.png)
Towards a Cure for Diamond–Blackfan Anemia: Views on Gene Therapy


Abstract
:1. Introduction
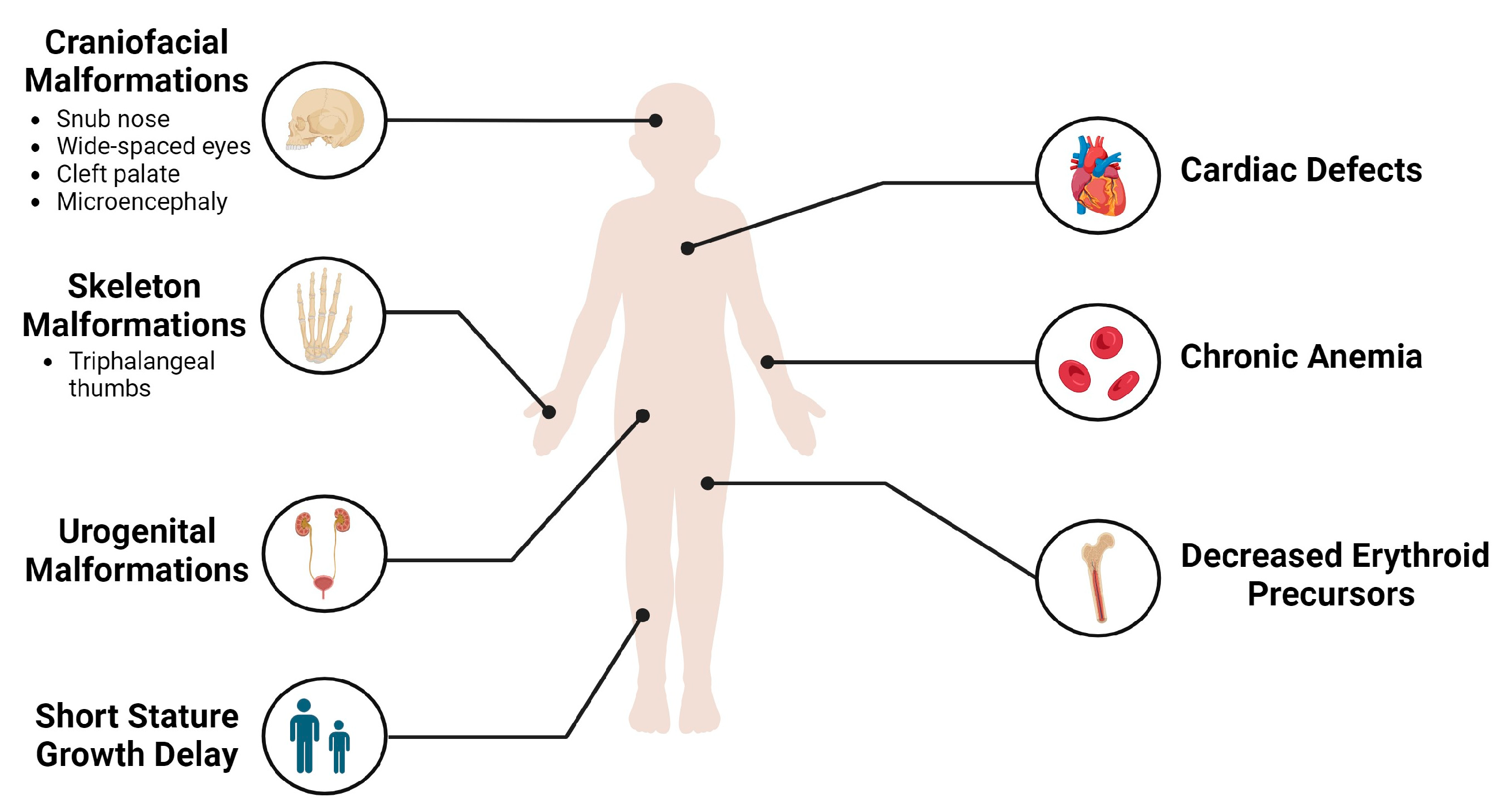
2. Clinical Presentation of DBA and Diagnosis
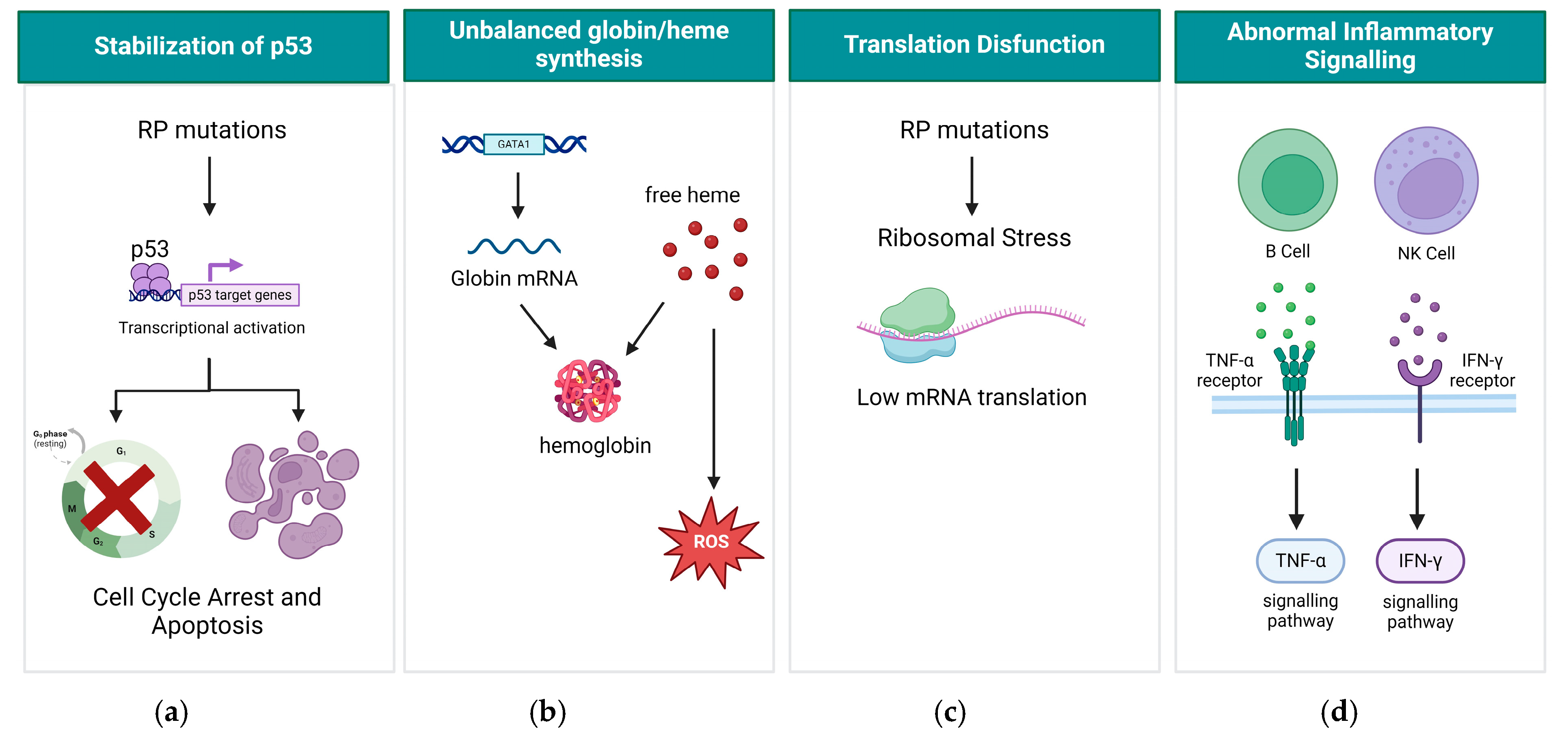
3. Molecular Mechanism of DBA
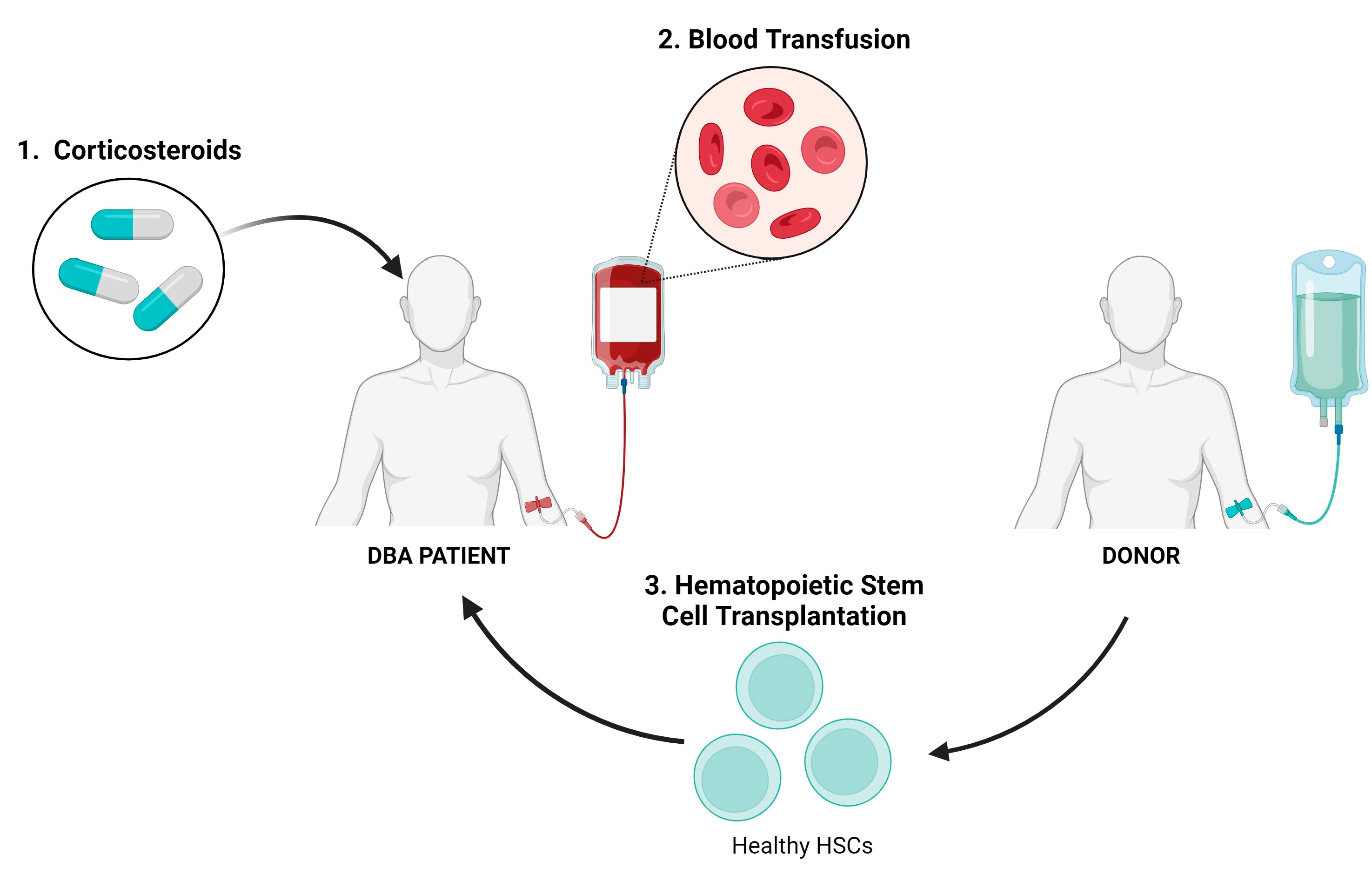
4. Existing Treatment Options for DBA
5. Gene Therapy for DBA—From Research Now to Clinic in the Future
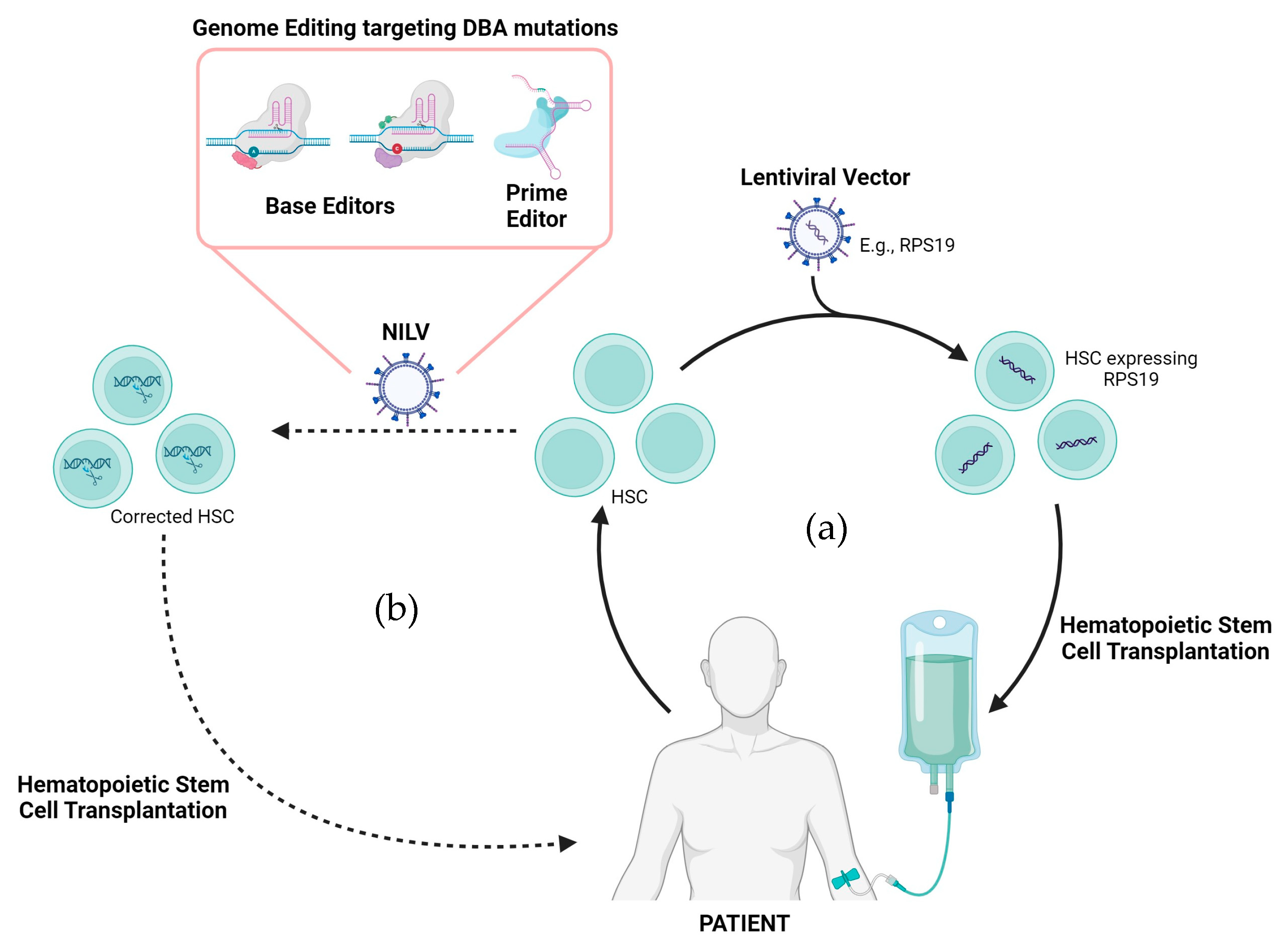
5.1. Lentiviral Vectors as a Potential Gene Therapy Approach for DBA
5.2. CRISPR/Cas9 Non-Integrating Lentiviral-Based Gene Therapy
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Liu, Y.; Karlsson, S. Perspectives of current understanding and therapeutics of Diamond-Blackfan anemia. Leukemia 2024, 38, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Brajanovski, N.; Chan, K.T.; Xuan, J.; Pearson, R.B.; Sanij, E. Ribosomal proteins and human diseases: Molecular mechanisms and targeted therapy. Signal Transduct. Target. Ther. 2021, 6, 323. [Google Scholar] [CrossRef] [PubMed]
- Ulirsch, J.C.; Verboon, J.M.; Kazerounian, S.; Guo, M.H.; Yuan, D.; Ludwig, L.S.; Handsaker, R.E.; Abdulhay, N.J.; Fiorini, C.; Genovese, G.; et al. The Genetic Landscape of Diamond-Blackfan Anemia. Am. J. Hum. Genet. 2018, 103, 930–947. [Google Scholar] [CrossRef] [PubMed]
- Maceckova, Z.; Kubickova, A.; De Sanctis, J.B.; Hajduch, M. Effect of Glucocorticosteroids in Diamond-Blackfan Anaemia: Maybe Not as Elusive as It Seems. Int. J. Mol. Sci. 2022, 23, 1886. [Google Scholar] [CrossRef] [PubMed]
- Bartels, M.; Bierings, M. How I manage children with Diamond-Blackfan anaemia. Br. J. Haematol. 2019, 184, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Strahm, B.; Loewecke, F.; Niemeyer, C.M.; Albert, M.; Ansari, M.; Bader, P.; Bertrand, Y.; Burkhardt, B.; Da Costa, L.M.; Ferster, A.; et al. Favorable outcomes of hematopoietic stem cell transplantation in children and adolescents with Diamond-Blackfan anemia. Blood Adv. 2020, 4, 1760–1769. [Google Scholar] [CrossRef] [PubMed]
- Vlachos, A.; Muir, E. How I treat Diamond-Blackfan anemia. Blood 2010, 116, 3715–3723. [Google Scholar] [CrossRef] [PubMed]
- Da Costa, L.; Leblanc, T.; Mohandas, N. Diamond-Blackfan Anemia. Blood 2020, 136, 1262–1273. [Google Scholar] [CrossRef] [PubMed]
- Lipton, J.M.; Ellis, S.R. Diamond-Blackfan anemia: Diagnosis, treatment, and molecular pathogenesis. Hematol. Oncol. Clin. North. Am. 2009, 23, 261–282. [Google Scholar] [CrossRef]
- Jahan, D.; Al Hasan, M.M.; Haque, M. Diamond-Blackfan anemia with mutation in RPS19: A case report and an overview of published pieces of literature. J. Pharm. Bioallied Sci. 2020, 12, 163–170. [Google Scholar] [CrossRef]
- Danilova, N.; Sakamoto, K.M.; Lin, S. Ribosomal protein S19 deficiency in zebrafish leads to developmental abnormalities and defective erythropoiesis through activation of p53 protein family. Blood 2008, 112, 5228–5237. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, A.; Uechi, T.; Higa, S.; Torihara, H.; Kenmochi, N. Loss of ribosomal protein L11 affects zebrafish embryonic development through a p53-dependent apoptotic response. PLoS ONE 2009, 4, e4152. [Google Scholar] [CrossRef] [PubMed]
- Moniz, H.; Gastou, M.; Leblanc, T.; Hurtaud, C.; Cretien, A.; Lecluse, Y.; Raslova, H.; Larghero, J.; Croisille, L.; Faubladier, M.; et al. Primary hematopoietic cells from DBA patients with mutations in RPL11 and RPS19 genes exhibit distinct erythroid phenotype in vitro. Cell Death Dis. 2012, 3, e356. [Google Scholar] [CrossRef] [PubMed]
- Rio, S.; Gastou, M.; Karboul, N.; Derman, R.; Suriyun, T.; Manceau, H.; Leblanc, T.; El Benna, J.; Schmitt, C.; Azouzi, S.; et al. Regulation of globin-heme balance in Diamond-Blackfan anemia by HSP70/GATA1. Blood 2019, 133, 1358–1370. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.W.; Green, R. Ribosomopathies: There’s strength in numbers. Science 2017, 358, eaan2755. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, L.S.; Gazda, H.T.; Eng, J.C.; Eichhorn, S.W.; Thiru, P.; Ghazvinian, R.; George, T.I.; Gotlib, J.R.; Beggs, A.H.; Sieff, C.A.; et al. Altered translation of GATA1 in Diamond-Blackfan anemia. Nat. Med. 2014, 20, 748–753. [Google Scholar] [CrossRef] [PubMed]
- Iskander, D.; Wang, G.; Heuston, E.F.; Christodoulidou, C.; Psaila, B.; Ponnusamy, K.; Ren, H.; Mokhtari, Z.; Robinson, M.; Chaidos, A.; et al. Single-cell profiling of human bone marrow progenitors reveals mechanisms of failing erythropoiesis in Diamond-Blackfan anemia. Sci. Transl. Med. 2021, 13, eabf0113. [Google Scholar] [CrossRef]
- Narla, A.; Vlachos, A.; Nathan, D.G. Diamond Blackfan anemia treatment: Past, present, and future. Semin. Hematol. 2011, 48, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Bhoopalan, S.V.; Suryaprakash, S.; Sharma, A.; Wlodarski, M.W. Hematopoietic cell transplantation and gene therapy for Diamond-Blackfan anemia: State of the art and science. Front. Oncol. 2023, 13, 1236038. [Google Scholar] [CrossRef]
- Sutton, R.E.; Reitsma, M.J.; Uchida, N.; Brown, P.O. Transduction of Human Progenitor Hematopoietic Stem Cells by Human Immunodeficiency Virus Type 1-Based Vectors Is Cell Cycle Dependent. J. Virol. 1999, 73, 3649–3660. [Google Scholar] [CrossRef]
- Peluffo, H.; Foster, E.; Ahmed, S.G.; Lago, N.; Hutson, T.H.; Moon, L.; Yip, P.; Wanisch, K.; Caraballo-Miralles, V.; Olmos, G.; et al. Efficient gene expression from integration-deficient lentiviral vectors in the spinal cord. Gene Ther. 2013, 20, 645–657. [Google Scholar] [CrossRef] [PubMed]
- Kalidasan, V.; Ng, W.H.; Ishola, O.A.; Ravichantar, N.; Tan, J.J.; Das, K.T. A guide in lentiviral vector production for hard-to-transfect cells, using cardiac-derived c-kit expressing cells as a model system. Sci. Rep. 2021, 11, 19265. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.; Kim, Y.S.; Wielgosz, M.M.; Ferrara, F.; Ma, Z.; Condori, J.; Palmer, L.E.; Zhao, X.; Kang, G.; Rawlings, D.J.; et al. Optimizing lentiviral vector transduction of hematopoietic stem cells for gene therapy. Gene Ther. 2020, 27, 545–556. [Google Scholar] [CrossRef] [PubMed]
- Schlimgen, R.; Howard, J.; Wooley, D.; Thompson, M.; Baden, L.R.; Yang, O.O.; Christiani, D.C.; Mostoslavsky, G.; Diamond, D.V.; Duane, E.G.; et al. Risks Associated with Lentiviral Vector Exposures and Prevention Strategies. J. Occup. Env. Med. 2016, 58, 1159–1166. [Google Scholar] [CrossRef] [PubMed]
- Howe, S.J.; Mansour, M.R.; Schwarzwaelder, K.; Bartholomae, C.; Hubank, M.; Kempski, H.; Brugman, M.H.; Pike-Overzet, K.; Chatters, S.J.; de Ridder, D.; et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J. Clin. Investig. 2008, 118, 3143–3150. [Google Scholar] [CrossRef] [PubMed]
- Flygare, J.; Aspesi, A.; Bailey, J.C.; Miyake, K.; Caffrey, J.M.; Karlsson, S.; Ellis, S.R. Human RPS19, the gene mutated in Diamond-Blackfan anemia, encodes a ribosomal protein required for the maturation of 40S ribosomal subunits. Blood 2006, 109, 980–986. [Google Scholar] [CrossRef] [PubMed]
- Hamaguchi, I.; Flygare, J.; Nishiura, H.; Brun, A.C.; Ooka, A.; Kiefer, T.; Ma, Z.; Dahl, N.; Richter, J.; Karlsson, S. Proliferation deficiency of multipotent hematopoietic progenitors in ribosomal protein S19 (RPS19)-deficient diamond-Blackfan anemia improves following RPS19 gene transfer. Mol. Ther. 2003, 7, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Jaako, P.; Debnath, S.; Olsson, K.; Modlich, U.; Rothe, M.; Schambach, A.; Flygare, J.; Karlsson, S. Gene therapy cures the anemia and lethal bone marrow failure in a mouse model of RPS19-deficient Diamond-Blackfan anemia. Haematologica 2014, 99, 1792–1798. [Google Scholar] [CrossRef] [PubMed]
- Debnath, S.; Jaako, P.; Siva, K.; Rothe, M.; Chen, J.; Dahl, M.; Gaspar, H.B.; Flygare, J.; Schambach, A.; Karlsson, S. Lentiviral Vectors with Cellular Promoters Correct Anemia and Lethal Bone Marrow Failure in a Mouse Model for Diamond-Blackfan Anemia. Mol. Ther. 2017, 25, 1805–1814. [Google Scholar] [CrossRef]
- Liu, Y.; Dahl, M.; Debnath, S.; Rothe, M.; Smith, E.M.; Grahn, T.H.M.; Warsi, S.; Chen, J.; Flygare, J.; Schambach, A.; et al. Successful gene therapy of Diamond-Blackfan anemia in a mouse model and human CD34(+) cord blood hematopoietic stem cells using a clinically applicable lentiviral vector. Haematologica 2022, 107, 446–456. [Google Scholar] [CrossRef]
- Voit, R.A.; Liao, X.; Cohen, B.; Armant, M.; Kamal, E.; Huang, M.-M.; Clarke, W.; Williams, D.A.; Shimamura, A.; Sankaran, V.G. Regulated Expression of GATA1 As a Gene Therapy Cure for Diamond-Blackfan Anemia. Blood 2022, 140, 986–987. [Google Scholar] [CrossRef]
- Gene Transfer For Patients with Sickle Cell Disease Using A Gamma Globin Lentivirus Vector: An Open-Label Phase 1/2 Pilot Study, NCT02186418. Available online: https://clinicaltrials.gov/study/NCT02186418?cond=Sickle%20Cell%20Disease&term=lentivirus%20gene%20therapy%20&rank=1 (accessed on 17 April 2024).
- Pilot and Feasibility Study of Hematopoietic Stem Cell Gene Transfer for Sickle Cell Disease, NCT03282656. Available online: https://clinicaltrials.gov/study/NCT03282656?cond=Sickle%20Cell%20Disease&term=lentivirus%20gene%20therapy%20&rank=2 (accessed on 17 April 2024).
- A Multi-Center, Phase 2 Gene Transfer Study Inducing Fetal Hemoglobin in Sickle Cell (GRASP, BMT CTN 2001), NCT05353647. Available online: https://clinicaltrials.gov/study/NCT05353647?cond=Sickle%20Cell%20Disease&term=lentivirus%20gene%20therapy%20&rank=3 (accessed on 17 April 2024).
- Clinical Research Study of Autologous Stem Cell Transplantation for Sickle Cell Disease (SCD) Using Peripheral Blood CD34+ Cells Modified with the Lenti/G-βAS3-FB Lentiviral Vector, NCT02247843. Available online: https://clinicaltrials.gov/study/NCT02247843?cond=Sickle%20Cell%20Disease&term=lentivirus%20gene%20therapy%20&rank=4 (accessed on 17 April 2024).
- A Phase 1/2 Open Label Study Evaluating the Safety and Efficacy of Gene Therapy of the Sickle Cell Disease by Transplantation of an Autologous CD34+ Enriched Cell Fraction That Contains CD34+ Cells Transduced Ex Vivo with the GLOBE1 Lentiviral Vector Expressing the βAS3 Globin Gene (GLOBE1 βAS3 Modified Autologous CD34+ Cells) in Patients with Sickle Cell Disease (SCD), NCT03964792. Available online: https://clinicaltrials.gov/study/NCT03964792?cond=Sickle%20Cell%20Disease&term=lentivirus%20gene%20therapy%20&rank=5 (accessed on 17 April 2024).
- A Phase 1/2 Open Label Study Evaluating the Safety and Efficacy of Gene Therapy of the β-Hemoglobinopathies (Sickle Cell Anemia and β-Thalassemia Major) by Transplantation of Autologous CD34+ Stem Cells Transduced Ex Vivo with a Lentiviral β-A-T87Q Globin Vector (LentiGlobin BB305 Drug Product), NCT02151526. Available online: https://clinicaltrials.gov/study/NCT02151526?cond=Sickle%20Cell%20Disease&term=lentivirus%20gene%20therapy%20&rank=6 (accessed on 17 April 2024).
- Evaluation of Safety and Efficacy of Transplantation of Autologous Hematopoietic Stem Cell Genetically Modified in Beta-Thalassemia Major, NCT03276455. Available online: https://clinicaltrials.gov/study/NCT03276455?cond=Thalassemia&term=lentivirus%20gene%20therapy%20&rank=3 (accessed on 17 April 2024).
- A Phase I/II Study Evaluating Safety and Efficacy of Autologous Hematopoietic Stem Cells Genetically Modified with GLOBE Lentiviral Vector Encoding for the Human Beta-Globin Gene for the Treatment of Patients Affected by Transfusion Dependent Beta-Thalassemia, NCT02453477. Available online: https://clinicaltrials.gov/study/NCT02453477?cond=Thalassemia&term=lentivirus%20gene%20therapy%20&rank=4 (accessed on 17 April 2024).
- Evaluation of the Safety and Efficacy of KL003 Gene Therapy in Patients with Transfusion-Dependent β-Thalassemia with No Conditioning Regimen, NCT06219239. Available online: https://clinicaltrials.gov/study/NCT06219239?cond=Thalassemia&term=lentivirus%20gene%20therapy%20&rank=6 (accessed on 17 April 2024).
- Safety and Efficacy Evaluation of β-globin Restored Autologous Hematopoietic Stem Cells in β-Thalassemia Major Patients, NCT05745532. Available online: https://clinicaltrials.gov/study/NCT05745532?cond=Thalassemia&term=lentivirus%20gene%20therapy%20&rank=7 (accessed on 17 April 2024).
- A Phase I/II Clinical Study Evaluating the Safety and Efficacy of KL003 Cell Injection in Transfusion-Dependent β-Thalassemia, NCT06280378. Available online: https://clinicaltrials.gov/study/NCT06280378?cond=Thalassemia&term=lentivirus%20gene%20therapy%20&rank=8 (accessed on 17 April 2024).
- A Phase I Clinical Trial for the Treatment of ß-Thalassemia Major with Autologous CD34+ Hematopoietic Progenitor Cells Transduced with TNS9.3.55 a Lentiviral Vector Encoding the Normal Human ß-Globin Gene, NCT01639690. Available online: https://clinicaltrials.gov/study/NCT01639690?cond=Thalassemia&term=lentivirus%20gene%20therapy%20&rank=9 (accessed on 17 April 2024).
- An Open Label Study Evaluating the Safety and Efficacy of Gene Therapy for Transfusion-Dependent β-Thalassemia by Transplantation of Autologous CD34+ Stem Cells Transduced Ex Vivo with a LentiRed Lentiviral Vector (GMCN-508B Drug Product, Also Called LentiRed), NCT05762510. Available online: https://clinicaltrials.gov/study/NCT05762510?cond=Thalassemia&term=lentivirus%20gene%20therapy%20&rank=11 (accessed on 17 April 2024).
- A Phase 1 Open Label Study Evaluating the Safety and Efficacy of Gene Therapy in Subjects with Transfusion-Dependent α-Thalassemia by Transplantation of Autologous CD34+ Cells Transduced Ex Vivo with a Lentiviral Vector (GMCN-508A Drug Product), NCT05757245. Available online: https://clinicaltrials.gov/study/NCT05757245?cond=Thalassemia&term=lentivirus%20gene%20therapy%20&rank=12 (accessed on 17 April 2024).
- A Phase 1 Open Label Study Evaluating the Safety and Efficacy of Gene Therapy in Subjects with β-Thalassemia Major by Transplantation of Autologous CD34+Stem Cells Transduced with a Lentiviral Vector Encoding βA-T87Q-Globin, NCT05015920. Available online: https://clinicaltrials.gov/study/NCT05015920?cond=Thalassemia&term=lentivirus%20gene%20therapy%20&rank=13 (accessed on 17 April 2024).
- Gene Transfer for Patients with Fanconi Anemia Complementation Group A (FANCA), NCT01331018. Available online: https://clinicaltrials.gov/study/NCT01331018?cond=Fanconi%20Anemia&term=lentivirus%20gene%20therapy%20&rank=1 (accessed on 17 April 2024).
- Long-Term Follow-up: Phase I/II Clinical Study to Evaluate the Safety and Efficacy of the Infusion of Autologous CD34+ Cells Transduced with a Lentiviral Vector Carrying the FANCA Gene in Patients with Fanconi Anaemia Subtype A: FANCOLEN-I, NCT04437771. Available online: https://clinicaltrials.gov/study/NCT04437771?cond=Fanconi%20Anemia&term=lentivirus%20gene%20therapy%20&rank=2 (accessed on 17 April 2024).
- Luis, A. The Old and the New: Prospects for Non-Integrating Lentiviral Vector Technology. Viruses 2020, 12, 1103. [Google Scholar] [CrossRef] [PubMed]
- Gurumoorthy, N.; Nordin, F.; Tye, G.J.; Wan Kamarul Zaman, W.S.; Ng, M.H. Non-Integrating Lentiviral Vectors in Clinical Applications: A Glance Through. Biomedicines 2022, 10, 107. [Google Scholar] [CrossRef] [PubMed]
- Milone, M.C.; O’Doherty, U. Clinical use of lentiviral vectors. Leukemia 2018, 32, 1529–1541. [Google Scholar] [CrossRef]
- Shaw, A.; Cornetta, K. Design and Potential of Non-Integrating Lentiviral Vectors. Biomedicines 2014, 2, 14–35. [Google Scholar] [CrossRef] [PubMed]
- Wienert, B.; Cromer, M.K. CRISPR nuclease off-target activity and mitigation strategies. Front. Genome Ed. 2022, 4, 1050507. [Google Scholar] [CrossRef] [PubMed]
- Hunt, J.M.T.; Samson, C.A.; Rand, A.D.; Sheppard, H.M. Unintended CRISPR-Cas9 editing outcomes: A review of the detection and prevalence of structural variants generated by gene-editing in human cells. Hum. Genet. 2023, 142, 705–720. [Google Scholar] [CrossRef] [PubMed]
- Kantor, A.; McClements, M.E.; MacLaren, R.E. CRISPR-Cas9 DNA Base-Editing and Prime-Editing. Int. J. Mol. Sci. 2020, 21, 6240. [Google Scholar] [CrossRef] [PubMed]
- Hryhorowicz, M.; Lipinski, D.; Zeyland, J. Evolution of CRISPR/Cas Systems for Precise Genome Editing. Int. J. Mol. Sci. 2023, 24, 4233. [Google Scholar] [CrossRef]
- Jeong, Y.K.; Song, B.; Bae, S. Current Status and Challenges of DNA Base Editing Tools. Mol. Ther. 2020, 28, 1938–1952. [Google Scholar] [CrossRef]
- Lu, C.; Kuang, J.; Shao, T.; Xie, S.; Li, M.; Zhu, L.; Zhu, L. Prime Editing: An All-Rounder for Genome Editing. Int. J. Mol. Sci. 2022, 23, 9862. [Google Scholar] [CrossRef] [PubMed]
- Mishra, R.; Joshi, R.K.; Zhao, K. Base editing in crops: Current advances, limitations and future implications. Plant Biotechnol. J. 2020, 18, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Porto, E.M.; Komor, A.C.; Slaymaker, I.M.; Yeo, G.W. Base editing: Advances and therapeutic opportunities. Nat. Rev. Drug Discov. 2020, 19, 839–859. [Google Scholar] [CrossRef] [PubMed]
- Collias, D.; Beisel, C.L. CRISPR technologies and the search for the PAM-free nuclease. Nat. Commun. 2021, 12, 555. [Google Scholar] [CrossRef] [PubMed]
- Aldag, P.; Welzel, F.; Jakob, L.; Schmidbauer, A.; Rutkauskas, M.; Fettes, F.; Grohmann, D.; Seidel, R. Probing the stability of the SpCas9-DNA complex after cleavage. Nucleic Acids Res. 2021, 49, 12411–12421. [Google Scholar] [CrossRef] [PubMed]
- Uchida, N.; Drysdale, C.M.; Nassehi, T.; Gamer, J.; Yapundich, M.; DiNicola, J.; Shibata, Y.; Hinds, M.; Gudmundsdottir, B.; Haro-Mora, J.J.; et al. Cas9 protein delivery non-integrating lentiviral vectors for gene correction in sickle cell disease. Mol. Ther. Methods Clin. Dev. 2021, 21, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Clinical Study on the Safety and Efficacy of a Single Intravenous Dose of CRISPR/Cas9-Edited Autologous CD34+ Hematopoietic Stem/Progenitor Cells (BRL-101) in the Treatment of Sickle Cell Disease, NCT06287099. Available online: https://clinicaltrials.gov/study/NCT06287099?cond=Sickle%20Cell%20Disease&intr=CRISPR%2FCas9&rank=1 (accessed on 17 April 2024).
- A Phase I/II Study of Nula-cel in Autologous CD34+ Hematopoietic Stem Cells to Convert HbS to HbA for Treating Severe Sickle Cell Disease, NCT04819841. Available online: https://clinicaltrials.gov/study/NCT04819841?cond=Sickle%20Cell%20Disease&intr=CRISPR%2FCas9&rank=4 (accessed on 17 April 2024).
- A Phase 1/2/3 Study to Evaluate the Safety and Efficacy of a Single Dose of Autologous CRISPR-Cas9 Modified CD34+ Human Hematopoietic Stem and Progenitor Cells (CTX001) in Subjects with Severe Sickle Cell Disease, NCT03745287. Available online: https://clinicaltrials.gov/study/NCT03745287?cond=Sickle%20Cell%20Disease&intr=CRISPR%2FCas9&rank=5 (accessed on 17 April 2024).
- A Phase 3 Study to Evaluate Efficacy and Safety of a Single Dose of Exa-cel in Subjects with Severe Sickle Cell Disease, βS/βC Genotype, NCT05951205. Available online: https://clinicaltrials.gov/study/NCT05951205?cond=Sickle%20Cell%20Disease&intr=CRISPR%2FCas9&rank=7 (accessed on 17 April 2024).
- Transplantation of CRISPRCas9 Corrected Hematopoietic Stem Cells (CRISPR_SCD001) in Patients with Severe Sickle Cell Disease, NCT04774536. Available online: https://clinicaltrials.gov/study/NCT04774536?cond=Sickle%20Cell%20Disease&intr=CRISPR%2FCas9&rank=8 (accessed on 17 April 2024).
- A Phase 3 Study to Evaluate the Safety and Efficacy of a Single Dose of CTX001 in Pediatric Subjects with Severe Sickle Cell Disease, NCT05329649. Available online: https://clinicaltrials.gov/study/NCT05329649?cond=Sickle%20Cell%20Disease&intr=CRISPR%2FCas9&rank=10 (accessed on 17 April 2024).
- A Phase 3b Study to Evaluate Efficacy and Safety of a Single Dose of Autologous CRISPR Cas9 Modified CD34+ Human Hematopoietic Stem and Progenitor Cells (CTX001) in Subjects with Transfusion-Dependent β-Thalassemia or Severe Sickle Cell Disease, NCT05477563. Available online: https://clinicaltrials.gov/study/NCT05477563?cond=Thalassemia&intr=CRISPR%2FCas9&rank=2 (accessed on 17 April 2024).
- A Long-term Follow-up Study of Subjects with β-Thalassemia or Sickle Cell Disease Treated with Autologous CRISPR-Cas9 Modified Hematopoietic Stem Cells (CTX001), NCT04208529. Available online: https://clinicaltrials.gov/study/NCT04208529?cond=Thalassemia&intr=CRISPR%2FCas9&rank=7 (accessed on 17 April 2024).
- A Phase 1/2/3 Study of the Safety and Efficacy of a Single Dose of Autologous CRISPR-Cas9 Modified CD34+ Human Hematopoietic Stem and Progenitor Cells (hHSPCs) in Subjects with Transfusion-Dependent β-Thalassemia, NCT03655678. Available online: https://clinicaltrials.gov/study/NCT03655678?cond=Thalassemia&intr=CRISPR%2FCas9&rank=1 (accessed on 17 April 2024).
- A Multicenter, Open Label Phase 1 Study to Evaluate the Safety and Efficacy of a Single Dose of Autologous CRISPR-Cas9 Modified CD34+ Human Hematopoietic Stem and Progenitor Cells (hHSPCs) in Subjects with Transfusion Dependent β-Thalassaemia, NCT04925206. Available online: https://clinicaltrials.gov/study/NCT04925206?cond=Thalassemia&intr=CRISPR%2FCas9&rank=3 (accessed on 17 April 2024).
- A Phase 1/2 Clinical Study to Evaluate the Safety and Efficacy of Single Dose Intravenous Infusion of BRL-101 in Subjects with Transfusion-Dependent β-Thalassemia, NCT05577312. Available online: https://clinicaltrials.gov/study/NCT05577312?cond=Thalassemia&intr=CRISPR%2FCas9&rank=4 (accessed on 17 April 2024).
- A Phase 3 Study to Evaluate the Safety and Efficacy of a Single Dose of CTX001 in Pediatric Subjects with Transfusion-Dependent β-Thalassemia, NCT05356195. Available online: https://clinicaltrials.gov/study/NCT05356195?cond=Thalassemia&intr=CRISPR%2FCas9&rank=5 (accessed on 17 April 2024).
- A Safety and Efficacy Study of a Single Center, Open-Label, Single Arm About the Gene Correction of HBB in Patient-Specific iHSCs Using CRISPR/Cas9 That Intervent Subjests with β-Thalassemia Mutations, NCT03728322. Available online: https://clinicaltrials.gov/study/NCT03728322?cond=Thalassemia&intr=CRISPR%2FCas9&rank=6 (accessed on 17 April 2024).
- Frangoul, H.; Altshuler, D.; Cappellini, M.D.; Chen, Y.S.; Domm, J.; Eustace, B.K.; Foell, J.; de la Fuente, J.; Grupp, S.; Handgretinger, R.; et al. CRISPR-Cas9 Gene Editing for Sickle Cell Disease and beta-Thalassemia. N. Engl. J. Med. 2021, 384, 252–260. [Google Scholar] [CrossRef]
- Newby, G.A.; Yen, J.S.; Woodard, K.J.; Mayuranathan, T.; Lazzarotto, C.R.; Li, Y.; Sheppard-Tillman, H.; Porter, S.N.; Yao, Y.; Mayberry, K.; et al. Base editing of haematopoietic stem cells rescues sickle cell disease in mice. Nature 2021, 595, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Georgakopoulou, A.; Newby, G.A.; Chen, P.J.; Everette, K.A.; Paschoudi, K.; Vlachaki, E.; Gil, S.; Anderson, A.K.; Koob, T.; et al. In vivo HSC prime editing rescues sickle cell disease in a mouse model. Blood 2023, 141, 2085–2099. [Google Scholar] [CrossRef]
- FDA. FDA Approves First Gene Therapies to Treat Patients with Sickle Cell Disease. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-gene-therapies-treat-patients-sickle-cell-disease (accessed on 14 May 2024).
- Mohammadian Gol, T.; Zahedipour, F.; Trosien, P.; Urena-Bailen, G.; Kim, M.; Antony, J.S.; Mezger, M. Gene therapy in pediatrics—Clinical studies and approved drugs (as of 2023). Life Sci. 2024, 348, 122685. [Google Scholar] [CrossRef]
- ZYNTEGLO. ZYNTEGLO™ Mechanism of Action. Available online: https://www.zynteglohcp.com/mechanism-of-action (accessed on 14 May 2024).
- FDA. Long Term Follow-up After Administration of Human Gene Therapy Products. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/long-term-follow-after-administration-human-gene-therapy-products (accessed on 14 May 2024).
- Lee, N.K.; Chang, J.W. Manufacturing Cell and Gene Therapies: Challenges in Clinical Translation. Ann. Lab. Med. 2024, 44, 314–323. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Clinical Trial ID | Intervention/Treatment | Ref. |
|---|---|---|---|
| Sickle Cell Disease (SCD) | NCT02186418 | CD34+ cells transduced with gamma-globin lentiviral vector. | [32] |
| NCT03282656 | Single infusion of autologous bone marrow derived CD34+ HSC cells transduced with the lentiviral vector containing a short-hairpin RNA targeting BCL11a. | [33] | |
| NCT05353647 | Autologous transplantation of CD34+ HSC cells transduced with the lentiviral vector containing a shRNA targeting BCL11a. | [34] | |
| NCT02247843 | Autologous transplantation of peripheral blood CD34+ cells transduced ex vivo by the Lenti/G-βAS3-FB lentiviral vector to express an anti-sickling (βAS3) gene. | [35] | |
| NCT03964792 | Transplantation of an autologous CD34+ enriched cell fraction that contains CD34+ cells transduced ex vivo with the GLOBE1 lentiviral vector expressing the βAS3 globin gene (GLOBE1 βAS3 modified autologous CD34+ cells). | [36] | |
| SCD and β-Thalassemia | NCT02151526 | Administration of LentiGlobin BB305 drug product to participants with either transfusion dependent β-thalassemia (TDT) or sickle cell disease (SCD). | [37] |
| β-Thalassemia | NCT03276455 | Autologous transplantation of HSCs transduced with lentiviral vector encoding for beta-globin gene. | [38] |
| NCT02453477 | Autologous transplantation of HSCs genetically modified with GLOBE lentiviral vector encoding for the human beta-globin gene. | [39] | |
| NCT06219239 | Autologous transplantation of HSCs transduced with lentiviral vector encoding βA-T87Q-globin gene. | [40] | |
| NCT05745532 | Autologous transplantation of HSCs transduced with LentiHBBT87Q system to restore β-globin expression. | [41] | |
| NCT06280378 | Autologous transplantation of CD34+ stem cells transduced ex vivo with a lentiviral vector encoding βA-T87Q-globin. | [42] | |
| NCT01639690 | Autologous transplantation of CD34+ cells transduced with TNS9.3.55 lentiviral vector encoding the normal human ß-globin gene. | [43] | |
| NCT05762510 | Autologous transplantation of CD34+ HSCs transduced with LentiRed lentiviral vector. | [44] | |
| NCT05757245 | Autologous HSCT using GMCN-508A drug product (autologous CD34+ HSCs transduced with GMCN-508A lentiviral vector encoding the human α-globin gene). | [45] | |
| NCT05015920 | Transplantation of autologous CD34+ stem cells transduced with a lentiviral vector encoding βA-T87Q-globin. | [46] | |
| Fanconi Anemia (FA) | NCT01331018 | Transplantation of autologous patient blood stem cells that have been corrected in the laboratory by introduction of the normal FANCA gene. | [47] |
| NCT04437771 | Transplantation of autologous CD34+ cells transduced with lentiviral vector carrying the FANCA gene. | [48] |
| Aspect | Integrating Lentiviral Vectors (ILVs) | Non-Integrating Lentiviral Vectors (NILVs) |
|---|---|---|
| Integration | Integrates the transgene into the host genome [22,50] | Does not integrate the transgene into the host genome [52] Expresses the transgene from episomal DNA in non-dividing cells or transiently in dividing cells [49] |
| Expression | Stable integration of the transgene into the host genome [49,50] | Enables transient expression or sustained episomal expression [50] |
| Safety | Higher risk of insertional mutagenesis and malignant transformation [50] | Reduced risk of insertional mutagenesis and malignant transformation [50] |
| Applications | Gene therapy for long-term gene expression [50,52] Recombinant protein production [50] Vaccination [50] Cell imaging [50] | Gene therapy for mutation correction [50,52] Cytotoxic cancer therapies [49,50] Cellular differentiation [49] Vaccination [49,52] Immunotherapies [49,50] |
| Limitations | Insertional mutagenesis [24] Oncogenic potential [24] | Residual integration risks [50] Transient expression is not suitable for all applications |
| Tools | Components | Applicability | Advantages | Drawbacks | Ref. |
|---|---|---|---|---|---|
| CRISPR/Cas9 | Cas9, sgRNA, and donor DNA (for HDR) | Point mutations Large DNA insertions and deletions Gene knock-out | Versatility in gene insertion, deletion, and modification | DSB induction Off-target effects Low efficiency for HDR | [56] |
| CBE | dCa9-cytosine deaminase, and sgRNA | Transitions mutations (C→T, G→A, A→G, T→C) | Induction of SSBs | Requires precise positioning of editing window Off-target DNA and RNA editing Bystander edits Only capable of four transition mutations | [56,57] |
| ABE | dCas9-adenine deaminase and sgRNA | ||||
| PE | dCas9(H840A)-M-MLV RT and pegRNA | Point mutations Small deletions and insertions | Induction of SSBs Allows for all precise modifications | Genomic scope constraints Variable efficiency in different cell types Delivery challenges due to large size of PE | [56,58] |
| Disease | Clinical Trial ID | Intervention/Treatment | Ref |
|---|---|---|---|
| Sickle Cell Disease (SCD) | NCT06287099 | Autologous CRISPR/Cas9 modified CD34+ hHSPCs (BRL-101) | [64] |
| NCT04819841 | Gene correction in autologous CD34+ HSCs (HbS to HbA) to treat severe SCD | [65] | |
| NCT03745287 | Autologous CRISPR/Cas9 modified CD34+ hHSPCs using CTX001 | [66] | |
| NCT05951205 | Single dose of CTX001 in subjects with severe SCD with βS/βC genotype | [67] | |
| NCT04774536 | Transplantation of CRISPR/Cas9 corrected HSCs (CRISPR_SCD001) in patients with severe SCD | [68] | |
| NCT05329649 | Administration of a single dose of CTX001 in pediatric subjects with severe SCD | [69] | |
| SCD and β-Thalassemia | NCT05477563 | Single dose of autologous CRISPR/Cas9 modified CD34+ hHSPCs (CTX001) in subjects with transfusion-dependent β-Thalassemia or severe SCD | [70] |
| NCT04208529 | A long-term follow-up study of subjects with β-thalassemia or SCD treated with autologous CRISPR/Cas9 modified HSCs (CTX001) | [71] | |
| β-Thalassemia | NCT03655678 | Autologous CRISPR/Cas9 modified CD34+ hHSPCs using CTX001 in subjects with transfusion-dependent β-Thalassemia | [72] |
| NCT04925206 | Autologous CRISPR/Cas9 modified CD34+ hHSPCs using ET-01 in subjects with transfusion-dependent β-Thalassemia | [73] | |
| NCT05577312 | Autologous CRISPR/Cas9 modified CD34+ hHSPCs (BRL-101) | [74] | |
| NCT05356195 | Autologous CRISPR/Cas9 modified CD34+ hHSPCs (CTX001) in pediatric subjects with transfusion-dependent β-Thalassemia | [75] | |
| NCT03728322 | Gene correction of HBB in patient-specific iHSCs using CRISPR/Cas9 | [76] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vale, M.; Prochazka, J.; Sedlacek, R. Towards a Cure for Diamond–Blackfan Anemia: Views on Gene Therapy. Cells 2024, 13, 920. https://doi.org/10.3390/cells13110920
Vale M, Prochazka J, Sedlacek R. Towards a Cure for Diamond–Blackfan Anemia: Views on Gene Therapy. Cells. 2024; 13(11):920. https://doi.org/10.3390/cells13110920
Chicago/Turabian StyleVale, Matilde, Jan Prochazka, and Radislav Sedlacek. 2024. "Towards a Cure for Diamond–Blackfan Anemia: Views on Gene Therapy" Cells 13, no. 11: 920. https://doi.org/10.3390/cells13110920
APA StyleVale, M., Prochazka, J., & Sedlacek, R. (2024). Towards a Cure for Diamond–Blackfan Anemia: Views on Gene Therapy. Cells, 13(11), 920. https://doi.org/10.3390/cells13110920