Cell Disease. J. Clin. Investig. 2017, 127, 750–760. [Google Scholar] [CrossRef]
- Andemariam, B.; Sheehan, V.A.; Hamza, M.; Kelly, K.; Climax, J. A Phase 2, Open-Label Study to Assess the Pharmacokinetics, Pharmacodynamics and Safety of Orally Administered Epeleuton in Patients with Sickle Cell Disease. Blood 2023, 142, 1159. [Google Scholar] [CrossRef]
- Federti, E.; Matte’, A.; Hamza, M.; Lafferty, A.; Coughlan, D.; Weissbach, M.; Bhatt, D.L.; Riccardi, V.; Perissinotto, R.; Siciliano, A.; et al. P1483: Epeleuton, a Novel Synthetic Second Generation W-3 Fatty Acid, Protects Humanized Sickle Cell Mice against Hypoxia/Reoxygenation Organ Damage. Hemasphere 2022, 6, 1365–1366. [Google Scholar] [CrossRef]
- Andemariam, B.; Inati, A.; Colombatti, R.; Minniti, C.; Brown, C.; Hottmann, M.; Gray, S.; Hoppe, C.; Davis, M.; Yue, P. P1486: Trials in Progress: The Thrive Studies Evaluating the Efficacy, Safety, and Long-Term Treatment with Inclacumab, A P-Selectin Inhibitor, in Patients with Sickle Cell Disease. Hemasphere 2022, 6, 1368–1369. [Google Scholar] [CrossRef]
- Chang, J.; Shi, P.A.; Chiang, E.Y.; Frenette, P.S. Intravenous Immunoglobulins Reverse Acute Vaso-Occlusive Crises in Sickle Cell Mice through Rapid Inhibition of Neutrophil Adhesion. Blood 2008, 111, 915–923. [Google Scholar] [CrossRef]
- Manwani, D.; Chen, G.; Carullo, V.; Serban, S.; Olowokure, O.; Jang, J.; Huggins, M.; Cohen, H.W.; Billett, H.; Atweh, G.F.; et al. Single-Dose Intravenous Gammaglobulin Can Stabilize Neutrophil Mac-1 Activation in Sickle Cell Pain Crisis. Am. J. Hematol. 2015, 90, 381–385. [Google Scholar] [CrossRef]
- Manwani, D.; Xu, C.; Lee, S.K.; Amatuni, G.; Cohen, H.W.; Carullo, V.; Morrone, K.; Davila, J.; Shi, P.A.; Ireland, K.; et al. Randomized Phase 2 Trial of Intravenous Gamma Globulin (IVIG) for the Treatment of Acute Vaso-Occlusive Crisis in Patients with Sickle Cell Disease: Lessons Learned from the Midpoint Analysis. Complement. Ther. Med. 2020, 52, 102481. [Google Scholar] [CrossRef]
- AbuSamra, D.B.; Aleisa, F.A.; Al-Amoodi, A.S.; Ahmed, H.M.J.; Chin, C.J.; Abuelela, A.F.; Bergam, P.; Sougrat, R.; Merzaban, J.S. Not Just a Marker: CD34 on Human Hematopoietic Stem/Progenitor Cells Dominates Vascular Selectin Binding along with CD44. Blood Adv. 2017, 1, 2799–2816. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.A.; Walters, M.C.; Kwiatkowski, J.; Rasko, J.E.J.; Ribeil, J.-A.; Hongeng, S.; Magrin, E.; Schiller, G.J.; Payen, E.; Semeraro, M.; et al. Gene Therapy in Patients with Transfusion-Dependent β-Thalassemia. New Engl. J. Med. 2018, 378, 1479–1493. [Google Scholar] [CrossRef]
- Ribeil, J.-A.; Hacein-Bey-Abina, S.; Payen, E.; Magnani, A.; Semeraro, M.; Magrin, E.; Caccavelli, L.; Neven, B.; Bourget, P.; El Nemer, W.; et al. Gene Therapy in a Patient with Sickle Cell Disease. New Engl. J. Med. 2017, 376, 848–855. [Google Scholar] [CrossRef]
- Sankaran, V.G.; Menne, T.F.; Xu, J.; Akie, T.E.; Lettre, G.; Van Handel, B.; Mikkola, H.K.A.; Hirschhorn, J.N.; Cantor, A.B.; Orkin, S.H. Human Fetal Hemoglobin Expression Is Regulated by the Developmental Stage-Specific Repressor BCL11A. Science 2008, 322, 1839–1842. [Google Scholar] [CrossRef]
- Esrick, E.B.; Lehmann, L.E.; Biffi, A.; Achebe, M.; Brendel, C.; Ciuculescu, M.F.; Daley, H.; MacKinnon, B.; Morris, E.; Federico, A.; et al. Post-Transcriptional Genetic Silencing of BCL11A to Treat Sickle Cell Disease. New Engl. J. Med. 2021, 384, 205–215. [Google Scholar] [CrossRef]
- Frangoul, H.; Altshuler, D.; Cappellini, M.D.; Chen, Y.S.; Domm, J.; Eustace, B.K.; Foell, J.; De La Fuente, J.; Grupp, S.; Handgretinger, R.; et al. CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia. Obstet. Gynecol. Surv. 2021, 76, 327–329. [Google Scholar] [CrossRef]
- Kanter, J.; Walters, M.C.; Krishnamurti, L.; Mapara, M.Y.; Kwiatkowski, J.L.; Rifkin-Zenenberg, S.; Aygun, B.; Kasow, K.A.; Pierciey, F.J.; Bonner, M.; et al. Biologic and Clinical Efficacy of LentiGlobin for Sickle Cell Disease. New Engl. J. Med. 2022, 386, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Magrin, E.; Semeraro, M.; Hebert, N.; Joseph, L.; Magnani, A.; Chalumeau, A.; Gabrion, A.; Roudaut, C.; Marouene, J.; Lefrere, F.; et al. Long-Term Outcomes of Lentiviral Gene Therapy for the β-Hemoglobinopathies: The HGB-205 Trial. Nat. Med. 2022, 28, 81–88. [Google Scholar] [CrossRef]
- De Dreuzy, E.; Heath, J.; Zuris, J.A.; Sousa, P.; Viswanathan, R.; Scott, S.; Da Silva, J.; Ta, T.; Capehart, S.; Wang, T.; et al. EDIT-301: An Experimental Autologous Cell Therapy Comprising Cas12a-RNP Modified MPB-CD34+ Cells for the Potential Treatment of SCD. Blood 2019, 134, 4636. [Google Scholar] [CrossRef]
- Hanna, R.; Frangoul, H.; Mckinney, C.; Pineiro, L.; Mapara, M.; Chang, K.-H.; Jaskolka, M.; Kim, K.; Rizk, M.; Afonja, O.; et al. S264: Edit-301 Shows Promising Preliminary Safety and Efficacy Results in the Phase I/Ii Clinical Trial (Ruby) of Patients with Severe Sickle Cell Disease Using Highly Specific and Efficient Ascas12a Enzyme. Hemasphere 2023, 7, e05170e0. [Google Scholar] [CrossRef]
- Zarghamian, P.; Klermund, J.; Cathomen, T. Clinical Genome Editing to Treat Sickle Cell Disease—A Brief Update. Front. Med. 2023, 9, 1065377. [Google Scholar] [CrossRef] [PubMed]
- Niihara, Y. L-Glutamine Therapy Reduces Hospitalization for Sickle Cell Anemia and Sickle Β°-Thalassemia Patients at Six Months—A Phase II Randomized Trial. Clin. Pharmacol. Biopharm. 2014, 3, 2. [Google Scholar] [CrossRef]
- Niihara, Y.; Zerez, C.R.; Akiyama, D.S.; Tanaka, K.R. Oral L-Glutamine Therapy for Sickle Cell Anemia: I. Subjective Clinical Improvement and Favorable Change in Red Cell NAD Redox Potential. Am. J. Hematol. 1998, 58, 117–121. [Google Scholar] [CrossRef]
- Niihara, Y.; Miller, S.T.; Kanter, J.; Lanzkron, S.; Smith, W.R.; Hsu, L.L.; Gordeuk, V.R.; Viswanathan, K.; Sarnaik, S.; Osunkwo, I.; et al. A Phase 3 Trial of l -Glutamine in Sickle Cell Disease. New Engl. J. Med. 2018, 379, 226–235. [Google Scholar] [CrossRef]
- Baker, D.E.; Demaris, K. Defibrotide. Hosp. Pharm. 2016, 51, 847–854. [Google Scholar] [CrossRef]
- Milner, J.; Hung, J.; Ayello, J.; Shi, Q.; Talano, J.-A.M.; Moore, T.B.; Friedman, D.; Dozor, A.J.; Klejmont, L.; Mahanti, H.; et al. Determining the Safety and Efficacy of Prophylactic Defibrotide Administration in Children, Adolescents, and Young Adults with Sickle Cell Disease Following Myeloimmunoablative Conditioning (MAC) and Haploidentical Stem Cell Transplantation Utilizing CD34+ Selection and T-Cell (CD3) Addback (IND127812). Biol. Blood Marrow Transplant. 2020, 26, S212–S213. [Google Scholar] [CrossRef]
- Milner, J.; Friedman, D.; D’Souza, M.; Sankaran, K.; Klejmont, L.; Morris, E.; Mahanti, H.; Otero, N.; Hochberg, J.C.; Brudnicki, A.; et al. Preliminary Results of a Phase II Study to Determine the Safety of Defibrotide in Children and Adolescents with Sickle Cell Disease-Associated Acute Chest Syndrome (IND 127812). Blood 2020, 136, 8–9. [Google Scholar] [CrossRef]
- Callaghan, M.; Ataga, K.; De Franceschi, L.; Minniti, C.; Balachandran, N.; Imbs, D.; Perretti, T.; Ramos, J.; Sostelly, A.; Bartolucci, P. P120: Trial in Progress: The Randomized, Double-Blind, Placebo-Controlled Phase 2a Crosswalk-C Trial Evaluating the Efficacy of Crovalimab as Adjunct Treatment in the Prevention of Vaso-Occlusive Episodes (Voes) in Patients (Pts) with Sickle Cell Disease (Scd). Hemasphere 2022, 6, 27–28. [Google Scholar] [CrossRef]
- Varelas, C.; Tampaki, A.; Sakellari, I.; Anagnostopoulos, A.; Gavriilaki, E.; Vlachaki, E. Complement in Sickle Cell Disease: Are We Ready for Prime Time? J. Blood Med. 2021, 12, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Hill, K.; Harris, D.; Shen, T.; Yuan, C.-X.; Gasteyger, C. A Phase 2a, Randomized, Open-Label Study to Evaluate Multiple Dosing Regimens of Subcutaneous ALXN1820 in Adult Patients with Sickle Cell Disease. Blood 2022, 140, 8298–8299. [Google Scholar] [CrossRef]
- Röth, A.; Nishimura, J.I.; Nagy, Z.; Gaàl-Weisinger, J.; Panse, J.; Yoon, S.S.; Egyed, M.; Ichikawa, S.; Ito, Y.; Kim, J.S.; et al. The Complement C5 Inhibitor Crovalimab in Paroxysmal Nocturnal Hemoglobinuria. Blood 2020, 135, 912–920. [Google Scholar] [CrossRef] [PubMed]
- Purvis, S.H.; Keefer, J.R.; Fortenberry, Y.M.; Barron-Casella, E.A.; Casella, J.F. Identification of Aptamers That Bind to Sickle Hemoglobin and Inhibit Its Polymerization. Nucleic Acid. Ther. 2017, 27, 354–364. [Google Scholar] [CrossRef] [PubMed]
- Gutsaeva, D.R.; Parkerson, J.B.; Yerigenahally, S.D.; Kurz, J.C.; Schaub, R.G.; Ikuta, T.; Head, C.A. Inhibition of Cell Adhesion by Anti-P-Selectin Aptamer: A New Potential Therapeutic Agent for Sickle Cell Disease. Blood 2011, 117, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Cyr, A.R.; Huckaby, L.V.; Shiva, S.S.; Zuckerbraun, B.S. Nitric Oxide and Endothelial Dysfunction. Crit. Care Clin. 2020, 36, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Hallmark, L.; Almeida, L.E.F.; Kamimura, S.; Smith, M.; Quezado, Z.M.N. Nitric Oxide and Sickle Cell Disease—Is There a Painful Connection? Exp. Biol. Med. 2021, 246, 332–341. [Google Scholar] [CrossRef] [PubMed]
- Ambrusko, S.J.; Gunawardena, S.; Sakara, A.; Windsor, B.; Lanford, L.; Michelson, P.; Krishnamurti, L. Elevation of Tricuspid Regurgitant Jet Velocity, a Marker for Pulmonary Hypertension in Children with Sickle Cell Disease. Pediatr. Blood Cancer 2006, 47, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Liem, R.I.; Akinosun, M.; Muntz, D.S.; Thompson, A.A. Feasibility and Safety of Home Exercise Training in Children with Sickle Cell Anemia. Pediatr. Blood Cancer 2017, 64. [Google Scholar] [CrossRef]
- Gellen, B.; Messonnier, L.; Merlet, A.; Audureau, E.; Rupp, T.; Peyrot, S.; Martin, C.; Ribeil, J.-A.; Galactéros, F.; Feasson, L.; et al. Prospective Controlled Trial on Endurance Exercise Training in Adult Sickle Cell Disease Patients. Blood 2017, 130, 991. [Google Scholar] [CrossRef]
- Setty, B.N.Y.; Betal, S.G.; Miller, R.E.; Brown, D.S.; Meier, M.; Cahill, M.; Lerner, N.B.; Apollonsky, N.; Stuart, M.J. Relationship of Omega-3 Fatty Acids DHA and EPA with the Inflammatory Biomarker Hs-CRP in Children with Sickle Cell Anemia. Prostaglandins Leukot. Essent. Fat. Acids 2019, 146, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Abdelhalim, S.M.; Murphy, J.E.; Meabed, M.H.; Elberry, A.A.; Gamaleldin, M.M.; Shaalan, M.S.; Hussein, R.R.S. Comparative Effectiveness of Adding Omega-3 or Vitamin D to Standard Therapy in Preventing and Treating Episodes of Painful Crisis in Pediatric Sickle Cell Patients. Eur. Rev. Med. Pharmacol. Sci. 2022, 26, 5043–5052. [Google Scholar] [CrossRef] [PubMed]
- Daak, A.A.; Ghebremeskel, K.; Hassan, Z.; Attallah, B.; Azan, H.H.; Elbashir, M.I.; Crawford, M. Effect of Omega-3 (n-3) Fatty Acid Supplementation in Patients with Sickle Cell Anemia: Randomized, Double-Blind, Placebo-Controlled Trial. Am. J. Clin. Nutr. 2013, 97, 37–44. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barak, M.; Hu, C.; Matthews, A.; Fortenberry, Y.M. Current and Future Therapeutics for Treating Patients with Sickle Cell Disease. Cells 2024, 13, 848. https://doi.org/10.3390/cells13100848
Barak M, Hu C, Matthews A, Fortenberry YM. Current and Future Therapeutics for Treating Patients with Sickle Cell Disease. Cells. 2024; 13(10):848. https://doi.org/10.3390/cells13100848
Chicago/Turabian StyleBarak, Mariam, Christopher Hu, Alicia Matthews, and Yolanda M. Fortenberry. 2024. "Current and Future Therapeutics for Treating Patients with Sickle Cell Disease" Cells 13, no. 10: 848. https://doi.org/10.3390/cells13100848
APA StyleBarak, M., Hu, C., Matthews, A., & Fortenberry, Y. M. (2024). Current and Future Therapeutics for Treating Patients with Sickle Cell Disease. Cells, 13(10), 848. https://doi.org/10.3390/cells13100848