

Inhibition of PDIs Downregulates Core LINC Complex Proteins, Promoting the Invasiveness of MDA-MB-231 Breast Cancer Cells in Confined Spaces In Vitro
 , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. MCF10A and MDA-MB-231 Cell Culture
2.2. Immunofluorescence Staining and Microscopy Imaging
2.3. EdU Cell Proliferation Assay
2.4. SDS-PAGE and Western Blotting
2.5. 2D Scratch Wound Assay
2.6. Cell Polarisation Assay
2.7. Space Restrictive Cell Migration/Chemotaxis Assay
2.8. MTT Cytotoxicity Assay
2.9. SUN2 Bioimage Analysis
2.10. Statistical Analysis
3. Results
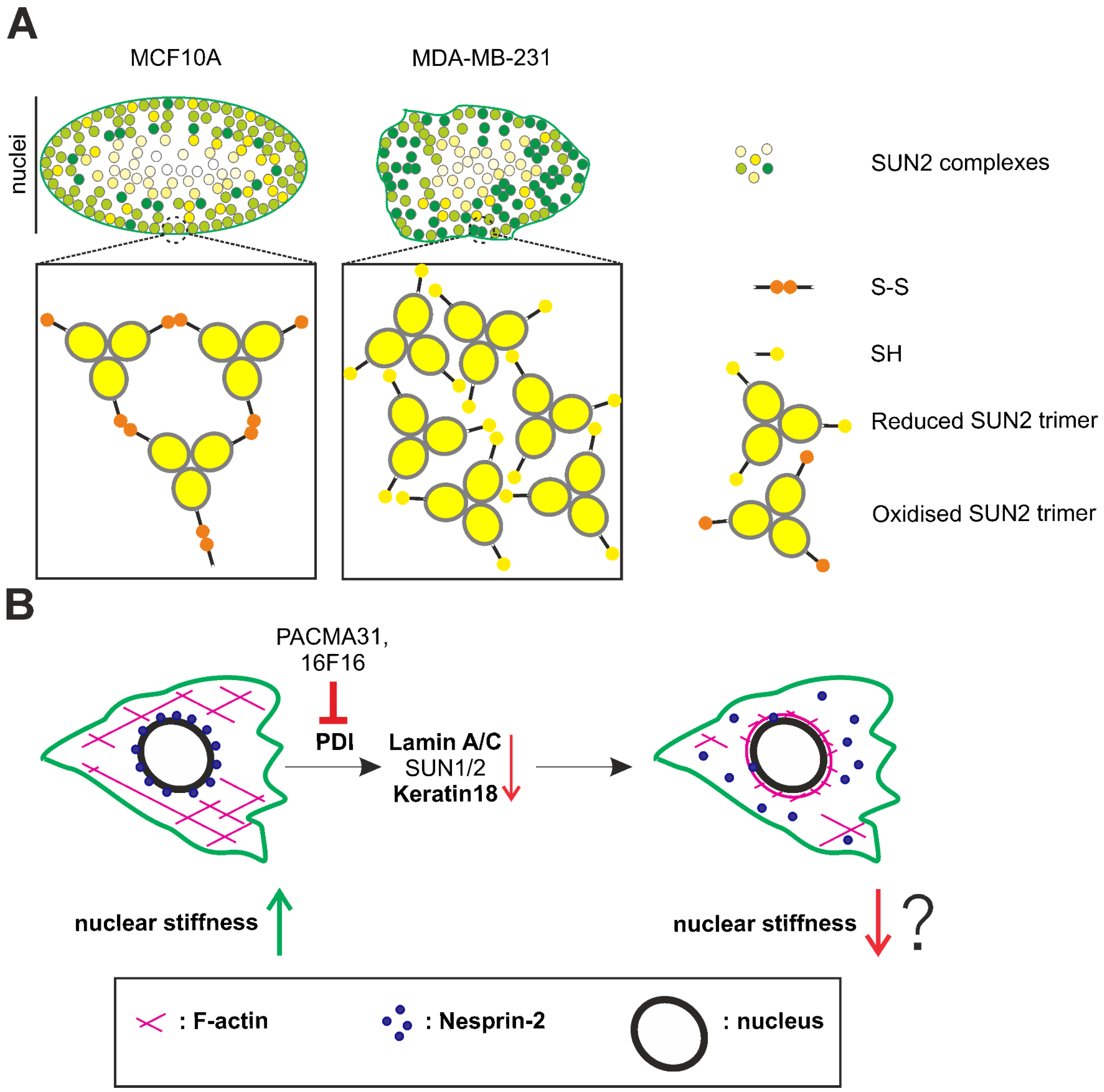
3.1. Disulphide Bond-Mediated Oligomerisation of SUN2 and LAMIN B1 Is Prominent in MCF10A Cells, but Not in MDA-MB-231 Cells
3.2. Super-Resolution Imaging Suggests the Presence of Higher-Order Molecular Structures for SUN2 at the Periphery of the MCF10A Nucleus, Which Are Absent in MDA-MB-231 Cells
3.3. PDI inhibitors, 16F16 and PACMA31, Upregulate HO-1 Protein Levels and Modulate the Expression of Core LINC Complex Proteins and Associated Nuclear Lamina Proteins in MDA-MB-231 Cells
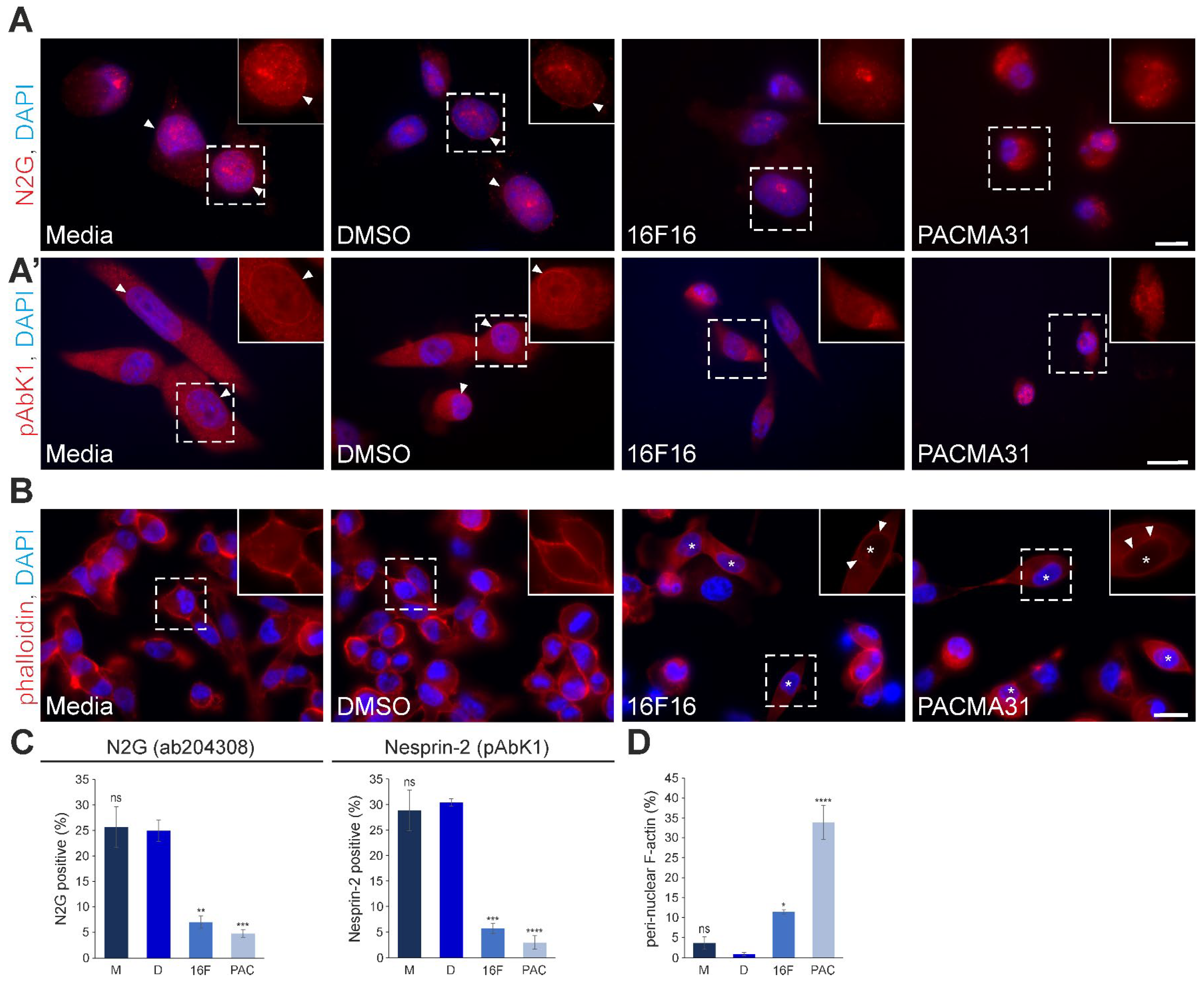
3.4. Pharmacological Inhibition of PDIs by 16F16 and PACMA31 Displaces Nesprin-2 from the Nuclear Envelope and Alters Perinuclear F-actin Architecture in MDA-MB-231 Cells
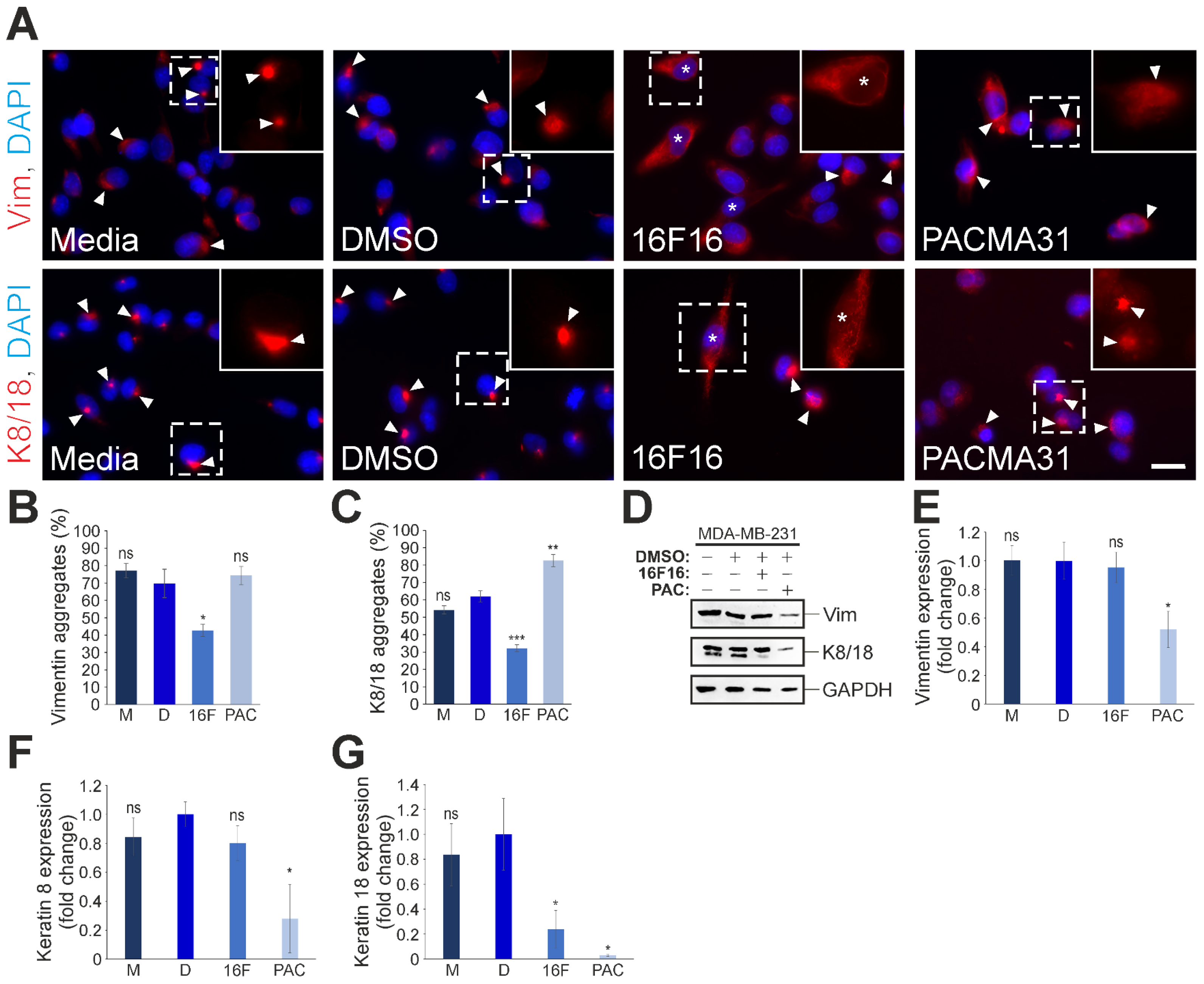
3.5. Differential Effects of 16F16 and PACMA31 Treatment on the Expression and Organisation of Cytoplasmic Intermediate Filaments in MDA-MB-231 Cells
3.6. Inhibition of PDIs by 16F16 and PACMA31 Reduces Cell Proliferation, Cell Motility, and Polarity in 2D Scratch Wound Assays, While Facilitating the Migration of MDA-MB-231 Cells in Space Restrictive in Vitro Environments
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Crisp, M.; Liu, Q.; Roux, K.; Rattner, J.B.; Shanahan, C.; Burke, B.; Stahl, P.D.; Hodzic, D. Coupling of the nucleus and cytoplasm: Role of the LINC complex. J. Cell Biol. 2006, 172, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Padmakumar, V.C.; Libotte, T.; Lu, W.; Zaim, H.; Abraham, S.; Noegel, A.A.; Gotzmann, J.; Foisner, R.; Karakesisoglou, I. The inner nuclear membrane protein Sun1 mediates the anchorage of Nesprin-2 to the nuclear envelope. J. Cell Sci. 2005, 118, 3419–3430. [Google Scholar] [CrossRef] [PubMed]
- Haque, F.; Lloyd, D.J.; Smallwood, D.T.; Dent, C.L.; Shanahan, C.M.; Fry, A.M.; Trembath, R.C.; Shackleton, S. SUN1 interacts with nuclear lamin A and cytoplasmic nesprins to provide a physical connection between the nuclear lamina and the cytoskeleton. Mol. Cell Biol. 2006, 26, 3738–3751. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Skepper, J.N.; Yang, F.; Davies, J.D.; Hegyi, L.; Roberts, R.G.; Weissberg, P.L.; Ellis, J.A.; Shanahan, C.M. Nesprins: A novel family of spectrin-repeat-containing proteins that localize to the nuclear membrane in multiple tissues. J. Cell Sci. 2001, 114, 4485–4498. [Google Scholar] [CrossRef] [PubMed]
- Rajgor, D.; Shanahan, C.M. Nesprins: From the nuclear envelope and beyond. Expert. Rev. Mol. Med. 2013, 15, e5. [Google Scholar] [CrossRef] [PubMed]
- Cartwright, S.; Karakesisoglou, I. Nesprins in health and disease. Semin. Cell Dev. Biol. 2014, 29, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Zhen, Y.Y.; Libotte, T.; Munck, M.; Noegel, A.A.; Korenbaum, E. NUANCE, a giant protein connecting the nucleus and actin cytoskeleton. J. Cell Sci. 2002, 115, 3207–3222. [Google Scholar] [CrossRef] [PubMed]
- Padmakumar, V.C.; Abraham, S.; Braune, S.; Noegel, A.A.; Tunggal, B.; Karakesisoglou, I.; Korenbaum, E. Enaptin, a giant actin-binding protein, is an element of the nuclear membrane and the actin cytoskeleton. Exp. Cell Res. 2004, 295, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Wilhelmsen, K.; Litjens, S.H.; Kuikman, I.; Tshimbalanga, N.; Janssen, H.; van den Bout, I.; Raymond, K.; Sonnenberg, A. Nesprin-3, a novel outer nuclear membrane protein, associates with the cytoskeletal linker protein plectin. J. Cell Biol. 2005, 171, 799–810. [Google Scholar] [CrossRef]
- Roux, K.J.; Crisp, M.L.; Liu, Q.; Kim, D.; Kozlov, S.; Stewart, C.L.; Burke, B. Nesprin 4 is an outer nuclear membrane protein that can induce kinesin-mediated cell polarization. Proc. Natl. Acad. Sci. USA 2009, 106, 2194–2199. [Google Scholar] [CrossRef]
- Libotte, T.; Zaim, H.; Abraham, S.; Padmakumar, V.C.; Schneider, M.; Lu, W.; Munck, M.; Hutchison, C.; Wehnert, M.; Fahrenkrog, B.; et al. Lamin A/C-dependent localization of Nesprin-2, a giant scaffolder at the nuclear envelope. Mol. Biol. Cell 2005, 16, 3411–3424. [Google Scholar] [CrossRef] [PubMed]
- Mislow, J.M.; Holaska, J.M.; Kim, M.S.; Lee, K.K.; Segura-Totten, M.; Wilson, K.L.; McNally, E.M. Nesprin-1alpha self-associates and binds directly to emerin and lamin A in vitro. FEBS Lett. 2002, 525, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Lei, K.; Zhang, X.; Ding, X.; Guo, X.; Chen, M.; Zhu, B.; Xu, T.; Zhuang, Y.; Xu, R.; Han, M. SUN1 and SUN2 play critical but partially redundant roles in anchoring nuclei in skeletal muscle cells in mice. Proc. Natl. Acad. Sci. USA 2009, 106, 10207–10212. [Google Scholar] [CrossRef] [PubMed]
- Hodzic, D.M.; Yeater, D.B.; Bengtsson, L.; Otto, H.; Stahl, P.D. Sun2 is a novel mammalian inner nuclear membrane protein*. J. Biol. Chem. 2004, 279, 25805–25812. [Google Scholar] [CrossRef] [PubMed]
- Göb, E.; Schmitt, J.; Benavente, R.; Alsheimer, M. Mammalian sperm head formation involves different polarization of two novel LINC complexes. PLoS ONE 2010, 5, e12072. [Google Scholar] [CrossRef] [PubMed]
- Frohnert, C.; Schweizer, S.; Hoyer-Fender, S. SPAG4L/SPAG4L-2 are testis-specific SUN domain proteins restricted to the apical nuclear envelope of round spermatids facing the acrosome. Mol. Hum. Reprod. 2011, 17, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Pasch, E.; Link, J.; Beck, C.; Scheuerle, S.; Alsheimer, M. The LINC complex component Sun4 plays a crucial role in spermhead formation and fertility. Biol. Open 2015, 4, 1792–1802. [Google Scholar] [CrossRef] [PubMed]
- Calvi, A.; Wong, A.S.W.; Wright, G.; Wong, E.S.M.; Loo, T.H.; Stewart, C.L.; Burke, B. SUN4 is essential for nuclear remodeling during mammalian spermiogenesis. Dev. Biol. 2015, 407, 321–330. [Google Scholar] [CrossRef]
- Thoma, H.; Grünewald, L.; Braune, S.; Pasch, E.; Alsheimer, M. SUN4 is a spermatid type II inner nuclear membrane protein that forms heteromeric assemblies with SUN3 and interacts with lamin B3. J. Cell Sci. 2023, 136, jcs260155. [Google Scholar] [CrossRef]
- Tzur, Y.B.; Wilson, K.L.; Gruenbaum, Y. SUN-domain proteins: ‘Velcro’ that links the nucleoskeleton to the cytoskeleton. Nat. Rev. Mol. Cell Biol. 2006, 7, 782–788. [Google Scholar] [CrossRef]
- Lu, W.; Gotzmann, J.; Sironi, L.; Jaeger, V.M.; Schneider, M.; Lüke, Y.; Uhlén, M.; Szigyarto, C.A.; Brachner, A.; Ellenberg, J.; et al. Sun1 forms immobile macromolecular assemblies at the nuclear envelope. Biochim. Biophys. Acta 2008, 1783, 2415–2426. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.; Rivero, F.; Euteneuer, U.; Mondal, S.; Mana-Capelli, S.; Larochelle, D.; Vogel, A.; Gassen, B.; Noegel, A.A. Dictyostelium Sun-1 connects the centrosome to chromatin and ensures genome stability. Traffic 2008, 9, 708–724. [Google Scholar] [CrossRef] [PubMed]
- Sosa, B.A.; Rothballer, A.; Kutay, U.; Schwartz, T.U. LINC complexes form by binding of three KASH peptides to domain interfaces of trimeric SUN proteins. Cell 2012, 149, 1035–1047. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Du, X.; Cai, Z.; Song, X.; Zhang, H.; Mizuno, T.; Suzuki, E.; Yee, M.R.; Berezov, A.; Murali, R.; et al. Structure of Sad1-UNC84 homology (SUN) domain defines features of molecular bridge in nuclear envelope. J. Biol. Chem. 2012, 287, 5317–5326. [Google Scholar] [CrossRef]
- Nie, S.; Ke, H.; Gao, F.; Ren, J.; Wang, M.; Huo, L.; Gong, W.; Feng, W. Coiled-Coil Domains of SUN Proteins as Intrinsic Dynamic Regulators. Structure 2016, 24, 80–91. [Google Scholar] [CrossRef]
- Gurusaran, M.; Davies, O.R. A molecular mechanism for LINC complex branching by structurally diverse SUN-KASH 6:6 assemblies. Elife 2021, 10, e60175. [Google Scholar] [CrossRef]
- Benham, A.M. The protein disulfide isomerase family: Key players in health and disease. Antioxid. Redox Signal 2012, 16, 781–789. [Google Scholar] [CrossRef]
- Powell, L.E.; Foster, P.A. Protein disulphide isomerase inhibition as a potential cancer therapeutic strategy. Cancer Med. 2021, 10, 2812–2825. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, D.M.; Söling, H.D. The protein disulphide-isomerase family: Unravelling a string of folds. Biochem. J. 1999, 339, 1–10. [Google Scholar] [CrossRef]
- Gromov, P.; Gromova, I.; Bunkenborg, J.; Cabezon, T.; Moreira, J.M.; Timmermans-Wielenga, V.; Roepstorff, P.; Rank, F.; Celis, J.E. Up-regulated proteins in the fluid bathing the tumour cell microenvironment as potential serological markers for early detection of cancer of the breast. Mol. Oncol. 2010, 4, 65–89. [Google Scholar] [CrossRef]
- Thongwatchara, P.; Promwikorn, W.; Srisomsap, C.; Chokchaichamnankit, D.; Boonyaphiphat, P.; Thongsuksai, P. Differential protein expression in primary breast cancer and matched axillary node metastasis. Oncol. Rep. 2011, 26, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.H.; Lim, C.A.; Cheong, Y.T.; Singh, M.; Gam, L.H. Comparison of protein expression profiles of different stages of lymph nodes metastasis in breast cancer. Int. J. Biol. Sci. 2012, 8, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Stojak, M.; Milczarek, M.; Kurpinska, A.; Suraj-Prazmowska, J.; Kaczara, P.; Wojnar-Lason, K.; Banach, J.; Stachowicz-Suhs, M.; Rossowska, J.; Kalviņš, I.; et al. Protein Disulphide Isomerase A1 Is Involved in the Regulation of Breast Cancer Cell Adhesion and Transmigration via Lung Microvascular Endothelial Cells. Cancers 2020, 12, 2850. [Google Scholar] [CrossRef] [PubMed]
- Wise, R.; Duhachek-Muggy, S.; Qi, Y.; Zolkiewski, M.; Zolkiewska, A. Protein disulfide isomerases in the endoplasmic reticulum promote anchorage-independent growth of breast cancer cells. Breast Cancer Res. Treat. 2016, 157, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Song, M.N.; Moon, P.G.; Lee, J.E.; Na, M.; Kang, W.; Chae, Y.S.; Park, J.Y.; Park, H.; Baek, M.C. Proteomic analysis of breast cancer tissues to identify biomarker candidates by gel-assisted digestion and label-free quantification methods using LC-MS/MS. Arch. Pharm. Res. 2012, 35, 1839–1847. [Google Scholar] [CrossRef] [PubMed]
- Ramos, F.S.; Serino, L.T.; Carvalho, C.M.; Lima, R.S.; Urban, C.A.; Cavalli, I.J.; Ribeiro, E.M. PDIA3 and PDIA6 gene expression as an aggressiveness marker in primary ductal breast cancer. Genet. Mol. Res. 2015, 14, 6960–6967. [Google Scholar] [CrossRef] [PubMed]
- Da Costa, G.G.; Gomig, T.H.; Kaviski, R.; Santos Sousa, K.; Kukolj, C.; De Lima, R.S.; De Andrade Urban, C.; Cavalli, I.J.; Ribeiro, E.M. Comparative Proteomics of Tumor and Paired Normal Breast Tissue Highlights Potential Biomarkers in Breast Cancer. Cancer Genom. Proteom. 2015, 12, 251–261. [Google Scholar]
- Germon, A.; Heesom, K.J.; Amoah, R.; Adams, J.C. Protein disulfide isomerase A3 activity promotes extracellular accumulation of proteins relevant to basal breast cancer outcomes in human MDA-MB-A231 breast cancer cells. Am. J. Physiol. Cell Physiol. 2023, 324, C113–C132. [Google Scholar] [CrossRef] [PubMed]
- Young, H.S.; McGowan, L.M.; Jepson, K.A.; Adams, J.C. Impairment of cell adhesion and migration by inhibition of protein disulphide isomerases in three breast cancer cell lines. Biosci. Rep. 2020, 40, BSR20193271. [Google Scholar] [CrossRef]
- Matsumoto, A.; Hieda, M.; Yokoyama, Y.; Nishioka, Y.; Yoshidome, K.; Tsujimoto, M.; Matsuura, N. Global loss of a nuclear lamina component, lamin A/C, and LINC complex components SUN1, SUN2, and nesprin-2 in breast cancer. Cancer Med. 2015, 4, 1547–1557. [Google Scholar] [CrossRef]
- Lv, X.B.; Liu, L.; Cheng, C.; Yu, B.; Xiong, L.; Hu, K.; Tang, J.; Zeng, L.; Sang, Y. SUN2 exerts tumor suppressor functions by suppressing the Warburg effect in lung cancer. Sci. Rep. 2015, 5, 17940. [Google Scholar] [CrossRef] [PubMed]
- Hoffstrom, B.G.; Kaplan, A.; Letso, R.; Schmid, R.S.; Turmel, G.J.; Lo, D.C.; Stockwell, B.R. Inhibitors of protein disulfide isomerase suppress apoptosis induced by misfolded proteins. Nat. Chem. Biol. 2010, 6, 900–906. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Butkevich, A.N.; Yamada, R.; Zhou, Y.; Debnath, B.; Duncan, R.; Zandi, E.; Petasis, N.A.; Neamati, N. Discovery of an orally active small-molecule irreversible inhibitor of protein disulfide isomerase for ovarian cancer treatment. Proc. Natl. Acad. Sci. USA 2012, 109, 16348–16353. [Google Scholar] [CrossRef] [PubMed]
- Bekendam, R.H.; Bendapudi, P.K.; Lin, L.; Nag, P.P.; Pu, J.; Kennedy, D.R.; Feldenzer, A.; Chiu, J.; Cook, K.M.; Furie, B.; et al. A substrate-driven allosteric switch that enhances PDI catalytic activity. Nat. Commun. 2016, 7, 12579. [Google Scholar] [CrossRef] [PubMed]
- Young, N.; Asif, M.; Jackson, M.; Fernández-Mayoralas, D.M.; de la Peña, M.J.; Calleja-Pérez, B.; Álvarez, S.; Hunter-Featherstone, E.; Noegel, A.A.; Höhne, W.; et al. Biallelic SYNE2 Missense Mutations Leading to Nesprin-2 Giant Hypo-Expression Are Associated with Intellectual Disability and Autism. Genes 2021, 12, 1294. [Google Scholar] [CrossRef] [PubMed]
- Wolfram Research, Laplacian Gaussian Filter, Wolfram Language function. Available online: https://reference.wolfram.com/language/ref/LaplacianGaussianFilter.html (accessed on 15 April 2024).
- Otsu, N. A Threshold Selection Method from Gray-Level Histograms. IEEE Trans. Syst. Man Cybern. 1979, 9, 62–66. [Google Scholar] [CrossRef]
- Soille, P. Morphological Image Analysis, 1st ed.; Springer: Heidelberg, Germany, 1999; pp. 49–88. [Google Scholar]
- Borgefors, G. Distance Transformations in Arbitrary Dimensions. Comput. Vis. Graph. Image Process 1984, 27, 321–345. [Google Scholar] [CrossRef]
- Felzenszwalb, P.F.; Huttenlocher, D.P. Distance transforms of sampled functions. Theory Comput. 2012, 8, 415–428. [Google Scholar] [CrossRef]
- Jahed, Z.; Fadavi, D.; Vu, U.T.; Asgari, E.; Luxton, G.W.G.; Mofrad, M.R.K. Molecular Insights into the Mechanisms of SUN1 Oligomerization in the Nuclear Envelope. Biophys. J. 2018, 114, 1190–1203. [Google Scholar] [CrossRef]
- Lherbette, M.; Dos Santos, Á.; Hari-Gupta, Y.; Fili, N.; Toseland, C.P.; Schaap, I.A.T. Atomic Force Microscopy micro-rheology reveals large structural inhomogeneities in single cell-nuclei. Sci. Rep. 2017, 7, 8116. [Google Scholar] [CrossRef]
- Arsenovic, P.T.; Ramachandran, I.; Bathula, K.; Zhu, R.; Narang, J.D.; Noll, N.A.; Lemmon, C.A.; Gundersen, G.G.; Conway, D.E. Nesprin-2G, a Component of the Nuclear LINC Complex, Is Subject to Myosin-Dependent Tension. Biophys. J. 2016, 110, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, L.K.; Ju, Z.; Ghagre, A.; Ehrlicher, A.J. Spatial distribution of lamin A/C determines nuclear stiffness and stress-mediated deformation. J. Cell Sci. 2021, 134, jcs248559. [Google Scholar] [CrossRef] [PubMed]
- Ihalainen, T.O.; Aires, L.; Herzog, F.A.; Schwartlander, R.; Moeller, J.; Vogel, V. Differential basal-to-apical accessibility of lamin A/C epitopes in the nuclear lamina regulated by changes in cytoskeletal tension. Nat. Mater. 2015, 14, 1252–1261. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Wirtz, D. Cytoskeletal tension induces the polarized architecture of the nucleus. Biomaterials 2015, 48, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Ruan, J.; Xu, C.; Bian, C.; Lam, R.; Wang, J.P.; Kania, J.; Min, J.; Zang, J. Crystal structures of the coil 2B fragment and the globular tail domain of human lamin B1. FEBS Lett. 2012, 586, 314–318. [Google Scholar] [CrossRef] [PubMed]
- Nishioka, Y.; Imaizumi, H.; Imada, J.; Katahira, J.; Matsuura, N.; Hieda, M. SUN1 splice variants, SUN1_888, SUN1_785, and predominant SUN1_916, variably function in directional cell migration. Nucleus 2016, 7, 572–584. [Google Scholar] [CrossRef] [PubMed]
- Haque, F.; Mazzeo, D.; Patel, J.T.; Smallwood, D.T.; Ellis, J.A.; Shanahan, C.M.; Shackleton, S. Mammalian SUN protein interaction networks at the inner nuclear membrane and their role in laminopathy disease processes. J. Biol. Chem. 2010, 285, 3487–3498. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Chiu, P.H.; Yip, K.Y.; Chan, S.Y. Subcellular localization of SUN2 is regulated by lamin A and Rab5. PLoS ONE 2011, 6, e20507. [Google Scholar] [CrossRef] [PubMed]
- Maremonti, M.I.; Panzetta, V.; Dannhauser, D.; Netti, P.A.; Causa, F. Wide-range viscoelastic compression forces in microfluidics to probe cell-dependent nuclear structural and mechanobiological responses. J. R. Soc. Interface 2022, 19, 20210880. [Google Scholar] [CrossRef]
- Sharma, R.; Hetzer, M.W. Disulfide bond in SUN2 regulates dynamic remodeling of LINC complexes at the nuclear envelope. Life Sci. Alliance 2023, 6, e202302031. [Google Scholar] [CrossRef]
- Jahed, Z.; Shams, H.; Mofrad, M.R. A Disulfide Bond Is Required for the Transmission of Forces through SUN-KASH Complexes. Biophys. J. 2015, 109, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Kutomi, G.; Kajiwara, T.; Kukita, K.; Kochin, V.; Kanaseki, T.; Tsukahara, T.; Hirohashi, Y.; Torigoe, T.; Okamoto, Y.; et al. Cancer-associated oxidoreductase ERO1-α drives the production of VEGF via oxidative protein folding and regulating the mRNA level. Br. J. Cancer 2016, 114, 1227–1234. [Google Scholar] [CrossRef] [PubMed]
- Kutomi, G.; Tamura, Y.; Tanaka, T.; Kajiwara, T.; Kukita, K.; Ohmura, T.; Shima, H.; Takamaru, T.; Satomi, F.; Suzuki, Y.; et al. Human endoplasmic reticulum oxidoreductin 1-α is a novel predictor for poor prognosis of breast cancer. Cancer Sci. 2013, 104, 1091–1096. [Google Scholar] [CrossRef] [PubMed]
- Jozkowicz, A.; Was, H.; Dulak, J. Heme oxygenase-1 in tumors: Is it a false friend? Antioxid. Redox Signal 2007, 9, 2099–2117. [Google Scholar] [CrossRef]
- Schuck, S.; Prinz, W.A.; Thorn, K.S.; Voss, C.; Walter, P. Membrane expansion alleviates endoplasmic reticulum stress independently of the unfolded protein response. J. Cell Biol. 2009, 187, 525–536. [Google Scholar] [CrossRef]
- Buchwalter, A.; Schulte, R.; Tsai, H.; Capitanio, J.; Hetzer, M. Selective clearance of the inner nuclear membrane protein emerin by vesicular transport during ER stress. Elife 2019, 8, e49796. [Google Scholar] [CrossRef]
- Kucińska, M.K.; Fedry, J.; Galli, C.; Morone, D.; Raimondi, A.; Soldà, T.; Förster, F.; Molinari, M. TMX4-driven LINC complex disassembly and asymmetric autophagy of the nuclear envelope upon acute ER stress. Nat. Commun. 2023, 14, 3497. [Google Scholar] [CrossRef] [PubMed]
- Lüke, Y.; Zaim, H.; Karakesisoglou, I.; Jaeger, V.M.; Sellin, L.; Lu, W.; Schneider, M.; Neumann, S.; Beijer, A.; Munck, M.; et al. Nesprin-2 Giant (NUANCE) maintains nuclear envelope architecture and composition in skin. J. Cell Sci. 2008, 121, 1887–1898. [Google Scholar] [CrossRef]
- Luxton, G.W.; Gomes, E.R.; Folker, E.S.; Worman, H.J.; Gundersen, G.G. TAN lines: A novel nuclear envelope structure involved in nuclear positioning. Nucleus 2011, 2, 173–181. [Google Scholar] [CrossRef]
- Popielarski, M.; Ponamarczuk, H.; Stasiak, M.; Watała, C.; Świątkowska, M. Modifications of disulfide bonds in breast cancer cell migration and invasiveness. Am. J. Cancer Res. 2019, 9, 1554–1582. [Google Scholar]
- Roncato, F.; Regev, O.; Feigelson, S.W.; Yadav, S.K.; Kaczmarczyk, L.; Levi, N.; Drago-Garcia, D.; Ovadia, S.; Kizner, M.; Addadi, Y.; et al. Reduced Lamin A/C Does Not Facilitate Cancer Cell Transendothelial Migration but Compromises Lung Metastasis. Cancers 2021, 13, 2383. [Google Scholar] [CrossRef] [PubMed]
- Bell, E.S.; Shah, P.; Zuela-Sopilniak, N.; Kim, D.; Varlet, A.A.; Morival, J.L.P.; McGregor, A.L.; Isermann, P.; Davidson, P.M.; Elacqua, J.J.; et al. Low lamin A levels enhance confined cell migration and metastatic capacity in breast cancer. Oncogene 2022, 41, 4211–4230. [Google Scholar] [CrossRef] [PubMed]
- Stewart-Hutchinson, P.J.; Hale, C.M.; Wirtz, D.; Hodzic, D. Structural requirements for the assembly of LINC complexes and their function in cellular mechanical stiffness. Exp. Cell Res. 2008, 314, 1892–1905. [Google Scholar] [CrossRef]
- Seltmann, K.; Fritsch, A.W.; Käs, J.A.; Magin, T.M. Keratins significantly contribute to cell stiffness and impact invasive behavior. Proc. Natl. Acad. Sci. USA 2013, 110, 18507–18512. [Google Scholar] [CrossRef]
- Fortier, A.M.; Asselin, E.; Cadrin, M. Keratin 8 and 18 loss in epithelial cancer cells increases collective cell migration and cisplatin sensitivity through claudin1 up-regulation. J. Biol. Chem. 2013, 288, 11555–11571. [Google Scholar] [CrossRef]
- Shi, R.; Wang, C.; Fu, N.; Liu, L.; Zhu, D.; Wei, Z.; Zhang, H.; Xing, J.; Wang, Y. Downregulation of cytokeratin 18 enhances BCRP-mediated multidrug resistance through induction of epithelial-mesenchymal transition and predicts poor prognosis in breast cancer. Oncol. Rep. 2019, 41, 3015–3026. [Google Scholar] [CrossRef] [PubMed]
- Pogoda, K.; Byfield, F.; Deptuła, P.; Cieśluk, M.; Suprewicz, Ł.; Skłodowski, K.; Shivers, J.L.; van Oosten, A.; Cruz, K.; Tarasovetc, E.; et al. Unique Role of Vimentin Networks in Compression Stiffening of Cells and Protection of Nuclei from Compressive Stress. Nano Lett. 2022, 22, 4725–4732. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Coulombe, P.A. A role for disulfide bonding in keratin intermediate filament organization and dynamics in skin keratinocytes. J. Cell Biol. 2015, 209, 59–72. [Google Scholar] [CrossRef]
- Rogers, K.R.; Herrmann, H.; Franke, W.W. Characterization of disulfide crosslink formation of human vimentin at the dimer, tetramer, and intermediate filament levels. J. Struct. Biol. 1996, 117, 55–69. [Google Scholar] [CrossRef]
- Pérez-Sala, D.; Oeste, C.L.; Martínez, A.E.; Carrasco, M.J.; Garzón, B.; Cañada, F.J. Vimentin filament organization and stress sensing depend on its single cysteine residue and zinc binding. Nat. Commun. 2015, 6, 7287. [Google Scholar] [CrossRef]
- Kaus-Drobek, M.; Mücke, N.; Szczepanowski, R.H.; Wedig, T.; Czarnocki-Cieciura, M.; Polakowska, M.; Herrmann, H.; Wysłouch-Cieszyńska, A.; Dadlez, M. Vimentin S-glutathionylation at Cys328 inhibits filament elongation and induces severing of mature filaments in vitro. FEBS J. 2020, 287, 5304–5322. [Google Scholar] [CrossRef] [PubMed]
- Santana-Codina, N.; Carretero, R.; Sanz-Pamplona, R.; Cabrera, T.; Guney, E.; Oliva, B.; Clezardin, P.; Olarte, O.E.; Loza-Alvarez, P.; Méndez-Lucas, A.; et al. A transcriptome-proteome integrated network identifies endoplasmic reticulum thiol oxidoreductase (ERp57) as a hub that mediates bone metastasis. Mol. Cell Proteomics 2013, 12, 2111–2125. [Google Scholar] [CrossRef] [PubMed]
- Sobierajska, K.; Skurzynski, S.; Stasiak, M.; Kryczka, J.; Cierniewski, C.S.; Swiatkowska, M. Protein disulfide isomerase directly interacts with β-actin Cys374 and regulates cytoskeleton reorganization. J. Biol. Chem. 2014, 289, 5758–5773. [Google Scholar] [CrossRef] [PubMed]
- DalleDonne, I.; Milzani, A.; Colombo, R. The tert-butyl hydroperoxide-induced oxidation of actin Cys-374 is coupled with structural changes in distant regions of the protein. Biochemistry 1999, 38, 12471–12480. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Young, N.; Gui, Z.; Mustafa, S.; Papa, K.; Jessop, E.; Ruddell, E.; Bevington, L.; Quinlan, R.A.; Benham, A.M.; Goldberg, M.W.; et al. Inhibition of PDIs Downregulates Core LINC Complex Proteins, Promoting the Invasiveness of MDA-MB-231 Breast Cancer Cells in Confined Spaces In Vitro. Cells 2024, 13, 906. https://doi.org/10.3390/cells13110906
Young N, Gui Z, Mustafa S, Papa K, Jessop E, Ruddell E, Bevington L, Quinlan RA, Benham AM, Goldberg MW, et al. Inhibition of PDIs Downregulates Core LINC Complex Proteins, Promoting the Invasiveness of MDA-MB-231 Breast Cancer Cells in Confined Spaces In Vitro. Cells. 2024; 13(11):906. https://doi.org/10.3390/cells13110906
Chicago/Turabian StyleYoung, Natalie, Zizhao Gui, Suleiman Mustafa, Kleopatra Papa, Emily Jessop, Elizabeth Ruddell, Laura Bevington, Roy A. Quinlan, Adam M. Benham, Martin W. Goldberg, and et al. 2024. "Inhibition of PDIs Downregulates Core LINC Complex Proteins, Promoting the Invasiveness of MDA-MB-231 Breast Cancer Cells in Confined Spaces In Vitro" Cells 13, no. 11: 906. https://doi.org/10.3390/cells13110906