
Fibroblasts in Pulmonary Hypertension: Roles and Molecular Mechanisms


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Fibroblasts in Health and Disease
1.2. Challenges in Studying Fibroblast Pathology in Diseases
1.3. Advances in Studying Fibroblast Pathology in Diseases
2. Fibroblasts’ Contribution to Pulmonary Vascular Remodeling in PH
2.1. Disordered Fibroblast Growth in PH
2.2. Proinflammatory Phenotype of Fibroblasts in PH
2.3. Fibroblast’s Contribution to Fibrotic Responses in PH
2.4. Role of PH Fibroblasts in Activating Other Cell Types in PH
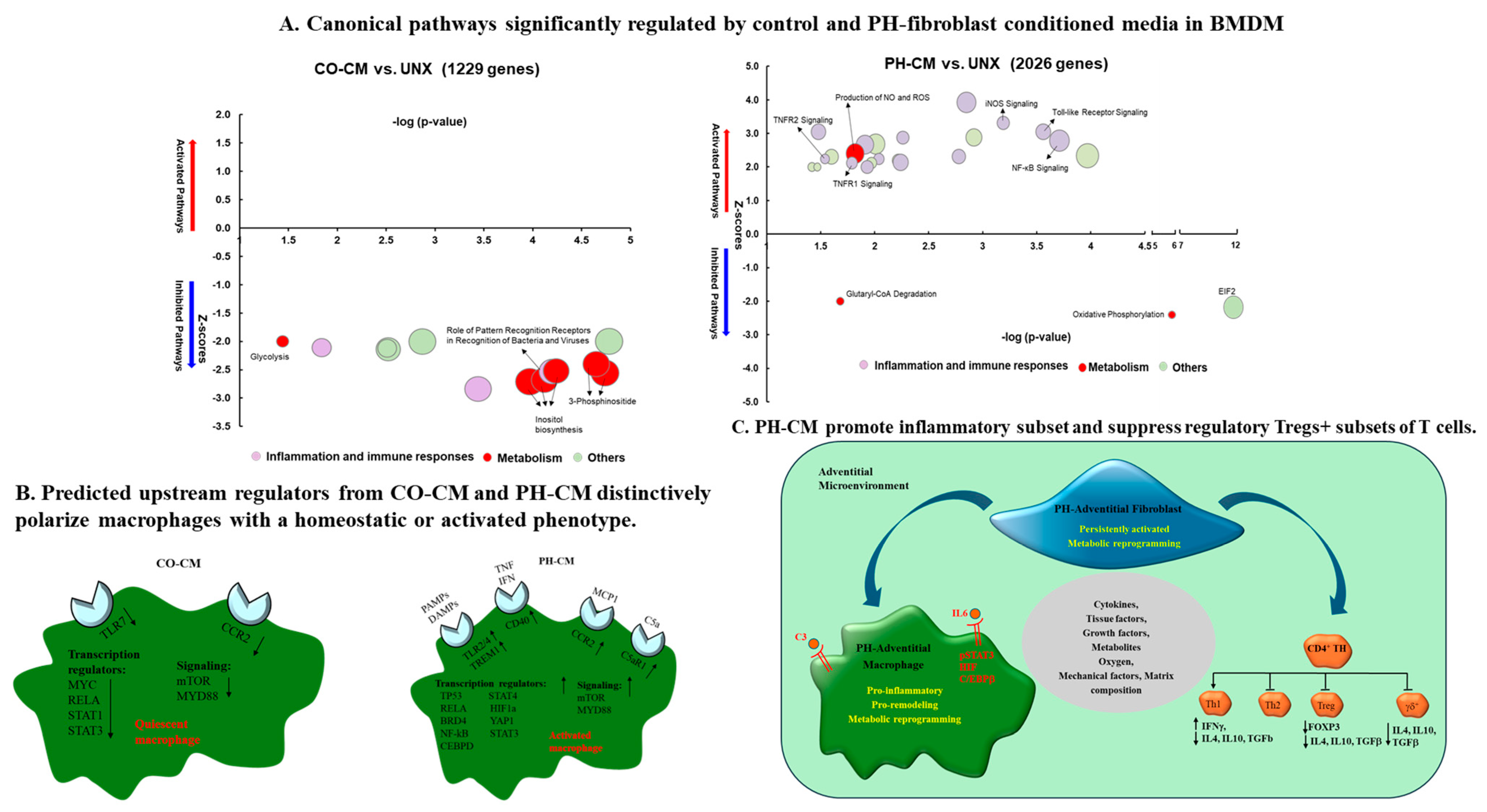
2.4.1. Fibroblasts’ Ability to Induce Macrophage Activation in PH
2.4.2. Fibroblasts Regulate T-Cell Function in PH
2.4.3. Fibroblasts Direct Smooth-Muscle-Cell-State Transition in PH
3. Mechanisms Underlying Fibroblast Abnormalities in PH
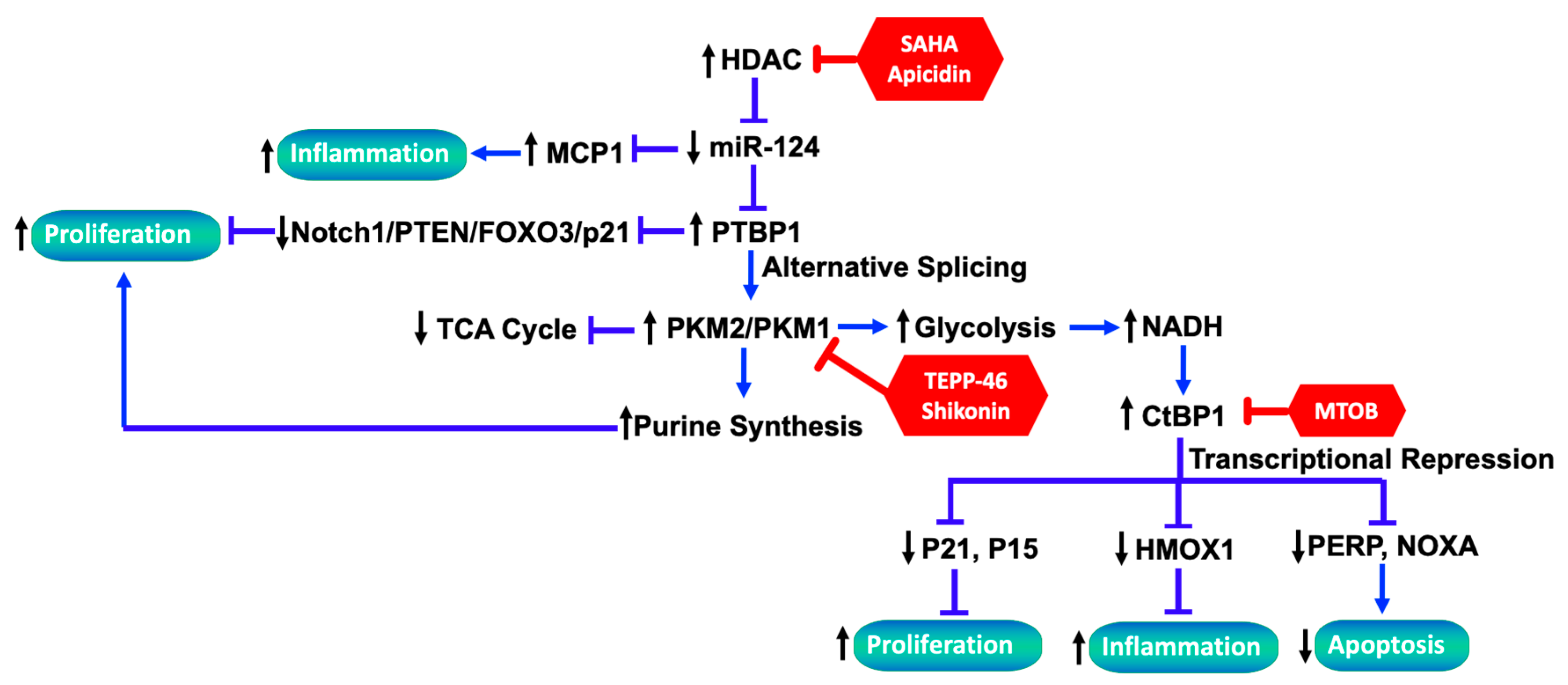
3.1. Role of Dysregulated miRNAs Underlying PH-Fib Abnormalities
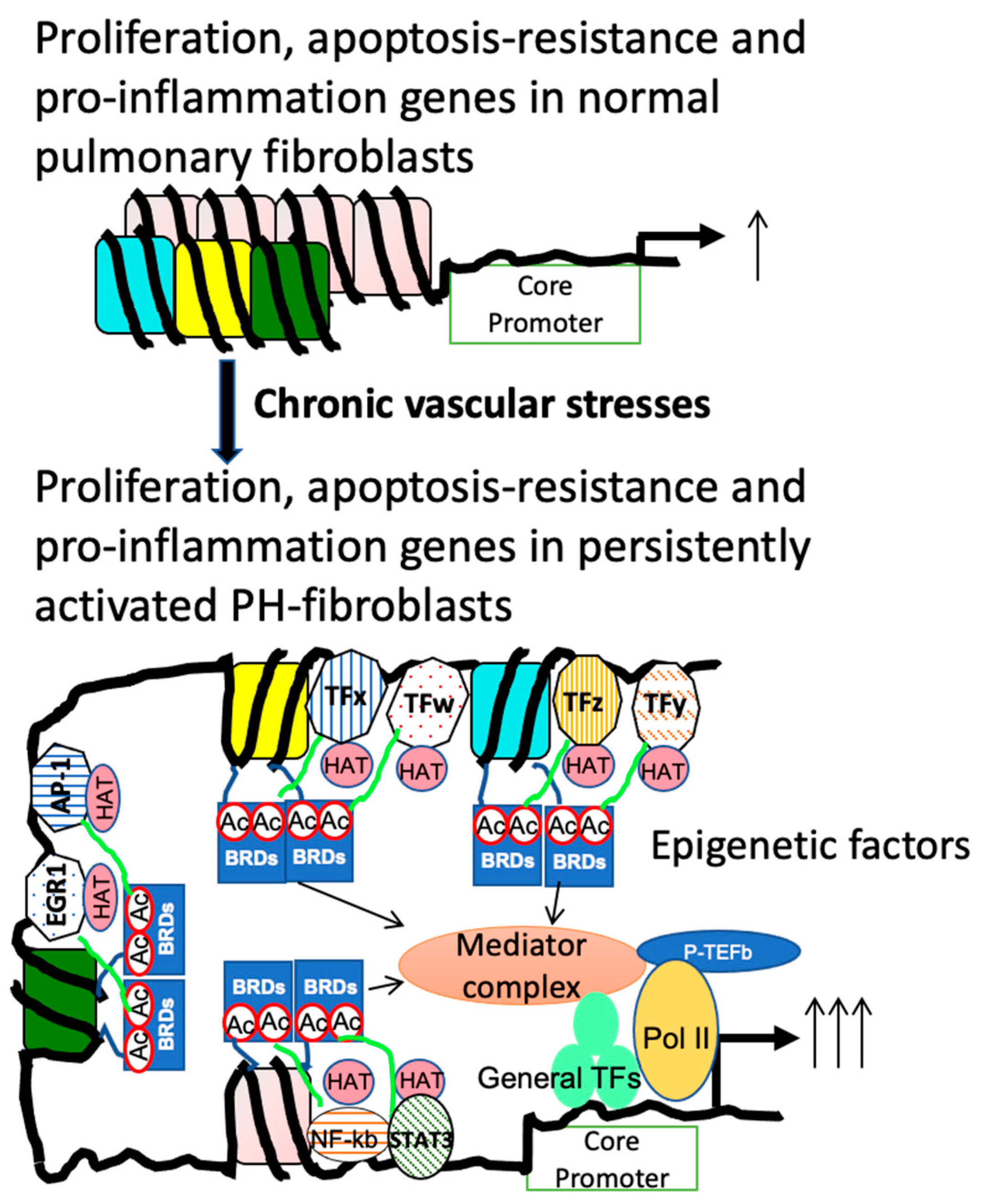
3.2. Role of Dysregulated HDAC Activity Underlying PH-Fib Abnormalities
3.3. The Link between Metabolic Reprogramming and Functionality of Fibroblasts in PH
3.3.1. Metabolic Reprograming in PH-Fibs
3.3.2. CtBP1 Senses Metabolic Changes and Links to Transcriptional Alterations
3.3.3. The Interplay between Altered Metabolites and Epigenetic Regulation
4. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Simonneau, G.; Gatzoulis, M.A.; Adatia, I.; Celermajer, D.; Denton, C.; Ghofrani, A.; Gomez Sanchez, M.A.; Krishna Kumar, R.; Landzberg, M.; Machado, R.F.; et al. Updated clinical classification of pulmonary hypertension. J. Am. Coll. Cardiol. 2013, 62, D34–D41. [Google Scholar] [CrossRef] [PubMed]
- Price, L.C.; Wort, S.J.; Perros, F.; Dorfmuller, P.; Huertas, A.; Montani, D.; Cohen-Kaminsky, S.; Humbert, M. Inflammation in pulmonary arterial hypertension. Chest 2012, 141, 210–221. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandro, A.; El Kasmi, K.C.; Plecita-Hlavata, L.; Jezek, P.; Li, M.; Zhang, H.; Gupte, S.A.; Stenmark, K.R. Hallmarks of Pulmonary Hypertension: Mesenchymal and Inflammatory Cell Metabolic Reprogramming. Antioxid. Redox Signal 2018, 28, 230–250. [Google Scholar] [CrossRef] [PubMed]
- Stenmark, K.R.; Meyrick, B.; Galie, N.; Mooi, W.J.; McMurtry, I.F. Animal models of pulmonary arterial hypertension: The hope for etiological discovery and pharmacological cure. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 297, L1013–L1032. [Google Scholar] [CrossRef] [PubMed]
- Das, M.; Burns, N.; Wilson, S.J.; Zawada, W.M.; Stenmark, K.R. Hypoxia exposure induces the emergence of fibroblasts lacking replication repressor signals of PKCzeta in the pulmonary artery adventitia. Cardiovasc. Res. 2008, 78, 440–448. [Google Scholar] [CrossRef] [PubMed]
- El Kasmi, K.C.; Pugliese, S.C.; Riddle, S.R.; Poth, J.M.; Anderson, A.L.; Frid, M.G.; Li, M.; Pullamsetti, S.S.; Savai, R.; Nagel, M.A.; et al. Adventitial fibroblasts induce a distinct proinflammatory/profibrotic macrophage phenotype in pulmonary hypertension. J. Immunol. 2014, 193, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Krick, S.; Hanze, J.; Eul, B.; Savai, R.; Seay, U.; Grimminger, F.; Lohmeyer, J.; Klepetko, W.; Seeger, W.; Rose, F. Hypoxia-driven proliferation of human pulmonary artery fibroblasts: Cross-talk between HIF-1alpha and an autocrine angiotensin system. FASEB J. 2005, 19, 857–859. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Riddle, S.R.; Frid, M.G.; El Kasmi, K.C.; McKinsey, T.A.; Sokol, R.J.; Strassheim, D.; Meyrick, B.; Yeager, M.E.; Flockton, A.R.; et al. Emergence of fibroblasts with a proinflammatory epigenetically altered phenotype in severe hypoxic pulmonary hypertension. J. Immunol. 2011, 187, 2711–2722. [Google Scholar] [CrossRef] [PubMed]
- Panzhinskiy, E.; Zawada, W.M.; Stenmark, K.R.; Das, M. Hypoxia induces unique proliferative response in adventitial fibroblasts by activating PDGFbeta receptor-JNK1 signalling. Cardiovasc. Res. 2012, 95, 356–365. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, H.; Li, M.; Frid, M.G.; Flockton, A.R.; McKeon, B.A.; Yeager, M.E.; Fini, M.A.; Morrell, N.W.; Pullamsetti, S.S.; et al. MicroRNA-124 controls the proliferative, migratory, and inflammatory phenotype of pulmonary vascular fibroblasts. Circ. Res. 2014, 114, 67–78. [Google Scholar] [CrossRef]
- Molenaar, J.C. From the library of the Netherlands Journal of Medicine. Rudolf Virchow: Die Cellularpathologie in ihrer Begrundung auf physiologische und pathologische Gewebelehre; 1858. Ned. Tijdschr. Geneeskd. 2003, 147, 2236–2244. [Google Scholar] [PubMed]
- Lendahl, U.; Muhl, L.; Betsholtz, C. Identification, discrimination and heterogeneity of fibroblasts. Nat. Commun. 2022, 13, 3409. [Google Scholar] [CrossRef] [PubMed]
- Bloom, W.; Fawcett, D. A Textbook of Histology; CRC Press: Boca Raton, FL, USA, 1998. [Google Scholar]
- Movat, H.Z.; Fernando, N.V. The fine structure of connective tissue. I. The fibroblast. Exp. Mol. Pathol. 1962, 1, 509–534. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O.; Naba, A. Overview of the matrisome—An inventory of extracellular matrix constituents and functions. Cold Spring Harb. Perspect. Biol. 2012, 4, a004903. [Google Scholar] [CrossRef] [PubMed]
- Karsdal, M. Biochemistry of Collagens, Laminins and Elastin: Structure, Function and Biomarkers; Leeming, D., Henriksen, K., Bay-Jensen, A.-C., Eds.; Academic Press: Cambridge, MA, USA, 2019; Volume 1. [Google Scholar]
- Lu, P.; Takai, K.; Weaver, V.M.; Werb, Z. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a005058. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.Y.; Chi, J.T.; Dudoit, S.; Bondre, C.; van de Rijn, M.; Botstein, D.; Brown, P.O. Diversity, topographic differentiation, and positional memory in human fibroblasts. Proc. Natl. Acad. Sci. USA 2002, 99, 12877–12882. [Google Scholar] [CrossRef] [PubMed]
- Schor, S.L.; Schor, A.M. Clonal heterogeneity in fibroblast phenotype: Implications for the control of epithelial-mesenchymal interactions. Bioessays 1987, 7, 200–204. [Google Scholar] [CrossRef] [PubMed]
- Gabbiani, G.; Ryan, G.B.; Majne, G. Presence of modified fibroblasts in granulation tissue and their possible role in wound contraction. Experientia 1971, 27, 549–550. [Google Scholar] [CrossRef] [PubMed]
- Pakshir, P.; Noskovicova, N.; Lodyga, M.; Son, D.O.; Schuster, R.; Goodwin, A.; Karvonen, H.; Hinz, B. The myofibroblast at a glance. J. Cell Sci. 2020, 133, 227900. [Google Scholar] [CrossRef]
- Kalluri, R. The biology and function of exosomes in cancer. J. Clin. Investig. 2016, 126, 1208–1215. [Google Scholar] [CrossRef]
- Pietras, K.; Ostman, A. Hallmarks of cancer: Interactions with the tumor stroma. Exp. Cell Res. 2010, 316, 1324–1331. [Google Scholar] [CrossRef] [PubMed]
- Costea, D.E.; Johannessen, A.C.; Vintermyr, O.K. Fibroblast control on epithelial differentiation is gradually lost during in vitro tumor progression. Differentiation 2005, 73, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Degirmenci, B.; Valenta, T.; Dimitrieva, S.; Hausmann, G.; Basler, K. GLI1-expressing mesenchymal cells form the essential Wnt-secreting niche for colon stem cells. Nature 2018, 558, 449–453. [Google Scholar] [CrossRef] [PubMed]
- El Ghalbzouri, A.; Lamme, E.; Ponec, M. Crucial role of fibroblasts in regulating epidermal morphogenesis. Cell Tissue Res. 2002, 310, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y. Cellular and molecular mechanisms of renal fibrosis. Nat. Rev. Nephrol. 2011, 7, 684–696. [Google Scholar] [CrossRef] [PubMed]
- Roulis, M.; Kaklamanos, A.; Schernthanner, M.; Bielecki, P.; Zhao, J.; Kaffe, E.; Frommelt, L.S.; Qu, R.; Knapp, M.S.; Henriques, A.; et al. Paracrine orchestration of intestinal tumorigenesis by a mesenchymal niche. Nature 2020, 580, 524–529. [Google Scholar] [CrossRef] [PubMed]
- Shoshkes-Carmel, M.; Wang, Y.J.; Wangensteen, K.J.; Toth, B.; Kondo, A.; Massasa, E.E.; Itzkovitz, S.; Kaestner, K.H. Subepithelial telocytes are an important source of Wnts that supports intestinal crypts. Nature 2018, 557, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Heruth, D.P.; Gibson, M.; Grigoryev, D.N.; Zhang, L.Q.; Ye, S.Q. RNA-seq analysis of synovial fibroblasts brings new insights into rheumatoid arthritis. Cell Biosci. 2012, 2, 43. [Google Scholar] [CrossRef] [PubMed]
- Lahiry, P.; Lee, L.J.; Frey, B.J.; Rupar, C.A.; Siu, V.M.; Blencowe, B.J.; Hegele, R.A. Transcriptional profiling of endocrine cerebro-osteodysplasia using microarray and next-generation sequencing. PLoS ONE 2011, 6, e25400. [Google Scholar] [CrossRef] [PubMed]
- Nota, B.; Ndika, J.D.; van de Kamp, J.M.; Kanhai, W.A.; van Dooren, S.J.; van de Wiel, M.A.; Pals, G.; Salomons, G.S. RNA sequencing of creatine transporter (SLC6A8) deficient fibroblasts reveals impairment of the extracellular matrix. Hum. Mutat. 2014, 35, 1128–1135. [Google Scholar] [CrossRef]
- Schaum, N.; Karkanias, J.; Neff, N.F.; May, A.P.; Quake, S.R.; Wyss-Coray, T.; Darmanis, S.; Batson, J.; Botvinnik, O.; Chen, M.B.; et al. Tabula Muris Consortium: Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 2018, 562, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Green, C.D.; Ma, Q.; Manske, G.L.; Shami, A.N.; Zheng, X.; Marini, S.; Moritz, L.; Sultan, C.; Gurczynski, S.J.; Moore, B.B.; et al. A Comprehensive Roadmap of Murine Spermatogenesis Defined by Single-Cell RNA-Seq. Dev. Cell 2018, 46, 651–667.e610. [Google Scholar] [CrossRef] [PubMed]
- Bartoschek, M.; Oskolkov, N.; Bocci, M.; Lovrot, J.; Larsson, C.; Sommarin, M.; Madsen, C.D.; Lindgren, D.; Pekar, G.; Karlsson, G.; et al. Spatially and functionally distinct subclasses of breast cancer-associated fibroblasts revealed by single cell RNA sequencing. Nat. Commun. 2018, 9, 5150. [Google Scholar] [CrossRef] [PubMed]
- Wei, K.; Korsunsky, I.; Marshall, J.L.; Gao, A.; Watts, G.F.M.; Major, T.; Croft, A.P.; Watts, J.; Blazar, P.E.; Lange, J.K.; et al. Notch signalling drives synovial fibroblast identity and arthritis pathology. Nature 2020, 582, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.Q. Fibroblasts in Rheumatoid Arthritis. N. Engl. J. Med. 2020, 383, 1679–1681. [Google Scholar] [CrossRef] [PubMed]
- Wei, K.; Nguyen, H.N.; Brenner, M.B. Fibroblast pathology in inflammatory diseases. J. Clin. Investig. 2021, 131, e149538. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Muhl, L.; Padberg, Y.; Dupont, L.; Peterson-Maduro, J.; Stehling, M.; Le Noble, F.; Colige, A.; Betsholtz, C.; Schulte-Merker, S.; et al. Specific fibroblast subpopulations and neuronal structures provide local sources of Vegfc-processing components during zebrafish lymphangiogenesis. Nat. Commun. 2020, 11, 2724. [Google Scholar] [CrossRef]
- Muhl, L.; Genove, G.; Leptidis, S.; Liu, J.; He, L.; Mocci, G.; Sun, Y.; Gustafsson, S.; Buyandelger, B.; Chivukula, I.V.; et al. Single-cell analysis uncovers fibroblast heterogeneity and criteria for fibroblast and mural cell identification and discrimination. Nat. Commun. 2020, 11, 3953. [Google Scholar] [CrossRef]
- Buechler, M.B.; Pradhan, R.N.; Krishnamurty, A.T.; Cox, C.; Calviello, A.K.; Wang, A.W.; Yang, Y.A.; Tam, L.; Caothien, R.; Roose-Girma, M.; et al. Cross-tissue organization of the fibroblast lineage. Nature 2021, 593, 575–579. [Google Scholar] [CrossRef]
- Zhang, X.; Shi, X.; Xie, F.; Liu, Y.; Wei, X.; Cai, Y.; Chao, J. Dissecting pulmonary fibroblasts heterogeneity in lung development, health and diseases. Heliyon 2023, 9, e19428. [Google Scholar] [CrossRef]
- Di Carlo, S.E.; Peduto, L. The perivascular origin of pathological fibroblasts. J. Clin. Investig. 2018, 128, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Plikus, M.V.; Wang, X.; Sinha, S.; Forte, E.; Thompson, S.M.; Herzog, E.L.; Driskell, R.R.; Rosenthal, N.; Biernaskie, J.; Horsley, V. Fibroblasts: Origins, definitions, and functions in health and disease. Cell 2021, 184, 3852–3872. [Google Scholar] [CrossRef] [PubMed]
- El Agha, E.; Kramann, R.; Schneider, R.K.; Li, X.; Seeger, W.; Humphreys, B.D.; Bellusci, S. Mesenchymal Stem Cells in Fibrotic Disease. Cell Stem. Cell 2017, 21, 166–177. [Google Scholar] [CrossRef] [PubMed]
- Croft, A.P.; Campos, J.; Jansen, K.; Turner, J.D.; Marshall, J.; Attar, M.; Savary, L.; Wehmeyer, C.; Naylor, A.J.; Kemble, S.; et al. Distinct fibroblast subsets drive inflammation and damage in arthritis. Nature 2019, 570, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Rinkevich, Y.; Walmsley, G.G.; Hu, M.S.; Maan, Z.N.; Newman, A.M.; Drukker, M.; Januszyk, M.; Krampitz, G.W.; Gurtner, G.C.; Lorenz, H.P.; et al. Skin fibrosis. Identification and isolation of a dermal lineage with intrinsic fibrogenic potential. Science 2015, 348, aaa2151. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, C.X.; Muller, S.; Keerthivasan, S.; Koeppen, H.; Hung, J.; Gierke, S.; Breart, B.; Foreman, O.; Bainbridge, T.W.; Castiglioni, A.; et al. Single-Cell RNA Sequencing Reveals Stromal Evolution into LRRC15(+) Myofibroblasts as a Determinant of Patient Response to Cancer Immunotherapy. Cancer Discov. 2020, 10, 232–253. [Google Scholar] [CrossRef] [PubMed]
- Chelladurai, P.; Kuenne, C.; Bourgeois, A.; Gunther, S.; Valasarajan, C.; Cherian, A.V.; Rottier, R.J.; Romanet, C.; Weigert, A.; Boucherat, O.; et al. Epigenetic reactivation of transcriptional programs orchestrating fetal lung development in human pulmonary hypertension. Sci. Transl. Med. 2022, 14, eabe5407. [Google Scholar] [CrossRef] [PubMed]
- Crnkovic, S.; Valzano, F.; Fliesser, E.; Gindlhuber, J.; Thekkekara Puthenparampil, H.; Basil, M.; Morley, M.P.; Katzen, J.; Gschwandtner, E.; Klepetko, W.; et al. Single-cell transcriptomics reveals skewed cellular communication and phenotypic shift in pulmonary artery remodeling. JCI Insight 2022, 7, e153471. [Google Scholar] [CrossRef] [PubMed]
- Belknap, J.K.; Orton, E.C.; Ensley, B.; Tucker, A.; Stenmark, K.R. Hypoxia increases bromodeoxyuridine labeling indices in bovine neonatal pulmonary arteries. Am. J. Respir. Cell Mol. Biol. 1997, 16, 366–371. [Google Scholar] [CrossRef] [PubMed]
- Duffield, J.S.; Lupher, M.; Thannickal, V.J.; Wynn, T.A. Host responses in tissue repair and fibrosis. Annu. Rev. Pathol. 2013, 8, 241–276. [Google Scholar] [CrossRef]
- Majesky, M.W.; Dong, X.R.; Hoglund, V.; Mahoney, W.M., Jr.; Daum, G. The adventitia: A dynamic interface containing resident progenitor cells. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1530–1539. [Google Scholar] [CrossRef]
- Meyrick, B.; Reid, L. Ultrastructural findings in lung biopsy material from children with congenital heart defects. Am. J. Pathol. 1980, 101, 527–542. [Google Scholar] [PubMed]
- Stenmark, K.R.; Fagan, K.A.; Frid, M.G. Hypoxia-induced pulmonary vascular remodeling: Cellular and molecular mechanisms. Circ. Res. 2006, 99, 675–691. [Google Scholar] [CrossRef] [PubMed]
- Stenmark, K.R.; Yeager, M.E.; El Kasmi, K.C.; Nozik-Grayck, E.; Gerasimovskaya, E.V.; Li, M.; Riddle, S.R.; Frid, M.G. The adventitia: Essential regulator of vascular wall structure and function. Annu. Rev. Physiol. 2013, 75, 23–47. [Google Scholar] [CrossRef]
- Buckley, C.D.; Pilling, D.; Lord, J.M.; Akbar, A.N.; Scheel-Toellner, D.; Salmon, M. Fibroblasts regulate the switch from acute resolving to chronic persistent inflammation. Trends Immunol. 2001, 22, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Sartore, S.; Chiavegato, A.; Faggin, E.; Franch, R.; Puato, M.; Ausoni, S.; Pauletto, P. Contribution of adventitial fibroblasts to neointima formation and vascular remodeling: From innocent bystander to active participant. Circ. Res. 2001, 89, 1111–1121. [Google Scholar] [CrossRef]
- Stenmark, K.R.; Davie, N.; Frid, M.; Gerasimovskaya, E.; Das, M. Role of the adventitia in pulmonary vascular remodeling. Physiology 2006, 21, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Das, M.; Dempsey, E.C.; Reeves, J.T.; Stenmark, K.R. Selective expansion of fibroblast subpopulations from pulmonary artery adventitia in response to hypoxia. Am. J. Physiol. Lung Cell Mol. Physiol. 2002, 282, L976–L986. [Google Scholar] [CrossRef]
- Das, M.; Dempsey, E.C.; Bouchey, D.; Reyland, M.E.; Stenmark, K.R. Chronic hypoxia induces exaggerated growth responses in pulmonary artery adventitial fibroblasts: Potential contribution of specific protein kinase c isozymes. Am. J. Respir. Cell Mol. Biol. 2000, 22, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Welsh, D.J.; Scott, P.H.; Peacock, A.J. p38 MAP kinase isoform activity and cell cycle regulators in the proliferative response of pulmonary and systemic artery fibroblasts to acute hypoxia. Pulm. Pharmacol. Ther. 2006, 19, 128–138. [Google Scholar] [CrossRef]
- Anwar, A.; Li, M.; Frid, M.G.; Kumar, B.; Gerasimovskaya, E.V.; Riddle, S.R.; McKeon, B.A.; Thukaram, R.; Meyrick, B.O.; Fini, M.A.; et al. Osteopontin is an endogenous modulator of the constitutively activated phenotype of pulmonary adventitial fibroblasts in hypoxic pulmonary hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 2012, 303, L1–L11. [Google Scholar] [CrossRef] [PubMed]
- Chelladurai, P.; Dabral, S.; Basineni, S.R.; Chen, C.N.; Schmoranzer, M.; Bender, N.; Feld, C.; Notzold, R.R.; Dobreva, G.; Wilhelm, J.; et al. Isoform-specific characterization of class I histone deacetylases and their therapeutic modulation in pulmonary hypertension. Sci. Rep. 2020, 10, 12864. [Google Scholar] [CrossRef]
- Li, M.; Riddle, S.; Zhang, H.; D’Alessandro, A.; Flockton, A.; Serkova, N.J.; Hansen, K.C.; Moldovan, R.; McKeon, B.A.; Frid, M.; et al. Metabolic Reprogramming Regulates the Proliferative and Inflammatory Phenotype of Adventitial Fibroblasts in Pulmonary Hypertension Through the Transcriptional Corepressor C-Terminal Binding Protein-1. Circulation 2016, 134, 1105–1121. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.J.; Laux, A.; Gandjeva, A.; Wang, L.; Li, M.; Brown, R.D.; Riddle, S.; Kheyfets, V.O.; Tuder, R.M.; Zhang, H.; et al. The Effect of Hypoxia-inducible Factor Inhibition on the Phenotype of Fibroblasts in Human and Bovine Pulmonary Hypertension. Am. J. Respir. Cell Mol. Biol. 2023, 69, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Ashek, A.; Spruijt, O.A.; Harms, H.J.; Lammertsma, A.A.; Cupitt, J.; Dubois, O.; Wharton, J.; Dabral, S.; Pullamsetti, S.S.; Huisman, M.C.; et al. 3′-Deoxy-3′-[18F]Fluorothymidine Positron Emission Tomography Depicts Heterogeneous Proliferation Pathology in Idiopathic Pulmonary Arterial Hypertension Patient Lung. Circ. Cardiovasc. Imaging 2018, 11, e007402. [Google Scholar] [CrossRef] [PubMed]
- Huertas, A.; Tu, L.; Humbert, M.; Guignabert, C. Chronic inflammation within the vascular wall in pulmonary arterial hypertension: More than a spectator. Cardiovasc. Res. 2020, 116, 885–893. [Google Scholar] [CrossRef] [PubMed]
- Savai, R.; Pullamsetti, S.S.; Kolbe, J.; Bieniek, E.; Voswinckel, R.; Fink, L.; Scheed, A.; Ritter, C.; Dahal, B.K.; Vater, A.; et al. Immune and inflammatory cell involvement in the pathology of idiopathic pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2012, 186, 897–908. [Google Scholar] [CrossRef] [PubMed]
- Stenmark, K.R.; Tuder, R.M.; El Kasmi, K.C. Metabolic reprogramming and inflammation act in concert to control vascular remodeling in hypoxic pulmonary hypertension. J. Appl. Physiol. 2015, 119, 1164–1172. [Google Scholar] [CrossRef]
- Marsh, L.J.; Kemble, S.; Reis Nisa, P.; Singh, R.; Croft, A.P. Fibroblast pathology in inflammatory joint disease. Immunol. Rev. 2021, 302, 163–183. [Google Scholar] [CrossRef]
- Henderson, N.C.; Rieder, F.; Wynn, T.A. Fibrosis: From mechanisms to medicines. Nature 2020, 587, 555–566. [Google Scholar] [CrossRef]
- Kennel, K.B.; Bozlar, M.; De Valk, A.F.; Greten, F.R. Cancer-Associated Fibroblasts in Inflammation and Antitumor Immunity. Clin. Cancer Res. 2023, 29, 1009–1016. [Google Scholar] [CrossRef] [PubMed]
- Jones, F.S.; Jones, P.L. The tenascin family of ECM glycoproteins: Structure, function, and regulation during embryonic development and tissue remodeling. Dev. Dyn. 2000, 218, 235–259. [Google Scholar] [CrossRef]
- Stenmark, K.R.; Mecham, R.P. Cellular and molecular mechanisms of pulmonary vascular remodeling. Annu. Rev. Physiol. 1997, 59, 89–144. [Google Scholar] [CrossRef] [PubMed]
- Thenappan, T.; Chan, S.Y.; Weir, E.K. Role of extracellular matrix in the pathogenesis of pulmonary arterial hypertension. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1322–H1331. [Google Scholar] [CrossRef] [PubMed]
- Kohn, J.C.; Lampi, M.C.; Reinhart-King, C.A. Age-related vascular stiffening: Causes and consequences. Front. Genet. 2015, 6, 112. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.F.; Nambiar Veetil, N.; Li, Q.; Kucherenko, M.M.; Knosalla, C.; Kuebler, W.M. Pulmonary hypertension: Linking inflammation and pulmonary arterial stiffening. Front. Immunol. 2022, 13, 959209. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Valdez-Jasso, D. Cellular mechanosignaling in pulmonary arterial hypertension. Biophys. Rev. 2021, 13, 747–756. [Google Scholar] [CrossRef] [PubMed]
- Bertero, T.; Cottrill, K.A.; Lu, Y.; Haeger, C.M.; Dieffenbach, P.; Annis, S.; Hale, A.; Bhat, B.; Kaimal, V.; Zhang, Y.Y.; et al. Matrix Remodeling Promotes Pulmonary Hypertension through Feedback Mechanoactivation of the YAP/TAZ-miR-130/301 Circuit. Cell Rep. 2015, 13, 1016–1032. [Google Scholar] [CrossRef] [PubMed]
- Mueller, M.C.; Du, Y.; Walker, L.A.; Magin, C.M. Dynamically stiffening biomaterials reveal age- and sex-specific differences in pulmonary arterial adventitial fibroblast activation. bioRxiv 2024. preprint. [Google Scholar] [CrossRef]
- Jandl, K.; Radic, N.; Zeder, K.; Kovacs, G.; Kwapiszewska, G. Pulmonary vascular fibrosis in pulmonary hypertension—The role of the extracellular matrix as a therapeutic target. Pharmacol. Ther. 2023, 247, 108438. [Google Scholar] [CrossRef] [PubMed]
- Metchnikoff, O. Life of Elie Metchnikoff 1845–1916; Constable and Company, Ltd.: London, UK, 1921. [Google Scholar]
- Uderhardt, S.; Martins, A.J.; Tsang, J.S.; Lammermann, T.; Germain, R.N. Resident Macrophages Cloak Tissue Microlesions to Prevent Neutrophil-Driven Inflammatory Damage. Cell 2019, 177, 541–555.e517. [Google Scholar] [CrossRef]
- Zhou, X.; Franklin, R.A.; Adler, M.; Jacox, J.B.; Bailis, W.; Shyer, J.A.; Flavell, R.A.; Mayo, A.; Alon, U.; Medzhitov, R. Circuit Design Features of a Stable Two-Cell System. Cell 2018, 172, 744–757.e717. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Riddle, S.; Kumar, S.; Poczobutt, J.; McKeon, B.A.; Frid, M.G.; Ostaff, M.; Reisz, J.A.; Nemkov, T.; Fini, M.A.; et al. Microenvironmental Regulation of Macrophage Transcriptomic and Metabolomic Profiles in Pulmonary Hypertension. Front. Immunol. 2021, 12, 640718. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Frid, M.G.; Zhang, H.; Li, M.; Riddle, S.; Brown, R.D.; Yadav, S.C.; Roy, M.K.; Dzieciatkowska, M.E.; D’Alessandro, A.; et al. Complement-containing small extracellular vesicles from adventitial fibroblasts induce proinflammatory and metabolic reprogramming in macrophages. JCI Insight. 2021, 6, e148382. [Google Scholar] [CrossRef] [PubMed]
- Guilliams, M.; Thierry, G.R.; Bonnardel, J.; Bajenoff, M. Establishment and Maintenance of the Macrophage Niche. Immunity 2020, 52, 434–451. [Google Scholar] [CrossRef] [PubMed]
- Kelly, B.; O’Neill, L.A. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015, 25, 771–784. [Google Scholar] [CrossRef]
- Larionova, I.; Kazakova, E.; Patysheva, M.; Kzhyshkowska, J. Transcriptional, Epigenetic and Metabolic Programming of Tumor-Associated Macrophages. Cancers 2020, 12, 1411. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A. A critical role for citrate metabolism in LPS signalling. Biochem. J. 2011, 438, e5–e6. [Google Scholar] [CrossRef] [PubMed]
- Worrell, J.C.; MacLeod, M.K.L. Stromal-immune cell crosstalk fundamentally alters the lung microenvironment following tissue insult. Immunology 2021, 163, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Taraseviciene-Stewart, L.; Nicolls, M.R.; Kraskauskas, D.; Scerbavicius, R.; Burns, N.; Cool, C.; Wood, K.; Parr, J.E.; Boackle, S.A.; Voelkel, N.F. Absence of T cells confers increased pulmonary arterial hypertension and vascular remodeling. Am. J. Respir. Crit. Care Med. 2007, 175, 1280–1289. [Google Scholar] [CrossRef]
- Maston, L.D.; Jones, D.T.; Giermakowska, W.; Howard, T.A.; Cannon, J.L.; Wang, W.; Wei, Y.; Xuan, W.; Resta, T.C.; Gonzalez Bosc, L.V. Central role of T helper 17 cells in chronic hypoxia-induced pulmonary hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 2017, 312, L609–L624. [Google Scholar] [CrossRef]
- Steiner, M.K.; Syrkina, O.L.; Kolliputi, N.; Mark, E.J.; Hales, C.A.; Waxman, A.B. Interleukin-6 overexpression induces pulmonary hypertension. Circ. Res. 2009, 104, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.; Jiang, S.Y.; Jiang, X.; Tamosiuniene, R.; Kim, D.; Guan, T.; Arsalane, S.; Pasupneti, S.; Voelkel, N.F.; Tang, Q.; et al. The Role of Regulatory T Cells in Pulmonary Arterial Hypertension. Front. Immunol. 2021, 12, 684657. [Google Scholar] [CrossRef] [PubMed]
- Itoh, M.; Takahashi, T.; Sakaguchi, N.; Kuniyasu, Y.; Shimizu, J.; Otsuka, F.; Sakaguchi, S. Thymus and autoimmunity: Production of CD25+CD4+ naturally anergic and suppressive T cells as a key function of the thymus in maintaining immunologic self-tolerance. J. Immunol. 1999, 162, 5317–5326. [Google Scholar] [CrossRef] [PubMed]
- Gaowa, S.; Zhou, W.; Yu, L.; Zhou, X.; Liao, K.; Yang, K.; Lu, Z.; Jiang, H.; Chen, X. Effect of Th17 and Treg axis disorder on outcomes of pulmonary arterial hypertension in connective tissue diseases. Mediators Inflamm. 2014, 2014, 247372. [Google Scholar] [CrossRef] [PubMed]
- Huertas, A.; Phan, C.; Bordenave, J.; Tu, L.; Thuillet, R.; Le Hiress, M.; Avouac, J.; Tamura, Y.; Allanore, Y.; Jovan, R.; et al. Regulatory T Cell Dysfunction in Idiopathic, Heritable and Connective Tissue-Associated Pulmonary Arterial Hypertension. Chest 2016, 149, 1482–1493. [Google Scholar] [CrossRef] [PubMed]
- Plecita-Hlavata, L.; Brazdova, A.; Krivonoskova, M.; Hu, C.J.; Phang, T.; Tauber, J.; Li, M.; Zhang, H.; Hoetzenecker, K.; Crnkovic, S.; et al. Microenvironmental regulation of T-cells in pulmonary hypertension. Front. Immunol. 2023, 14, 1223122. [Google Scholar] [CrossRef] [PubMed]
- Spin, J.M.; Maegdefessel, L.; Tsao, P.S. Vascular smooth muscle cell phenotypic plasticity: Focus on chromatin remodelling. Cardiovasc. Res. 2012, 95, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Crnkovic, S.; Puthenparampil, H.T.; Mulch, S.; Biasin, V.; Wilhelm, J.; Bartkuhn, M.; Rad, E.B.; Wawrzen, A.; Matzer, I.; Mitra, A.; et al. Adventitial fibroblasts direct smooth muscle cell-state transition in pulmonary vascular disease. bioRxiv 2024, preprint. [Google Scholar] [CrossRef]
- Humbert, M.; Guignabert, C.; Bonnet, S.; Dorfmuller, P.; Klinger, J.R.; Nicolls, M.R.; Olschewski, A.J.; Pullamsetti, S.S.; Schermuly, R.T.; Stenmark, K.R.; et al. Pathology and pathobiology of pulmonary hypertension: State of the art and research perspectives. Eur. Respir. J. 2019, 53, 1801887. [Google Scholar] [CrossRef]
- Esquela-Kerscher, A.; Slack, F.J. Oncomirs—microRNAs with a role in cancer. Nat. Rev. Cancer 2006, 6, 259–269. [Google Scholar] [CrossRef]
- Li, S.; Ran, Y.; Zhang, D.; Chen, J.; Li, S.; Zhu, D. MicroRNA-138 plays a role in hypoxic pulmonary vascular remodelling by targeting Mst1. Biochem. J. 2013, 452, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Santos-Ferreira, C.A.; Abreu, M.T.; Marques, C.I.; Goncalves, L.M.; Baptista, R.; Girao, H.M. Micro-RNA Analysis in Pulmonary Arterial Hypertension: Current Knowledge and Challenges. JACC Basic. Transl. Sci. 2020, 5, 1149–1162. [Google Scholar] [CrossRef] [PubMed]
- Small, E.M.; Olson, E.N. Pervasive roles of microRNAs in cardiovascular biology. Nature 2011, 469, 336–342. [Google Scholar] [CrossRef] [PubMed]
- White, K.; Loscalzo, J.; Chan, S.Y. Holding our breath: The emerging and anticipated roles of microRNA in pulmonary hypertension. Pulm. Circ. 2012, 2, 278–290. [Google Scholar] [CrossRef] [PubMed]
- Nakamachi, Y.; Kawano, S.; Takenokuchi, M.; Nishimura, K.; Sakai, Y.; Chin, T.; Saura, R.; Kurosaka, M.; Kumagai, S. MicroRNA-124a is a key regulator of proliferation and monocyte chemoattractant protein 1 secretion in fibroblast-like synoviocytes from patients with rheumatoid arthritis. Arthritis Rheum. 2009, 60, 1294–1304. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wang, D.; Li, M.; Plecita-Hlavata, L.; D’Alessandro, A.; Tauber, J.; Riddle, S.; Kumar, S.; Flockton, A.; McKeon, B.A.; et al. Metabolic and Proliferative State of Vascular Adventitial Fibroblasts in Pulmonary Hypertension Is Regulated Through a MicroRNA-124/PTBP1 (Polypyrimidine Tract Binding Protein 1)/Pyruvate Kinase Muscle Axis. Circulation 2017, 136, 2468–2485. [Google Scholar] [CrossRef] [PubMed]
- Dayton, T.L.; Jacks, T.; Vander Heiden, M.G. PKM2, cancer metabolism, and the road ahead. EMBO Rep. 2016, 17, 1721–1730. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; D’Alessandro, A.; Li, M.; Reisz, J.A.; Riddle, S.; Muralidhar, A.; Bull, T.; Zhao, L.; Gerasimovskaya, E.; Stenmark, K.R. Histone deacetylase inhibitors synergize with sildenafil to suppress purine metabolism and proliferation in pulmonary hypertension. Vascul. Pharmacol. 2023, 149, 107157. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Laux, A.; Stenmark, K.R.; Hu, C.J. Mechanisms Contributing to the Dysregulation of miRNA-124 in Pulmonary Hypertension. Int. J. Mol. Sci. 2021, 22, 3852. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Dong, H.Y.; Zhang, B.; Feng, Z.; Liu, Y.; Gao, Y.Q.; Dong, M.Q.; Li, Z.C. miR-29a-3p attenuates hypoxic pulmonary hypertension by inhibiting pulmonary adventitial fibroblast activation. Hypertension 2015, 65, 414–420. [Google Scholar] [CrossRef]
- Wei, C.; Henderson, H.; Spradley, C.; Li, L.; Kim, I.K.; Kumar, S.; Hong, N.; Arroliga, A.C.; Gupta, S. Circulating miRNAs as potential marker for pulmonary hypertension. PLoS ONE 2013, 8, e64396. [Google Scholar] [CrossRef]
- Adcock, I.M. HDAC inhibitors as anti-inflammatory agents. Br. J. Pharmacol. 2007, 150, 829–831. [Google Scholar] [CrossRef]
- Bush, E.W.; McKinsey, T.A. Protein acetylation in the cardiorenal axis: The promise of histone deacetylase inhibitors. Circ. Res. 2010, 106, 272–284. [Google Scholar] [CrossRef]
- Kawabata, T.; Nishida, K.; Takasugi, K.; Ogawa, H.; Sada, K.; Kadota, Y.; Inagaki, J.; Hirohata, S.; Ninomiya, Y.; Makino, H. Increased activity and expression of histone deacetylase 1 in relation to tumor necrosis factor-alpha in synovial tissue of rheumatoid arthritis. Arthritis Res. Ther. 2010, 12, R133. [Google Scholar] [CrossRef]
- Horiuchi, M.; Morinobu, A.; Chin, T.; Sakai, Y.; Kurosaka, M.; Kumagai, S. Expression and function of histone deacetylases in rheumatoid arthritis synovial fibroblasts. J. Rheumatol. 2009, 36, 1580–1589. [Google Scholar] [CrossRef]
- Chelladurai, P.; Boucherat, O.; Stenmark, K.; Kracht, M.; Seeger, W.; Bauer, U.M.; Bonnet, S.; Pullamsetti, S.S. Targeting histone acetylation in pulmonary hypertension and right ventricular hypertrophy. Br. J. Pharmacol. 2021, 178, 54–71. [Google Scholar] [CrossRef]
- Cavasin, M.A.; Demos-Davies, K.; Horn, T.R.; Walker, L.A.; Lemon, D.D.; Birdsey, N.; Weiser-Evans, M.C.; Harral, J.; Irwin, D.C.; Anwar, A.; et al. Selective class I histone deacetylase inhibition suppresses hypoxia-induced cardiopulmonary remodeling through an antiproliferative mechanism. Circ. Res. 2012, 110, 739–748. [Google Scholar] [CrossRef]
- Zhao, L.; Chen, C.N.; Hajji, N.; Oliver, E.; Cotroneo, E.; Wharton, J.; Wang, D.; Li, M.; McKinsey, T.A.; Stenmark, K.R.; et al. Histone deacetylation inhibition in pulmonary hypertension: Therapeutic potential of valproic acid and suberoylanilide hydroxamic acid. Circulation 2012, 126, 455–467. [Google Scholar] [CrossRef]
- Plecita-Hlavata, L.; Tauber, J.; Li, M.; Zhang, H.; Flockton, A.R.; Pullamsetti, S.S.; Chelladurai, P.; D’Alessandro, A.; El Kasmi, K.C.; Jezek, P.; et al. Constitutive Reprogramming of Fibroblast Mitochondrial Metabolism in Pulmonary Hypertension. Am. J. Respir. Cell Mol. Biol. 2016, 55, 47–57. [Google Scholar] [CrossRef]
- Ryan, J.; Dasgupta, A.; Huston, J.; Chen, K.H.; Archer, S.L. Mitochondrial dynamics in pulmonary arterial hypertension. J. Mol. Med. 2015, 93, 229–242. [Google Scholar] [CrossRef]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Michelakis, E.D.; Gurtu, V.; Webster, L.; Barnes, G.; Watson, G.; Howard, L.; Cupitt, J.; Paterson, I.; Thompson, R.B.; Chow, K.; et al. Inhibition of pyruvate dehydrogenase kinase improves pulmonary arterial hypertension in genetically susceptible patients. Sci. Transl. Med. 2017, 9, aao4583. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Plecita-Hlavata, L.; Dobrinskikh, E.; McKeon, B.A.; Gandjeva, A.; Riddle, S.; Laux, A.; Prasad, R.R.; Kumar, S.; Tuder, R.M.; et al. SIRT3 Is a Critical Regulator of Mitochondrial Function of Fibroblasts in Pulmonary Hypertension. Am. J. Respir. Cell Mol. Biol. 2023, 69, 570–583. [Google Scholar] [CrossRef]
- O’Neill, L.A.; Hardie, D.G. Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature 2013, 493, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Cho, E.J.; Kim, S.T.; Youn, H.D. CtBP represses p300-mediated transcriptional activation by direct association with its bromodomain. Nat. Struct. Mol. Biol. 2005, 12, 423–428. [Google Scholar] [CrossRef]
- Mirnezami, A.H.; Campbell, S.J.; Darley, M.; Primrose, J.N.; Johnson, P.W.; Blaydes, J.P. Hdm2 recruits a hypoxia-sensitive corepressor to negatively regulate p53-dependent transcription. Curr. Biol. 2003, 13, 1234–1239. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Piston, D.W.; Goodman, R.H. Regulation of corepressor function by nuclear NADH. Science 2002, 295, 1895–1897. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Yoshimatsu, Y.; Hildebrand, J.; Frisch, S.M.; Goodman, R.H. Homeodomain interacting protein kinase 2 promotes apoptosis by downregulating the transcriptional corepressor CtBP. Cell 2003, 115, 177–186. [Google Scholar] [CrossRef]
- Fjeld, C.C.; Birdsong, W.T.; Goodman, R.H. Differential binding of NAD+ and NADH allows the transcriptional corepressor carboxyl-terminal binding protein to serve as a metabolic sensor. Proc. Natl. Acad. Sci. USA 2003, 100, 9202–9207. [Google Scholar] [CrossRef]
- Chinnadurai, G. The transcriptional corepressor CtBP: A foe of multiple tumor suppressors. Cancer Res. 2009, 69, 731–734. [Google Scholar] [CrossRef]
- Tuder, R.M.; Archer, S.L.; Dorfmuller, P.; Erzurum, S.C.; Guignabert, C.; Michelakis, E.; Rabinovitch, M.; Schermuly, R.; Stenmark, K.R.; Morrell, N.W. Relevant issues in the pathology and pathobiology of pulmonary hypertension. J. Am. Coll Cardiol. 2013, 62, D4–D12. [Google Scholar] [CrossRef] [PubMed]
- Abraham, N.G.; Levere, R.D.; Lin, J.H.; Beru, N.; Hermine, O.; Goldwasser, E. Co-regulation of heme oxygenase and erythropoietin genes. J. Cell Biochem. 1991, 47, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Morita, T.; Mitsialis, S.A.; Koike, H.; Liu, Y.; Kourembanas, S. Carbon monoxide controls the proliferation of hypoxic vascular smooth muscle cells. J. Biol. Chem. 1997, 272, 32804–32809. [Google Scholar] [CrossRef] [PubMed]
- Morita, T.; Perrella, M.A.; Lee, M.E.; Kourembanas, S. Smooth muscle cell-derived carbon monoxide is a regulator of vascular cGMP. Proc. Natl. Acad. Sci. USA 1995, 92, 1475–1479. [Google Scholar] [CrossRef] [PubMed]
- Yet, S.F.; Perrella, M.A.; Layne, M.D.; Hsieh, C.M.; Maemura, K.; Kobzik, L.; Wiesel, P.; Christou, H.; Kourembanas, S.; Lee, M.E. Hypoxia induces severe right ventricular dilatation and infarction in heme oxygenase-1 null mice. J. Clin. Investig. 1999, 103, R23–R29. [Google Scholar] [CrossRef] [PubMed]
- Minamino, T.; Christou, H.; Hsieh, C.M.; Liu, Y.; Dhawan, V.; Abraham, N.G.; Perrella, M.A.; Mitsialis, S.A.; Kourembanas, S. Targeted expression of heme oxygenase-1 prevents the pulmonary inflammatory and vascular responses to hypoxia. Proc. Natl. Acad. Sci. USA 2001, 98, 8798–8803. [Google Scholar] [CrossRef] [PubMed]
- Blevins, M.A.; Kouznetsova, J.; Krueger, A.B.; King, R.; Griner, L.M.; Hu, X.; Southall, N.; Marugan, J.J.; Zhang, Q.; Ferrer, M.; et al. Small Molecule, NSC95397, Inhibits the CtBP1-Protein Partner Interaction and CtBP1-Mediated Transcriptional Repression. J. Biomol. Screen 2015, 20, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yang, W.S.; Yu, T.; Yi, Y.S.; Park, J.G.; Jeong, D.; Kim, J.H.; Oh, J.S.; Yoon, K.; Kim, J.H.; et al. Novel anti-inflammatory function of NSC95397 by the suppression of multiple kinases. Biochem. Pharmacol. 2014, 88, 201–215. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.; Ramesh, V.; Locasale, J.W. The evolving metabolic landscape of chromatin biology and epigenetics. Nat. Rev. Genet 2020, 21, 737–753. [Google Scholar] [CrossRef]
- Joshi, S.R.; Kitagawa, A.; Jacob, C.; Hashimoto, R.; Dhagia, V.; Ramesh, A.; Zheng, C.; Zhang, H.; Jordan, A.; Waddell, I.; et al. Hypoxic activation of glucose-6-phosphate dehydrogenase controls the expression of genes involved in the pathogenesis of pulmonary hypertension through the regulation of DNA methylation. Am. J. Physiol. Lung Cell Mol. Physiol. 2020, 318, L773–L786. [Google Scholar] [CrossRef]
- Kitagawa, A.; Jacob, C.; Jordan, A.; Waddell, I.; McMurtry, I.F.; Gupte, S.A. Inhibition of Glucose-6-Phosphate Dehydrogenase Activity Attenuates Right Ventricle Pressure and Hypertrophy Elicited by VEGFR Inhibitor + Hypoxia. J. Pharmacol. Exp. Ther. 2021, 377, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, A.; Jacob, C.; Gupte, S.A. Glucose-6-phosphate dehydrogenase and MEG3 controls hypoxia-induced expression of serum response factor (SRF) and SRF-dependent genes in pulmonary smooth muscle cell. J. Smooth Muscle Res. 2022, 58, 34–49. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Kumar, S.; Sharma, S.; Aggarwal, S.; Lu, Q.; Gross, C.; Rafikova, O.; Lee, S.G.; Dasarathy, S.; Hou, Y.; et al. Endothelin-1 induces a glycolytic switch in pulmonary arterial endothelial cells via the mitochondrial translocation of endothelial nitric oxide synthase. Am. J. Respir. Cell Mol. Biol. 2014, 50, 1084–1095. [Google Scholar] [CrossRef] [PubMed]
- Breault, N.M.; Wu, D.; Dasgupta, A.; Chen, K.H.; Archer, S.L. Acquired disorders of mitochondrial metabolism and dynamics in pulmonary arterial hypertension. Front. Cell Dev. Biol. 2023, 11, 1105565. [Google Scholar] [CrossRef] [PubMed]
- Culley, M.K.; Chan, S.Y. Mitochondrial metabolism in pulmonary hypertension: Beyond mountains there are mountains. J. Clin. Investig. 2018, 128, 3704–3715. [Google Scholar] [CrossRef]
- Hong, J.; Arneson, D.; Umar, S.; Ruffenach, G.; Cunningham, C.M.; Ahn, I.S.; Diamante, G.; Bhetraratana, M.; Park, J.F.; Said, E.; et al. Single-Cell Study of Two Rat Models of Pulmonary Arterial Hypertension Reveals Connections to Human Pathobiology and Drug Repositioning. Am. J. Respir. Crit. Care Med. 2021, 203, 1006–1022. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.; Li, M.; Hu, C.-J.; Stenmark, K.R. Fibroblasts in Pulmonary Hypertension: Roles and Molecular Mechanisms. Cells 2024, 13, 914. https://doi.org/10.3390/cells13110914
Zhang H, Li M, Hu C-J, Stenmark KR. Fibroblasts in Pulmonary Hypertension: Roles and Molecular Mechanisms. Cells. 2024; 13(11):914. https://doi.org/10.3390/cells13110914
Chicago/Turabian StyleZhang, Hui, Min Li, Cheng-Jun Hu, and Kurt R. Stenmark. 2024. "Fibroblasts in Pulmonary Hypertension: Roles and Molecular Mechanisms" Cells 13, no. 11: 914. https://doi.org/10.3390/cells13110914
APA StyleZhang, H., Li, M., Hu, C.-J., & Stenmark, K. R. (2024). Fibroblasts in Pulmonary Hypertension: Roles and Molecular Mechanisms. Cells, 13(11), 914. https://doi.org/10.3390/cells13110914