
Normal and Dysregulated Sphingolipid Metabolism: Contributions to Podocyte Injury and Beyond


Abstract
:1. Introduction
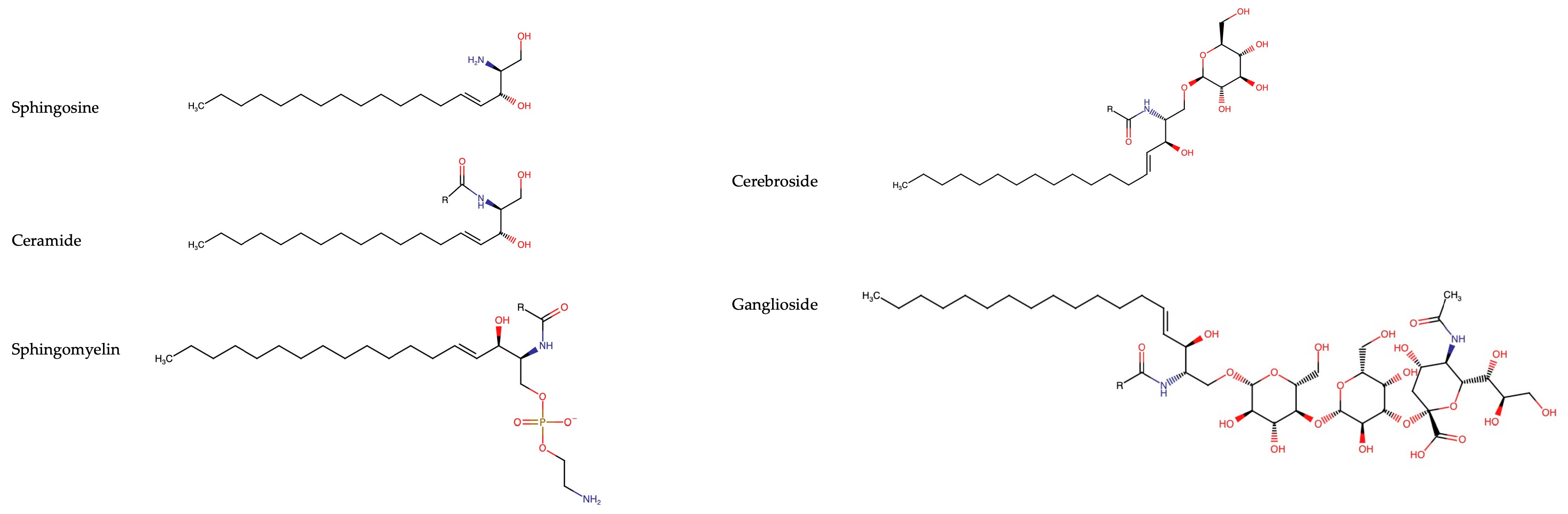
2. Overview of Sphingolipids
2.1. Sphingoid Bases
2.2. Ceramides
2.3. Sphingomyelins
2.4. Cerebrosides
2.5. Gangliosides
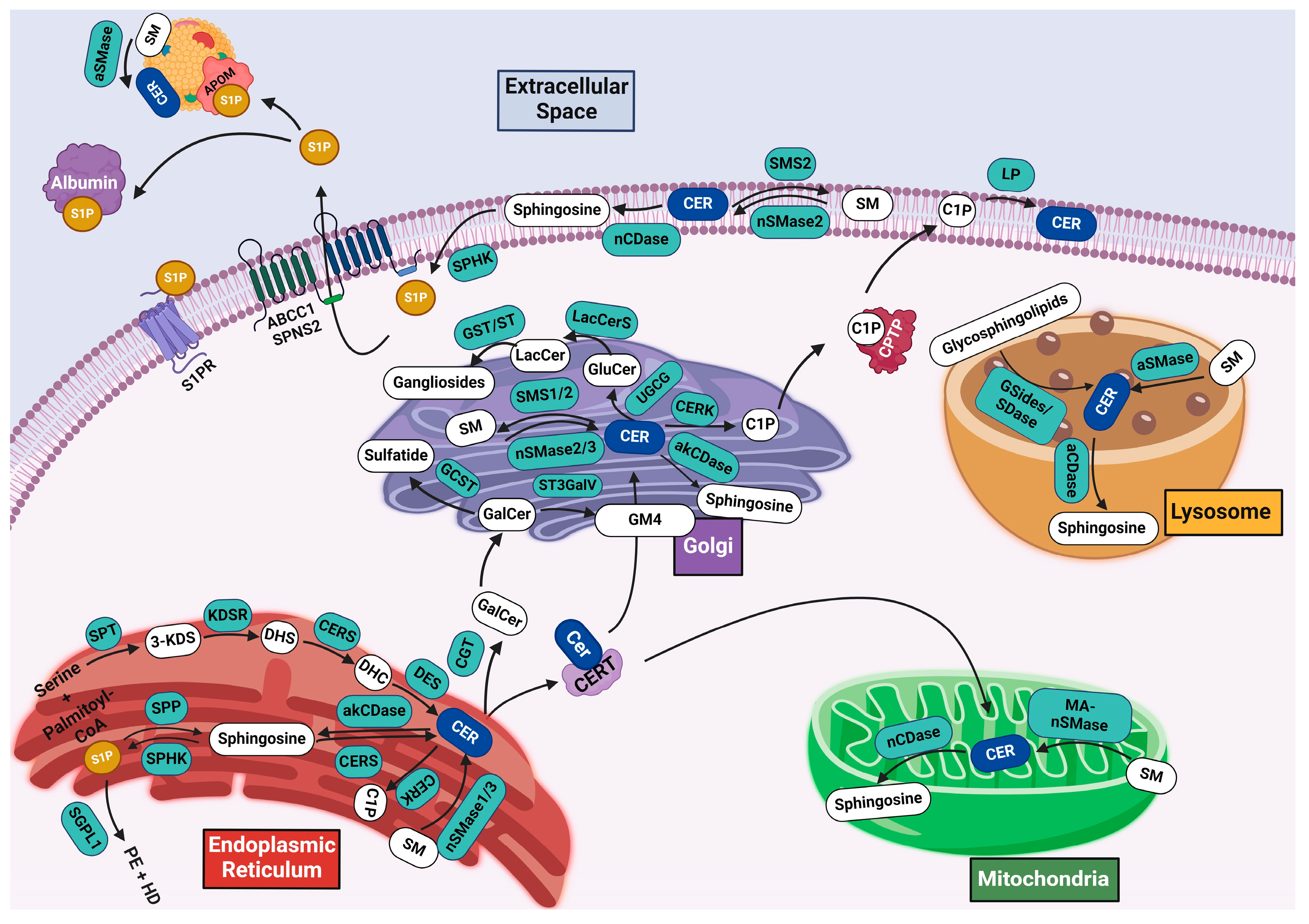
3. Sphingolipid Metabolism
3.1. De Novo Synthesis
3.2. Salvage Pathway
3.3. Ceramide Catabolism and Bioactive Sphingolipids
3.4. Synthesis of Complex Sphingolipids
3.4.1. Sphingomyelin Synthesis
3.4.2. Glycosphingolipid Synthesis
4. Sphingolipids in Podocytes
4.1. Gaucher Disease
4.2. Fabry Disease
4.3. Farber Disease
4.4. Niemann–Pick
4.5. Hereditary Inclusion Body Myopathy 2
4.6. Diabetic Kidney Disease
4.7. Puromycin Aminonucleoside (PAN)-Induced Nephropathy
4.8. Focal Segmental Glomerulosclerosis
4.9. S1P Lyase
4.10. APOL1 Genetic Alterations
4.11. Non-Alcoholic Steatohepatitis
4.12. Radiation Nephropathy
5. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pavenstadt, H.; Kriz, W.; Kretzler, M. Cell biology of the glomerular podocyte. Physiol. Rev. 2003, 83, 253–307. [Google Scholar] [CrossRef] [PubMed]
- Mundel, P.; Shankland, S.J. Podocyte biology and response to injury. J. Am. Soc. Nephrol. 2002, 13, 3005–3015. [Google Scholar] [CrossRef] [PubMed]
- Simons, M.; Schwarz, K.; Kriz, W.; Miettinen, A.; Reiser, J.; Mundel, P.; Holthofer, H. Involvement of lipid rafts in nephrin phosphorylation and organization of the glomerular slit diaphragm. Am. J. Pathol. 2001, 159, 1069–1077. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, K.; Simons, M.; Reiser, J.; Saleem, M.A.; Faul, C.; Kriz, W.; Shaw, A.S.; Holzman, L.B.; Mundel, P. Podocin, a raft-associated component of the glomerular slit diaphragm, interacts with CD2AP and nephrin. J. Clin. Investig. 2001, 108, 1621–1629. [Google Scholar] [CrossRef] [PubMed]
- Bartke, N.; Hannun, Y.A. Bioactive sphingolipids: Metabolism and function. J. Lipid Res. 2009, 50, S91–S96. [Google Scholar] [CrossRef]
- Hannun, Y.A.; Obeid, L.M. Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. [Google Scholar] [CrossRef]
- Merrill, A.H. Chapter 14—Sphingosine and Other Long-Chain Bases That Alter Cell Behavior. In Current Topics in Membranes; Hoekstra, D., Ed.; Academic Press: Cambridge, MA, USA, 1994; Volume 40, pp. 361–386. [Google Scholar]
- Hoekstra, D.; Maier, O.; van der Wouden, J.M.; Slimane, T.A.; van IJzendoorn, S.C. Membrane dynamics and cell polarity: The role of sphingolipids. J. Lipid Res. 2003, 44, 869–877. [Google Scholar] [CrossRef] [PubMed]
- Detournay, O.; Weis, V.M. Role of the sphingosine rheostat in the regulation of cnidarian-dinoflagellate symbioses. Biol. Bull. 2011, 221, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Pruett, S.T.; Bushnev, A.; Hagedorn, K.; Adiga, M.; Haynes, C.A.; Sullards, M.C.; Liotta, D.C.; Merrill, A.H., Jr. Biodiversity of sphingoid bases (“sphingosines”) and related amino alcohols. J. Lipid Res. 2008, 49, 1621–1639. [Google Scholar] [CrossRef] [PubMed]
- Quinville, B.M.; Deschenes, N.M.; Ryckman, A.E.; Walia, J.S. A Comprehensive Review: Sphingolipid Metabolism and Implications of Disruption in Sphingolipid Homeostasis. Int. J. Mol. Sci. 2021, 22, 5793. [Google Scholar] [CrossRef]
- Michaelson, L.V.; Napier, J.A.; Molino, D.; Faure, J.D. Plant sphingolipids: Their importance in cellular organization and adaption. Biochim. Biophys. Acta 2016, 1861, 1329–1335. [Google Scholar] [CrossRef]
- Maceyka, M.; Harikumar, K.B.; Milstien, S.; Spiegel, S. Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol. 2012, 22, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Leipelt, M.; Merrill, A.H. Sphingolipid Biosynthesis. In Encyclopedia of Biological Chemistry; Lane, M.D., Lennarz, W.J., Eds.; Elsevier: Amsterdam, The Netherlands, 2004; pp. 76–81. [Google Scholar]
- Simons, K.; Ikonen, E. Functional rafts in cell membranes. Nature 1997, 387, 569–572. [Google Scholar] [CrossRef] [PubMed]
- Siskind, L.J. Mitochondrial ceramide and the induction of apoptosis. J. Bioenerg. Biomembr. 2005, 37, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Arana, L.; Gangoiti, P.; Ouro, A.; Trueba, M.; Gomez-Munoz, A. Ceramide and ceramide 1-phosphate in health and disease. Lipids Health Dis. 2010, 9, 15. [Google Scholar] [CrossRef] [PubMed]
- Panevska, A.; Skocaj, M.; Krizaj, I.; Macek, P.; Sepcic, K. Ceramide phosphoethanolamine, an enigmatic cellular membrane sphingolipid. Biochim. Biophys. Acta Biomembr. 2019, 1861, 1284–1292. [Google Scholar] [CrossRef] [PubMed]
- Moskot, M.; Bochenska, K.; Jakobkiewicz-Banecka, J.; Banecki, B.; Gabig-Ciminska, M. Abnormal Sphingolipid World in Inflammation Specific for Lysosomal Storage Diseases and Skin Disorders. Int. J. Mol. Sci. 2018, 19, 247. [Google Scholar] [CrossRef] [PubMed]
- Fahy, E.; Subramaniam, S.; Murphy, R.C.; Nishijima, M.; Raetz, C.R.; Shimizu, T.; Spener, F.; van Meer, G.; Wakelam, M.J.; Dennis, E.A. Update of the LIPID MAPS comprehensive classification system for lipids. J. Lipid Res. 2009, 50, S9–S14. [Google Scholar] [CrossRef] [PubMed]
- Holleran, W.M.; Ginns, E.I.; Menon, G.K.; Grundmann, J.U.; Fartasch, M.; McKinney, C.E.; Elias, P.M.; Sidransky, E. Consequences of beta-glucocerebrosidase deficiency in epidermis. Ultrastructure and permeability barrier alterations in Gaucher disease. J. Clin. Investig. 1994, 93, 1756–1764. [Google Scholar] [CrossRef] [PubMed]
- Nyholm, P.G.; Pascher, I.; Sundell, S. The effect of hydrogen bonds on the conformation of glycosphingolipids. Methylated and unmethylated cerebroside studied by X-ray single crystal analysis and model calculations. Chem. Phys. Lipids 1990, 52, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Mamode Cassim, A.; Grison, M.; Ito, Y.; Simon-Plas, F.; Mongrand, S.; Boutte, Y. Sphingolipids in plants: A guidebook on their function in membrane architecture, cellular processes, and environmental or developmental responses. FEBS Lett. 2020, 594, 3719–3738. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z. Ganglioside GM1 and the Central Nervous System. Int. J. Mol. Sci. 2023, 24, 9558. [Google Scholar] [CrossRef] [PubMed]
- Lopez, P.H.; Schnaar, R.L. Gangliosides in cell recognition and membrane protein regulation. Curr. Opin. Struct. Biol. 2009, 19, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Schnaar, R.L. Gangliosides as Siglec ligands. Glycoconj. J. 2023, 40, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Rusnati, M.; Urbinati, C.; Tanghetti, E.; Dell’Era, P.; Lortat-Jacob, H.; Presta, M. Cell membrane GM1 ganglioside is a functional coreceptor for fibroblast growth factor 2. Proc. Natl. Acad. Sci. USA 2002, 99, 4367–4372. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Harrison, S.H.; Schoeniger, J.S. Gangliosides as receptors for biological toxins: Development of sensitive fluoroimmunoassays using ganglioside-bearing liposomes. Anal. Chem. 2000, 72, 6019–6024. [Google Scholar] [CrossRef] [PubMed]
- Yuki, N. Guillain-Barre syndrome and anti-ganglioside antibodies: A clinician-scientist’s journey. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2012, 88, 299–326. [Google Scholar] [CrossRef] [PubMed]
- Toro, C.; Zainab, M.; Tifft, C.J. The GM2 gangliosidoses: Unlocking the mysteries of pathogenesis and treatment. Neurosci. Lett. 2021, 764, 136195. [Google Scholar] [CrossRef] [PubMed]
- Han, G.; Gupta, S.D.; Gable, K.; Niranjanakumari, S.; Moitra, P.; Eichler, F.; Brown, R.H., Jr.; Harmon, J.M.; Dunn, T.M. Identification of small subunits of mammalian serine palmitoyltransferase that confer distinct acyl-CoA substrate specificities. Proc. Natl. Acad. Sci. USA 2009, 106, 8186–8191. [Google Scholar] [CrossRef]
- Li, J.; Yin, J.; Rong, C.; Li, K.E.; Wu, J.X.; Huang, L.Q.; Zeng, H.Y.; Sahu, S.K.; Yao, N. Orosomucoid Proteins Interact with the Small Subunit of Serine Palmitoyltransferase and Contribute to Sphingolipid Homeostasis and Stress Responses in Arabidopsis. Plant Cell 2016, 28, 3038–3051. [Google Scholar] [CrossRef] [PubMed]
- Lone, M.A.; Hulsmeier, A.J.; Saied, E.M.; Karsai, G.; Arenz, C.; von Eckardstein, A.; Hornemann, T. Subunit composition of the mammalian serine-palmitoyltransferase defines the spectrum of straight and methyl-branched long-chain bases. Proc. Natl. Acad. Sci. USA 2020, 117, 15591–15598. [Google Scholar] [CrossRef] [PubMed]
- Hannun, Y.A.; Obeid, L.M. Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 175–191. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, Y.; Kihara, A.; Igarashi, Y. Mammalian Lass6 and its related family members regulate synthesis of specific ceramides. Biochem. J. 2005, 390, 263–271. [Google Scholar] [CrossRef]
- Chung, L.H.; Liu, D.; Liu, X.T.; Qi, Y. Ceramide Transfer Protein (CERT): An Overlooked Molecular Player in Cancer. Int. J. Mol. Sci. 2021, 22, 13184. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, R.W.; Canals, D.; Hannun, Y.A. Roles and regulation of secretory and lysosomal acid sphingomyelinase. Cell Signal. 2009, 21, 836–846. [Google Scholar] [CrossRef]
- Ni, X.; Morales, C.R. The lysosomal trafficking of acid sphingomyelinase is mediated by sortilin and mannose 6-phosphate receptor. Traffic 2006, 7, 889–902. [Google Scholar] [CrossRef]
- Gault, C.R.; Obeid, L.M.; Hannun, Y.A. An overview of sphingolipid metabolism: From synthesis to breakdown. Adv. Exp. Med. Biol. 2010, 688, 1–23. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.S. Molecular genetics of GM1 beta-galactosidase. Clin. Genet. 1975, 8, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Zschoche, A.; Furst, W.; Schwarzmann, G.; Sanhoff, K. Hydrolysis of lactosylceramide by human galactosylceramidase and GM1-beta-galactosidase in a detergent-free system and its stimulation by sphingolipid activator proteins, sap-B and sap-C. Activator proteins stimulate lactosylceramide hydrolysis. Eur. J. Biochem. 1994, 222, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Momoi, T.; Ben-Yoseph, Y.; Nadler, H.L. Substrate-specificities of acid and alkaline ceramidases in fibroblasts from patients with Farber disease and controls. Biochem. J. 1982, 205, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Crivelli, S.M.; Giovagnoni, C.; Zhu, Z.; Tripathi, P.; Elsherbini, A.; Quadri, Z.; Pu, J.; Zhang, L.; Ferko, B.; Berkes, D.; et al. Function of ceramide transfer protein for biogenesis and sphingolipid composition of extracellular vesicles. J. Extracell. Vesicles 2022, 11, e12233. [Google Scholar] [CrossRef]
- Boath, A.; Graf, C.; Lidome, E.; Ullrich, T.; Nussbaumer, P.; Bornancin, F. Regulation and traffic of ceramide 1-phosphate produced by ceramide kinase: Comparative analysis to glucosylceramide and sphingomyelin. J. Biol. Chem. 2008, 283, 8517–8526. [Google Scholar] [CrossRef] [PubMed]
- Mitrofanova, A.; Mallela, S.K.; Ducasa, G.M.; Yoo, T.H.; Rosenfeld-Gur, E.; Zelnik, I.D.; Molina, J.; Varona Santos, J.; Ge, M.; Sloan, A.; et al. SMPDL3b modulates insulin receptor signaling in diabetic kidney disease. Nat. Commun. 2019, 10, 2692. [Google Scholar] [CrossRef] [PubMed]
- Pitson, S.M.; Moretti, P.A.; Zebol, J.R.; Lynn, H.E.; Xia, P.; Vadas, M.A.; Wattenberg, B.W. Activation of sphingosine kinase 1 by ERK1/2-mediated phosphorylation. EMBO J. 2003, 22, 5491–5500. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, M.; Shegogue, D.; Pei, H.; Bu, S.; Bielawska, A.; Bielawski, J.; Pettus, B.; Hannun, Y.A.; Obeid, L.; Trojanowska, M. Sphingosine kinase 1 (SPHK1) is induced by transforming growth factor-beta and mediates TIMP-1 up-regulation. J. Biol. Chem. 2004, 279, 53994–54001. [Google Scholar] [CrossRef] [PubMed]
- Olivera, A.; Spiegel, S. Sphingosine-1-phosphate as second messenger in cell proliferation induced by PDGF and FCS mitogens. Nature 1993, 365, 557–560. [Google Scholar] [CrossRef] [PubMed]
- Edsall, L.C.; Pirianov, G.G.; Spiegel, S. Involvement of sphingosine 1-phosphate in nerve growth factor-mediated neuronal survival and differentiation. J. Neurosci. 1997, 17, 6952–6960. [Google Scholar] [CrossRef] [PubMed]
- El-Shewy, H.M.; Johnson, K.R.; Lee, M.H.; Jaffa, A.A.; Obeid, L.M.; Luttrell, L.M. Insulin-like growth factors mediate heterotrimeric G protein-dependent ERK1/2 activation by transactivating sphingosine 1-phosphate receptors. J. Biol. Chem. 2006, 281, 31399–31407. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, Y.; Li, P.Y.; Wada, A.; Mitsutake, S.; Igarashi, Y. Identification of functional nuclear export sequences in human sphingosine kinase 1. Biochem. Biophys. Res. Commun. 2003, 311, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Ancellin, N.; Colmont, C.; Su, J.; Li, Q.; Mittereder, N.; Chae, S.S.; Stefansson, S.; Liau, G.; Hla, T. Extracellular export of sphingosine kinase-1 enzyme. Sphingosine 1-phosphate generation and the induction of angiogenic vascular maturation. J. Biol. Chem. 2002, 277, 6667–6675. [Google Scholar] [CrossRef]
- Maceyka, M.; Sankala, H.; Hait, N.C.; Le Stunff, H.; Liu, H.; Toman, R.; Collier, C.; Zhang, M.; Satin, L.S.; Merrill, A.H., Jr.; et al. SphK1 and SphK2, sphingosine kinase isoenzymes with opposing functions in sphingolipid metabolism. J. Biol. Chem. 2005, 280, 37118–37129. [Google Scholar] [CrossRef] [PubMed]
- Le Stunff, H.; Giussani, P.; Maceyka, M.; Lepine, S.; Milstien, S.; Spiegel, S. Recycling of sphingosine is regulated by the concerted actions of sphingosine-1-phosphate phosphohydrolase 1 and sphingosine kinase 2. J. Biol. Chem. 2007, 282, 34372–34380. [Google Scholar] [CrossRef] [PubMed]
- Donoviel, M.S.; Hait, N.C.; Ramachandran, S.; Maceyka, M.; Takabe, K.; Milstien, S.; Oravecz, T.; Spiegel, S. Spinster 2, a sphingosine-1-phosphate transporter, plays a critical role in inflammatory and autoimmune diseases. FASEB J. 2015, 29, 5018–5028. [Google Scholar] [CrossRef]
- Wang, P.; Yuan, Y.; Lin, W.; Zhong, H.; Xu, K.; Qi, X. Roles of sphingosine-1-phosphate signaling in cancer. Cancer Cell Int. 2019, 19, 295. [Google Scholar] [CrossRef] [PubMed]
- Mendelson, K.; Evans, T.; Hla, T. Sphingosine 1-phosphate signalling. Development 2014, 141, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Serra, M.; Saba, J.D. Sphingosine 1-phosphate lyase, a key regulator of sphingosine 1-phosphate signaling and function. Adv. Enzyme Regul. 2010, 50, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Hanada, K. Intracellular trafficking of ceramide by ceramide transfer protein. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2010, 86, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Huitema, K.; van den Dikkenberg, J.; Brouwers, J.F.; Holthuis, J.C. Identification of a family of animal sphingomyelin synthases. EMBO J. 2004, 23, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Chiang, Y.P.; Li, Z.; Chen, Y.; Cao, Y.; Jiang, X.C. Sphingomyelin synthase related protein is a mammalian phosphatidylethanolamine phospholipase C. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2021, 1866, 159017. [Google Scholar] [CrossRef]
- Sprong, H.; Kruithof, B.; Leijendekker, R.; Slot, J.W.; van Meer, G.; van der Sluijs, P. UDP-galactose:ceramide galactosyltransferase is a class I integral membrane protein of the endoplasmic reticulum. J. Biol. Chem. 1998, 273, 25880–25888. [Google Scholar] [CrossRef] [PubMed]
- Uemura, S.; Go, S.; Shishido, F.; Inokuchi, J. Expression machinery of GM4: The excess amounts of GM3/GM4S synthase (ST3GAL5) are necessary for GM4 synthesis in mammalian cells. Glycoconj. J. 2014, 31, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Allende, M.L.; Proia, R.L. Simplifying complexity: Genetically resculpting glycosphingolipid synthesis pathways in mice to reveal function. Glycoconj. J. 2014, 31, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.B.; Hou, J.; Bandaru, V.V.R.; Pezhouh, M.K.; Syed Rifat Mannan, A.A.; Sharma, R. Lactosylceramide synthase beta-1,4-GalT-V: A novel target for the diagnosis and therapy of human colorectal cancer. Biochem. Biophys. Res. Commun. 2019, 508, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Fornoni, A.; Merscher, S.; Kopp, J.B. Lipid biology of the podocyte--new perspectives offer new opportunities. Nat. Rev. Nephrol. 2014, 10, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Merscher, S.; Fornoni, A. Podocyte pathology and nephropathy—Sphingolipids in glomerular diseases. Front. Endocrinol. 2014, 5, 127. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Cui, S.; Hou, Y.; Yi, F. The Updates of Podocyte Lipid Metabolism in Proteinuric Kidney Disease. Kidney Dis. 2021, 7, 438–451. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Sison, K.; Li, C.; Tian, R.; Wnuk, M.; Sung, H.K.; Jeansson, M.; Zhang, C.; Tucholska, M.; Jones, N.; et al. Soluble FLT1 binds lipid microdomains in podocytes to control cell morphology and glomerular barrier function. Cell 2012, 151, 384–399. [Google Scholar] [CrossRef] [PubMed]
- Aflaki, E.; Westbroek, W.; Sidransky, E. The Complicated Relationship between Gaucher Disease and Parkinsonism: Insights from a Rare Disease. Neuron 2017, 93, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Chander, P.N.; Nurse, H.M.; Pirani, C.L. Renal involvement in adult Gaucher’s disease after splenectomy. Arch. Pathol. Lab. Med. 1979, 103, 440–445. [Google Scholar] [PubMed]
- Siegal, A.; Gutman, A.; Shapiro, M.S.; Griffel, B. Renal involvement in Gaucher’s disease. Postgrad. Med. J. 1981, 57, 398–401. [Google Scholar] [CrossRef] [PubMed]
- Santoro, D.; Rosenbloom, B.E.; Cohen, A.H. Gaucher disease with nephrotic syndrome: Response to enzyme replacement therapy. Am. J. Kidney Dis. 2002, 40, E4. [Google Scholar] [CrossRef] [PubMed]
- Alroy, J.; Sabnis, S.; Kopp, J.B. Renal pathology in Fabry disease. J. Am. Soc. Nephrol. 2002, 13, S134–S138. [Google Scholar] [CrossRef] [PubMed]
- Najafian, B.; Svarstad, E.; Bostad, L.; Gubler, M.C.; Tondel, C.; Whitley, C.; Mauer, M. Progressive podocyte injury and globotriaosylceramide (GL-3) accumulation in young patients with Fabry disease. Kidney Int. 2011, 79, 663–670. [Google Scholar] [CrossRef] [PubMed]
- Najafian, B.; Silvestroni, A.; Sokolovskiy, A.; Tondel, C.; Svarstad, E.; Obrisca, B.; Ismail, G.; Holida, M.D.; Mauer, M. A novel unbiased method reveals progressive podocyte globotriaosylceramide accumulation and loss with age in females with Fabry disease. Kidney Int. 2022, 102, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Trimarchi, H.; Ortiz, A.; Sanchez-Nino, M.D. Lyso-Gb3 Increases alphavbeta3 Integrin Gene Expression in Cultured Human Podocytes in Fabry Nephropathy. J. Clin. Med. 2020, 9, 3659. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Park, S.; Lee, S.W.; Lee, J.H.; Lee, E.S.; Kim, M.; Kim, Y.; Kang, J.S.; Chung, C.H.; Moon, J.S.; et al. RIPK3 Contributes to Lyso-Gb3-Induced Podocyte Death. Cells 2021, 10, 245. [Google Scholar] [CrossRef] [PubMed]
- Quinta, R.; Rodrigues, D.; Assuncao, M.; Macedo, M.F.; Azevedo, O.; Cunha, D.; Oliveira, P.; Sa Miranda, M.C. Reduced glucosylceramide in the mouse model of Fabry disease: Correction by successful enzyme replacement therapy. Gene 2014, 536, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Thurberg, B.L.; Rennke, H.; Colvin, R.B.; Dikman, S.; Gordon, R.E.; Collins, A.B.; Desnick, R.J.; O’Callaghan, M. Globotriaosylceramide accumulation in the Fabry kidney is cleared from multiple cell types after enzyme replacement therapy. Kidney Int. 2002, 62, 1933–1946. [Google Scholar] [CrossRef]
- Liebau, M.C.; Braun, F.; Hopker, K.; Weitbrecht, C.; Bartels, V.; Muller, R.U.; Brodesser, S.; Saleem, M.A.; Benzing, T.; Schermer, B.; et al. Dysregulated autophagy contributes to podocyte damage in Fabry’s disease. PLoS ONE 2013, 8, e63506. [Google Scholar] [CrossRef]
- Braun, F.; Abed, A.; Sellung, D.; Rogg, M.; Woidy, M.; Eikrem, O.; Wanner, N.; Gambardella, J.; Laufer, S.D.; Haas, F.; et al. Accumulation of alpha-synuclein mediates podocyte injury in Fabry nephropathy. J. Clin. Investig. 2023, 133. [Google Scholar] [CrossRef]
- Yu, F.P.S.; Amintas, S.; Levade, T.; Medin, J.A. Acid ceramidase deficiency: Farber disease and SMA-PME. Orphanet J. Rare Dis. 2018, 13, 121. [Google Scholar] [CrossRef] [PubMed]
- Beckmann, N.; Becker, K.A.; Kadow, S.; Schumacher, F.; Kramer, M.; Kuhn, C.; Schulz-Schaeffer, W.J.; Edwards, M.J.; Kleuser, B.; Gulbins, E.; et al. Acid Sphingomyelinase Deficiency Ameliorates Farber Disease. Int. J. Mol. Sci. 2019, 20, 6253. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Kidd, J.; Kaspar, C.; Dempsey, S.; Bhat, O.M.; Camus, S.; Ritter, J.K.; Gehr, T.W.B.; Gulbins, E.; Li, P.L. Podocytopathy and Nephrotic Syndrome in Mice with Podocyte-Specific Deletion of the Asah1 Gene: Role of Ceramide Accumulation in Glomeruli. Am. J. Pathol. 2020, 190, 1211–1223. [Google Scholar] [CrossRef] [PubMed]
- Grafft, C.A.; Fervenza, F.C.; Semret, M.H.; Orloff, S.; Sethi, S. Renal involvement in Neimann-Pick Disease. NDT Plus 2009, 2, 448–451. [Google Scholar] [CrossRef] [PubMed]
- Jerbi, M.; Sayhi, M.; Gaied, H.; Hedri, H.; Aoudia, R.; Goucha, R.; Abdallah, T.B. Renal Thrombotique microangiopathy: An unusual renal involvement in Niemann-Pick disease type B. Clin. Case Rep. 2020, 8, 3316–3321. [Google Scholar] [CrossRef]
- Kurochkina, N.; Yardeni, T.; Huizing, M. Molecular modeling of the bifunctional enzyme UDP-GlcNAc 2-epimerase/ManNAc kinase and predictions of structural effects of mutations associated with HIBM and sialuria. Glycobiology 2010, 20, 322–337. [Google Scholar] [CrossRef] [PubMed]
- Galeano, B.; Klootwijk, R.; Manoli, I.; Sun, M.; Ciccone, C.; Darvish, D.; Starost, M.F.; Zerfas, P.M.; Hoffmann, V.J.; Hoogstraten-Miller, S.; et al. Mutation in the key enzyme of sialic acid biosynthesis causes severe glomerular proteinuria and is rescued by N-acetylmannosamine. J. Clin. Investig. 2007, 117, 1585–1594. [Google Scholar] [CrossRef]
- White, K.E.; Bilous, R.W.; Marshall, S.M.; El Nahas, M.; Remuzzi, G.; Piras, G.; De Cosmo, S.; Viberti, G. Podocyte number in normotensive type 1 diabetic patients with albuminuria. Diabetes 2002, 51, 3083–3089. [Google Scholar] [CrossRef] [PubMed]
- Kremer, G.J.; Atzpodien, W.; Schnellbacher, E. Plasma glycosphingolipids in diabetics and normals. Klin. Wochenschr. 1975, 53, 637–638. [Google Scholar] [CrossRef]
- Haus, J.M.; Kashyap, S.R.; Kasumov, T.; Zhang, R.; Kelly, K.R.; Defronzo, R.A.; Kirwan, J.P. Plasma ceramides are elevated in obese subjects with type 2 diabetes and correlate with the severity of insulin resistance. Diabetes 2009, 58, 337–343. [Google Scholar] [CrossRef]
- Blachnio-Zabielska, A.U.; Pulka, M.; Baranowski, M.; Nikolajuk, A.; Zabielski, P.; Gorska, M.; Gorski, J. Ceramide metabolism is affected by obesity and diabetes in human adipose tissue. J. Cell Physiol. 2012, 227, 550–557. [Google Scholar] [CrossRef]
- Hammad, S.M.; Lopes-Virella, M.F. Circulating Sphingolipids in Insulin Resistance, Diabetes and Associated Complications. Int. J. Mol. Sci. 2023, 24, 14015. [Google Scholar] [CrossRef] [PubMed]
- Gorska, M.; Dobrzyn, A.; Baranowski, M. Concentrations of sphingosine and sphinganine in plasma of patients with type 2 diabetes. Med. Sci. Monit. 2005, 11, CR35–CR38. [Google Scholar] [PubMed]
- Geoffroy, K.; Troncy, L.; Wiernsperger, N.; Lagarde, M.; El Bawab, S. Glomerular proliferation during early stages of diabetic nephropathy is associated with local increase of sphingosine-1-phosphate levels. FEBS Lett. 2005, 579, 1249–1254. [Google Scholar] [CrossRef] [PubMed]
- Kurano, M.; Tsukamoto, K.; Shimizu, T.; Hara, M.; Yatomi, Y. Apolipoprotein M/sphingosine 1-phosphate protects against diabetic nephropathy. Transl. Res. 2023, 258, 16–34. [Google Scholar] [CrossRef] [PubMed]
- Kwak, D.H.; Rho, Y.I.; Kwon, O.D.; Ahan, S.H.; Song, J.H.; Choo, Y.K.; Kim, S.J.; Choi, B.K.; Jung, K.Y. Decreases of ganglioside GM3 in streptozotocin-induced diabetic glomeruli of rats. Life Sci. 2003, 72, 1997–2006. [Google Scholar] [CrossRef] [PubMed]
- Naito, S.; Nakayama, K.; Kawashima, N. Enhanced Levels of Glycosphingolipid GM3 Delay the Progression of Diabetic Nephropathy. Int. J. Mol. Sci. 2023, 24, 11355. [Google Scholar] [CrossRef] [PubMed]
- Yoo, T.H.; Pedigo, C.E.; Guzman, J.; Correa-Medina, M.; Wei, C.; Villarreal, R.; Mitrofanova, A.; Leclercq, F.; Faul, C.; Li, J.; et al. Sphingomyelinase-like phosphodiesterase 3b expression levels determine podocyte injury phenotypes in glomerular disease. J. Am. Soc. Nephrol. 2015, 26, 133–147. [Google Scholar] [CrossRef]
- Mallela, S.K.; Mitrofanova, A.; Merscher, S.; Fornoni, A. Regulation of the amount of ceramide-1-phosphate synthesized in differentiated human podocytes. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 158517. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Kidd, J.M.; Zou, Y.; Wu, X.; Gehr, T.W.B.; Li, P.L.; Li, G. Regulation of NLRP3 Inflammasome Activation and Inflammatory Exosome Release in Podocytes by Acid Sphingomyelinase During Obesity. Inflammation 2023, 46, 2037–2054. [Google Scholar] [CrossRef]
- Holthofer, H.; Reivinen, J.; Solin, M.L.; Haltia, A.; Miettinen, A. Decrease of glomerular disialogangliosides in puromycin nephrosis of the rat. Am. J. Pathol. 1996, 149, 1009–1015. [Google Scholar] [PubMed]
- Pawluczyk, I.Z.; Ghaderi Najafabadi, M.; Patel, S.; Desai, P.; Vashi, D.; Saleem, M.A.; Topham, P.S. Sialic acid attenuates puromycin aminonucleoside-induced desialylation and oxidative stress in human podocytes. Exp. Cell Res. 2014, 320, 258–268. [Google Scholar] [CrossRef] [PubMed]
- Kitiyakara, C.; Eggers, P.; Kopp, J.B. Twenty-one-year trend in ESRD due to focal segmental glomerulosclerosis in the United States. Am. J. Kidney Dis. 2004, 44, 815–825. [Google Scholar] [CrossRef]
- Fornoni, A.; Sageshima, J.; Wei, C.; Merscher-Gomez, S.; Aguillon-Prada, R.; Jauregui, A.N.; Li, J.; Mattiazzi, A.; Ciancio, G.; Chen, L.; et al. Rituximab targets podocytes in recurrent focal segmental glomerulosclerosis. Sci. Transl. Med. 2011, 3, 85ra46. [Google Scholar] [CrossRef] [PubMed]
- Naito, S.; Kawashima, N.; Ishii, D.; Fujita, T.; Iwamura, M.; Takeuchi, Y. Decreased GM3 correlates with proteinuria in minimal change nephrotic syndrome and focal segmental glomerulosclerosis. Clin. Exp. Nephrol. 2022, 26, 1078–1085. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, N.; Naito, S.; Nagane, M.; Yamashita, T.; Nakayama, K.I. Progression of albuminuria and podocyte injury in focal segmental glomerulosclerosis inhibited by enhanced glycosphingolipid GM3 via valproic acid. Sci. Rep. 2023, 13, 22487. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, N.; Naito, S.; Hanamatsu, H.; Nagane, M.; Takeuchi, Y.; Furukawa, J.I.; Iwasaki, N.; Yamashita, T.; Nakayama, K.I. Glycosphingolipid GM3 prevents albuminuria and podocytopathy induced by anti-nephrin antibody. Sci. Rep. 2022, 12, 16058. [Google Scholar] [CrossRef] [PubMed]
- Drexler, Y.; Molina, J.; Elfassy, T.; Ma, R.; Christoffersen, C.; Kurano, M.; Yatomi, Y.; Mariani, L.H.; Contreras, G.; Merscher, S.; et al. Identification of Glomerular and Plasma Apolipoprotein M as Novel Biomarkers in Glomerular Disease. Kidney Int. Rep. 2023, 8, 884–897. [Google Scholar] [CrossRef] [PubMed]
- Lovric, S.; Goncalves, S.; Gee, H.Y.; Oskouian, B.; Srinivas, H.; Choi, W.I.; Shril, S.; Ashraf, S.; Tan, W.; Rao, J.; et al. Mutations in sphingosine-1-phosphate lyase cause nephrosis with ichthyosis and adrenal insufficiency. J. Clin. Investig. 2017, 127, 912–928. [Google Scholar] [CrossRef] [PubMed]
- Schumann, J.; Grevot, A.; Ledieu, D.; Wolf, A.; Schubart, A.; Piaia, A.; Sutter, E.; Cote, S.; Beerli, C.; Pognan, F.; et al. Reduced Activity of Sphingosine-1-Phosphate Lyase Induces Podocyte-related Glomerular Proteinuria, Skin Irritation, and Platelet Activation. Toxicol. Pathol. 2015, 43, 694–703. [Google Scholar] [CrossRef]
- Imeri, F.; Stepanovska Tanturovska, B.; Manaila, R.; Pavenstadt, H.; Pfeilschifter, J.; Huwiler, A. Loss of S1P Lyase Expression in Human Podocytes Causes a Reduction in Nephrin Expression That Involves PKCdelta Activation. Int. J. Mol. Sci. 2023, 24, 3267. [Google Scholar] [CrossRef] [PubMed]
- Kopp, J.B.; Smith, M.W.; Nelson, G.W.; Johnson, R.C.; Freedman, B.I.; Bowden, D.W.; Oleksyk, T.; McKenzie, L.M.; Kajiyama, H.; Ahuja, T.S.; et al. MYH9 is a major-effect risk gene for focal segmental glomerulosclerosis. Nat. Genet. 2008, 40, 1175–1184. [Google Scholar] [CrossRef] [PubMed]
- Parsa, A.; Kao, W.H.; Xie, D.; Astor, B.C.; Li, M.; Hsu, C.Y.; Feldman, H.I.; Parekh, R.S.; Kusek, J.W.; Greene, T.H.; et al. APOL1 risk variants, race, and progression of chronic kidney disease. N. Engl. J. Med. 2013, 369, 2183–2196. [Google Scholar] [CrossRef] [PubMed]
- Kao, W.H.; Klag, M.J.; Meoni, L.A.; Reich, D.; Berthier-Schaad, Y.; Li, M.; Coresh, J.; Patterson, N.; Tandon, A.; Powe, N.R.; et al. MYH9 is associated with nondiabetic end-stage renal disease in African Americans. Nat. Genet. 2008, 40, 1185–1192. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Divers, J.; Freedman, B.I. Mechanisms of Injury in APOL1-associated Kidney Disease. Transplantation 2019, 103, 487–492. [Google Scholar] [CrossRef] [PubMed]
- Wakashin, H.; Heymann, J.; Roshanravan, H.; Daneshpajouhnejad, P.; Rosenberg, A.; Shin, M.K.; Hoek, M.; Kopp, J.B. APOL1 renal risk variants exacerbate podocyte injury by increasing inflammatory stress. BMC Nephrol. 2020, 21, 371. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.; Kumar, V.; Lan, X.; Shoshtari, S.S.M.; Eng, J.M.; Zhou, X.; Wang, F.; Wang, H.; Skorecki, K.; Xing, G.; et al. APOL1 risk variants cause podocytes injury through enhancing endoplasmic reticulum stress. Biosci. Rep. 2018, 38, BSR20171713. [Google Scholar] [CrossRef] [PubMed]
- Valsecchi, M.; Cazzetta, V.; Oriolo, F.; Lan, X.; Piazza, R.; Saleem, M.A.; Singhal, P.C.; Mavilio, D.; Mikulak, J.; Aureli, M. APOL1 polymorphism modulates sphingolipid profile of human podocytes. Glycoconj. J. 2020, 37, 729–744. [Google Scholar] [CrossRef] [PubMed]
- Rawat, S.S.; Johnson, B.T.; Puri, A. Sphingolipids: Modulators of HIV-1 infection and pathogenesis. Biosci. Rep. 2005, 25, 329–343. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Bhattacharya, D.; Yuan, Y.; Wei, C.; Zhong, F.; Ding, F.; D’Agati, V.D.; Lee, K.; Friedman, S.L.; He, J.C. Chronic kidney disease in a murine model of non-alcoholic steatohepatitis (NASH). Kidney Int. 2024, 105, 540–561. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Mitrofanova, A.; Bielawski, J.; Yang, Y.; Marples, B.; Fornoni, A.; Zeidan, Y.H. Sphingomyelinase-like phosphodiesterase 3b mediates radiation-induced damage of renal podocytes. FASEB J. 2017, 31, 771–780. [Google Scholar] [CrossRef] [PubMed]
- Azzam, P.; Francis, M.; Youssef, T.; Mroueh, M.; Daher, A.A.; Eid, A.A.; Fornoni, A.; Marples, B.; Zeidan, Y.H. Crosstalk Between SMPDL3b and NADPH Oxidases Mediates Radiation-Induced Damage of Renal Podocytes. Front. Med. 2021, 8, 732528. [Google Scholar] [CrossRef] [PubMed]
- Francis, M.; Ahmad, A.; Bodgi, L.; Azzam, P.; Youssef, T.; Abou Daher, A.; Eid, A.A.; Fornoni, A.; Pollack, A.; Marples, B.; et al. SMPDL3b modulates radiation-induced DNA damage response in renal podocytes. FASEB J. 2022, 36, e22545. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Disease | Genetic Mutation | Sphingolipid Alterations |
|---|---|---|
| Gaucher Disease | GBA1 (β-glucosylcerebrosidase) | Glucosylceramide accumulation |
| Fabry Disease | GLA (α-galactosidase) | Globotriaosylceramide accumulation |
| Farber Disease | ASAH1 (Acid ceramidase) | Ceramide accumulation |
| Niemann–Pick | SMPD1 (Acid sphingomyelinase) | Sphingomyelin accumulation |
| Hereditary Inclusion Body Myopathy 2 | GLE (UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase) | Reduced sialyation |
| S1P Lyase Mutation | SGPL1 (S1P Lyase) | Accumulation of S1P |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tolerico, M.; Merscher, S.; Fornoni, A. Normal and Dysregulated Sphingolipid Metabolism: Contributions to Podocyte Injury and Beyond. Cells 2024, 13, 890. https://doi.org/10.3390/cells13110890
Tolerico M, Merscher S, Fornoni A. Normal and Dysregulated Sphingolipid Metabolism: Contributions to Podocyte Injury and Beyond. Cells. 2024; 13(11):890. https://doi.org/10.3390/cells13110890
Chicago/Turabian StyleTolerico, Matthew, Sandra Merscher, and Alessia Fornoni. 2024. "Normal and Dysregulated Sphingolipid Metabolism: Contributions to Podocyte Injury and Beyond" Cells 13, no. 11: 890. https://doi.org/10.3390/cells13110890
APA StyleTolerico, M., Merscher, S., & Fornoni, A. (2024). Normal and Dysregulated Sphingolipid Metabolism: Contributions to Podocyte Injury and Beyond. Cells, 13(11), 890. https://doi.org/10.3390/cells13110890