
Apoptosis, a Metabolic “Head-to-Head” between Tumor and T Cells: Implications for Immunotherapy



Abstract
1. Introduction
2. Crosstalk between Apoptotic and Immune Cells Affects the Fate of Cancer
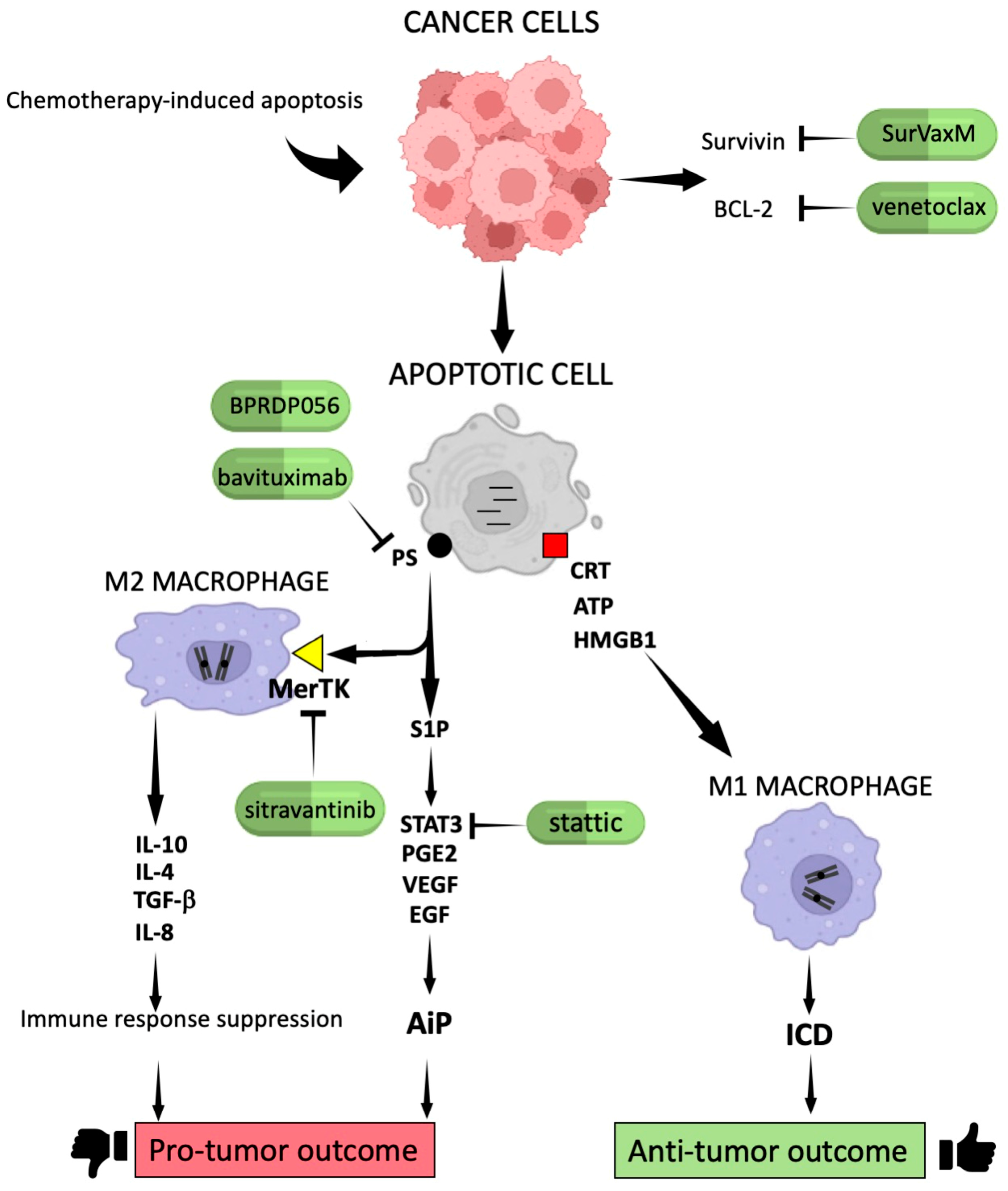
2.1. Molecular Signals Generated by Apoptotic Cells in Cancer
2.2. Mechanisms of Apoptosis Escape Devised by Cancer Cells
3. A More In-Depth Insight into the Role of Apoptosis in the Immune Landscape of the TME
3.1. Exploring Key Subsets in the TME
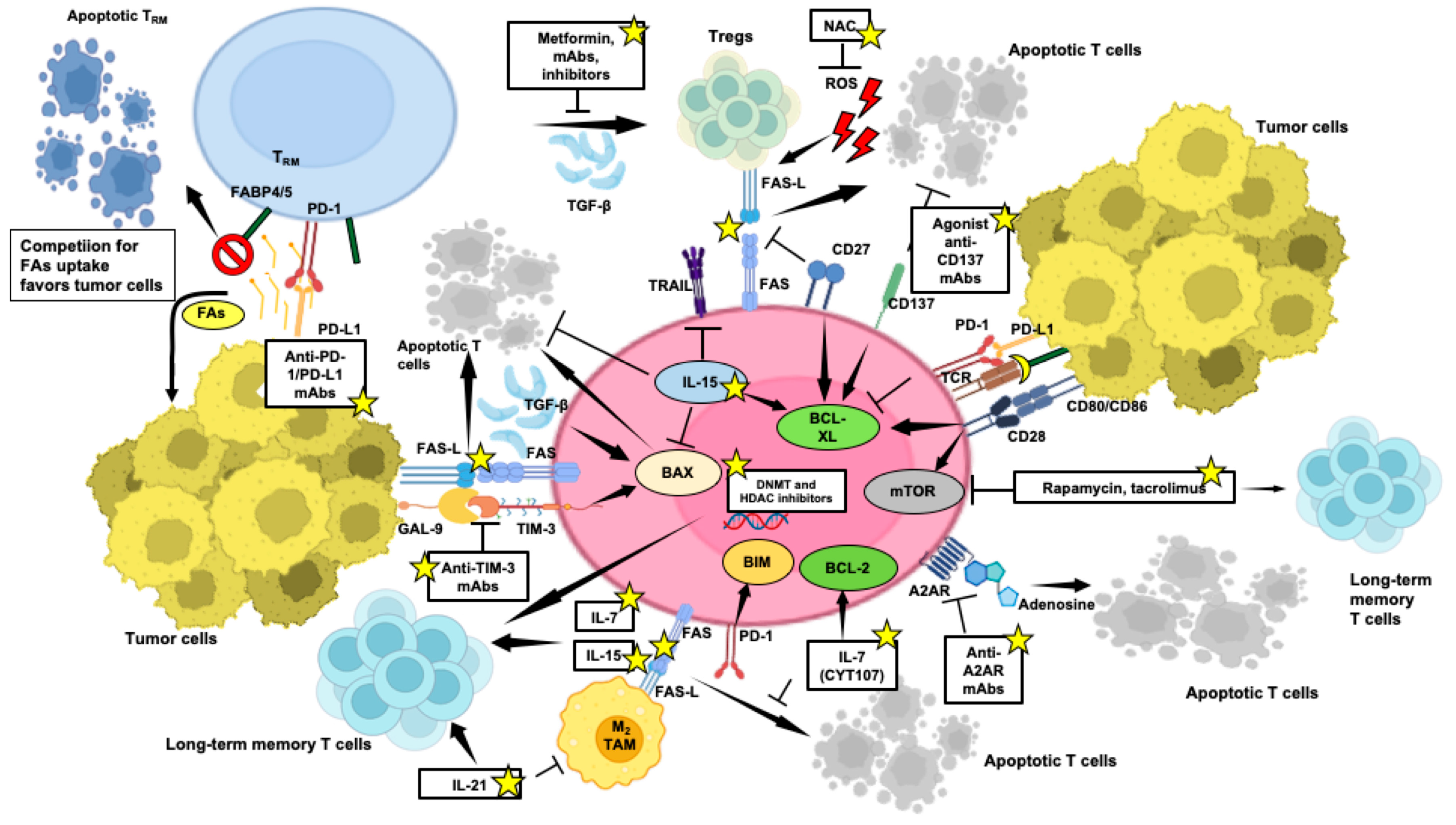
3.2. Role of Apoptosis in the Homeostatic Control of Anti-Tumor T-Cell-Mediated Responses
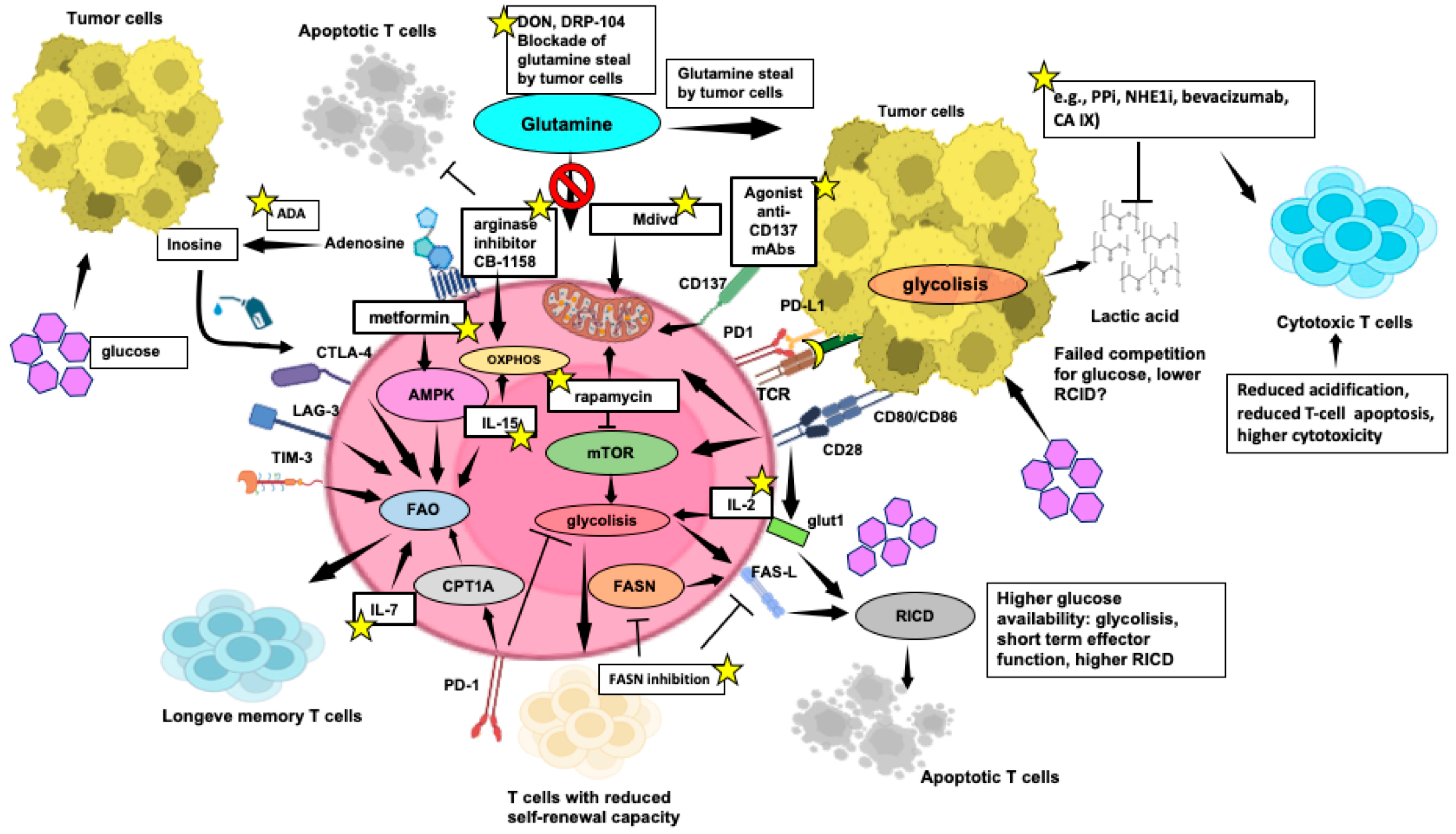
3.3. Nutrient Competition between Cytotoxic T Cells and Tumour Cells, and Impact on Anti-Tumor T-Cell Functions
4. Apoptosis Control to Bypass Immunotherapy Resistance
4.1. Unravelling Immunotherapy Challenges: Tumour-Infiltrating T-Cell Apoptosis, Implications for Treatment Resistance and Beneficial Strategies
4.2. Enhancing Anti-Tumor Response through Metabolic Reprogramming of T Cells
5. Interplay between Apoptosis and Metabolism
5.1. Metabolites Involved in the Anticancer Response: Escape from Apoptosis through Metabolic Renewal
5.2. Extracellular Metabolites and Their Relation with the TME
6. Conclusions and Future Perspective for Cancer Therapy
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN Estimates of incidence and mortality worldwide for 36 Cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Shukla, N.; Singh, S.S.; Kushwaha, S.; Shrivastava, R. Mechanism of interaction between autophagy and apoptosis in cancer. Apoptosis 2021, 26, 512–533. [Google Scholar] [CrossRef] [PubMed]
- Vitale, I.; Pietrocola, F.; Guilbaud, E.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostini, M.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; et al. Apoptotic cell death in disease-current understanding of the NCCD 2023. Cell Death Differ. 2023, 30, 1097–1154. [Google Scholar] [CrossRef] [PubMed]
- Alderson, M.R.; Tough, T.W.; Davis-Smith, T.; Braddy, S.; Falk, B.; Schooley, K.A.; Goodwin, R.G.; Smith, C.A.; Ramsdell, F.; Lynch, D.H. Fas Ligand mediates activation-induced cell death in human T lymphocytes. J. Exp. Med. 1995, 181, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Petit, P.F.; Van den Eynde, B.J. Apoptosis of tumor-infiltrating T lymphocytes: A new immune checkpoint mechanism. Cancer Immunol. Immunother. 2019, 68, 835–847. [Google Scholar] [CrossRef]
- Boselli, D.; Losana, G.; Bernabei, P.; Bosisio, D.; Drysdale, P.; Kiessling, R.; Gaston, J.S.; Lammas, D.; Casanova, J.L.; Kumararatne, D.S.; et al. IFN-gamma regulates Fas ligand expression in human CD4+ T lymphocytes and controls their anti-mycobacterial cytotoxic functions. Eur. J. Immunol. 2007, 37, 2196–2204. [Google Scholar] [CrossRef] [PubMed]
- Gadiyar, V.; Lahey, K.C.; Calianese, D.; Devoe, C.; Mehta, D.; Bono, K.; Desind, S.; Davra, V.; Birge, R.B. Cell death in the tumor microenvironment: Implications for cancer immunotherapy. Cells 2020, 9, 2207. [Google Scholar] [CrossRef] [PubMed]
- Dehne, N.; Mora, J.; Namgaladze, D.; Weigert, A.; Brüne, B. Cancer cell and macrophage cross-talk in the tumor microenvironment. Curr. Opin. Pharmacol. 2017, 35, 12–19. [Google Scholar] [CrossRef]
- Horton, B.L.; Williams, J.B.; Cabanov, A.; Spranger, S.; Gajewski, T.F. Intra-tumoral CD8+ T-cell apoptosis is a major component of T-cell dysfunction and impedes antitumor immunity. Cancer Immunol. Res. 2018, 6, 14–24. [Google Scholar] [CrossRef]
- Pan, Y.; Yu, Y.; Wang, X.; Zhang, T. Tumor-associated macrophages in tumor immunity. Front. Immunol. 2020, 11, 583084. [Google Scholar] [CrossRef]
- Frey, B.; Gaipl, U.S. The immune functions of phosphatidylserine in membranes of dying cells and microvesicles. Semin. Immunopathol. 2011, 33, 497–516. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Zhang, H.; Wang, Z.; Zhang, X.; Dai, Z.; Zhang, J.; Luo, P.; Zhang, L.; Hu, J.; Liu, Z.; et al. JMJD8 is an M2 macrophage biomarker, and it associates with DNA damage repair to facilitate stemness maintenance, chemoresistance, and immunosuppression in pan-cancer. Front. Immunol. 2022, 13, 875786. [Google Scholar] [CrossRef]
- Tanzer, M.C.; Frauenstein, A.; Stafford, C.A.; Phulphagar, K.; Mann, M.; Meissner, F. Quantitative and dynamic catalogs of proteins released during apoptotic and necroptotic cell death. Cell Rep. 2020, 30, 1260–1270. [Google Scholar] [CrossRef] [PubMed]
- Schimek, V.; Strasser, K.; Beer, A.; Göber, S.; Walterskirchen, N.; Brostjan, C.; Müller, C.; Bachleitner-Hofmann, T.; Bergmann, M.; Dolznig, H.; et al. Tumour cell apoptosis modulates the colorectal cancer immune microenvironment via interleukin-8-dependent neutrophil recruitment. Cell Death Dis. 2022, 13, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Gude, D.R.; Alvarez, S.E.; Paugh, S.W.; Mitra, P.; Yu, J.; Griffiths, R.; Barbour, S.E.; Milstien, S.; Spiegel, S. Apoptosis induces expression of sphingosine kinase 1 to release sphingosine-1-phosphate as a “come-and-get-me” signal. FASEB J. 2008, 22, 2629–2638. [Google Scholar] [CrossRef] [PubMed]
- Osman, R.; Tacnet-Delorme, P.; Kleman, J.P.; Millet, A.; Frachet, P. Calreticulin release at an early stage of death modulates the clearance by macrophages of apoptotic cells. Front. Immunol. 2017, 8, 1034–1046. [Google Scholar] [CrossRef]
- Fredly, H.; Ersvær, E.; Gjertsen, B.T.; Bruserud, O. Immunogenic apoptosis in human acute myeloid leukemia (AML): Primary human AML cells expose calreticulin and release heat shock protein (HSP) 70 and HSP90 during apoptosis. Oncol. Rep. 2011, 25, 1549–1556. [Google Scholar] [CrossRef] [PubMed]
- Krysko, D.V.; Garg, A.D.; Kaczmarek, A.; Krysko, O.; Agostinis, P.; Vandenabeele, P. Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Cancer 2012, 12, 860–875. [Google Scholar] [CrossRef]
- Martins, I.; Wang, Y.; Michaud, M.; Ma, Y.; Sukkurwala, A.Q.; Shen, S.; Kepp, O.; Métivier, D.; Galluzzi, L.; Perfettini, J.L.; et al. Molecular mechanisms of ATP secretion during immunogenic cell death. Cell Death Differ. 2014, 21, 79–91. [Google Scholar] [CrossRef]
- Smith, H.G.; Jamal, K.; Dayal, J.H.; Tenev, T.; Kyula-Currie, J.; Guppy, N.; Gazinska, P.; Roulstone, V.; Liccardi, G.; Davies, E.; et al. RIPK1-mediated immunogenic cell death promotes anti-tumour immunity against soft-tissue sarcoma. EMBO Mol. Med. 2020, 12, e10979. [Google Scholar] [CrossRef]
- Eek Mariampillai, A.; Hauge, S.; Kongsrud, K.; Syljuåsen, R.G. Immunogenic cell death after combined treatment with radiation and ATR inhibitors is dually regulated by apoptotic caspases. Front. Immunol. 2023, 14, 1138920. [Google Scholar] [CrossRef] [PubMed]
- Jafari, S.; Lavasanifar, A.; Hejazi, M.S.; Maleki-Dizaji, N.; Mesgari, M.; Molavi, O. STAT3 inhibitory stattic enhances immunogenic cell death induced by chemotherapy in cancer cells. Daru 2020, 28, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hu, R.; Song, Z.; Zhao, H.; Pan, Z.; Feng, Y.; Yu, Y.; Han, Q.; Zhang, J. Sorafenib combined with STAT3 knockdown triggers ER stress-induced HCC apoptosis and cGAS-STING-mediated anti-tumor immunity. Cancer Lett. 2022, 547, 215880–215894. [Google Scholar] [CrossRef]
- Matsushita, M.; Kashiwazaki, S.; Kamiko, S.; Kobori, M.; Osada, M.; Kunieda, H.; Hirao, M.; Ichikawa, D.; Hattori, Y. Immunomodulatory effect of proteasome inhibitors via the induction of immunogenic cell death in myeloma cells. Pharmaceuticals 2023, 16, 1367. [Google Scholar] [CrossRef] [PubMed]
- Spyridopoulou, K.; Tryfonopoulou, E.; Aindelis, G.; Ypsilantis, P.; Sarafidis, C.; Kalogirou, O.; Chlichlia, K. Biogenic selenium nanoparticles produced by Lactobacillus casei ATCC 393 inhibit colon cancer cell growth in vitro and in vivo. Nanoscale Adv. 2021, 3, 2516–2528. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, J.; Chen, Z.; Luo, J.; Guo, W.; Sun, L.; Lin, L. Targeting M2-like tumor-associated macrophages is a potential therapeutic approach to overcome antitumor drug resistance. NPJ Precis Oncol. 2024, 8, 31–49. [Google Scholar] [CrossRef]
- Poma, P.; Labbozzetta, M.; Notarbartolo, M. Patterns of innate or acquired resistance to anticancer drugs: Our experience to evercome it. Crit. Rev. Oncog. 2021, 26, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Steven, A.; Seliger, B. The role of immune escape and immune cell infiltration in breast cancer. Breast Care 2018, 13, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Ranjan, K.; Waghela, B.N.; Vaidya, F.U.; Pathak, C. Cell-penetrable peptide-conjugated FADD induces apoptosis and regulates inflammatory signaling in cancer cells. Int. J. Mol. Sci. 2020, 21, 6890. [Google Scholar] [CrossRef]
- Valentini, E.; D’Aguanno, S.; Di Martile, M.; Montesano, C.; Ferraresi, V.; Patsilinakos, A.; Sabatino, M.; Antonini, L.; Chiacchiarini, M.; Valente, S.; et al. Targeting the anti-apoptotic Bcl-2 family proteins: Machine learning virtual screening and biological evaluation of new small molecules. Theranostics 2022, 12, 2427–2444. [Google Scholar] [CrossRef]
- Chong, S.J.F.; Zhu, F.; Dashevsky, O.; Mizuno, R.; Lai, J.X.; Hackett, L.; Ryan, C.E.; Collins, M.C.; Iorgulescu, J.B.; Guièze, R.; et al. Hyperphosphorylation of BCL-2 family proteins underlies functional resistance to venetoclax in lymphoid malignancies. J. Clin. Investig. 2023, 133, e170169. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Liang, S.Q.; Yang, Z.; Yang, H.; Bruggmann, R.; Oberhaensli, S.; Berezowska, S.; Marti, T.M.; Sean, R.R.; Hall, S.R.R.; et al. Malignant pleural mesothelioma co-opts BCL-XL and autophagy to escape apoptosis. Cell Death Dis. 2021, 12, 406–419. [Google Scholar] [CrossRef]
- Bodaar, K.; Yamagata, N.; Barthe, A.; Landrigan, J.; Chonghaile, T.N.; Burns, M.; Stevenson, K.E.; Devidas, M.; Loh, M.L.; Hunger, S.P.; et al. JAK3 mutations and mitochondrial apoptosis resistance in T-cell acute lymphoblastic leukemia. Leukemia 2022, 36, 1499–1507. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Zhu, J.; Zhong, Y.; Geng, R.; Ji, Y.; Guan, Q.; Hong, C.; Wei, Y.; Min, N.; Qi, A.; et al. PIK3CA mutation confers resistance to chemotherapy in triple-negative breast cancer by inhibiting apoptosis and activating the PI3K/AKT/mTOR signaling pathway. Ann. Transl. Med. 2021, 9, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Hao, Q.; Lu, H. Mutant p53 in cancer therapy-the barrier or the path. J. Mol. Cell Biol. 2019, 11, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Roszkowska, K.A.; Piecuch, A.; Sady, M.; Gajewski, Z.; Flis, S. Gain of function (GOF) mutant p53 in cancer-current therapeutic approaches. Int. J. Mol. Sci. 2022, 23, 13287. [Google Scholar] [CrossRef] [PubMed]
- Peuget, S.; Zhou, X.; Selivanova, G. Translating p53-based therapies for cancer into the clinic. Nat. Rev. Cancer 2024, 24, 192–215. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Yu, M.; Xiao, W.; Celis, E.; Cui, Y. Local activation of p53 in the tumor microenvironment overcomes immune suppression and enhances antitumor immunity. Cancer Res. 2017, 77, 2292–2305. [Google Scholar] [CrossRef]
- Chauhan, S.; Jaiswal, S.; Jakhmola, V.; Singh, B.; Bhattacharya, S.; Garg, M.; Sengupta, S. Potential role of p53 deregulation in modulating immune responses in human malignancies: A paradigm to develop immunotherapy. Cancer Lett. 2024, 588, 216766–216780. [Google Scholar] [CrossRef]
- Ozyerli-Goknar, E.; Bagci-Onder, T. Epigenetic deregulation of apoptosis in cancers. Cancers 2021, 13, 3210. [Google Scholar] [CrossRef]
- Ruscito, I.; Gasparri, M.L.; De Marco, M.P.; Costanzi, F.; Besharat, A.R.; Papadia, A.; Kuehn, T.; Gentilini, O.D.; Bellati, F.; Caserta, D. The clinical and pathological profile of BRCA1 gene methylated breast cancer women: A meta-analysis. Cancers 2021, 13, 1391. [Google Scholar] [CrossRef] [PubMed]
- Qi, M.; Xiong, X. Promoter hypermethylation of RARβ2, DAPK, hMLH1, p14, and p15 is associated with progression of breast cancer: A PRISMA-compliant meta-analysis. Medicine 2018, 97, e13666. [Google Scholar] [CrossRef]
- Zhang, S.; Zhou, Y.F.; Cao, J.; Burley, S.K.; Wang, H.Y.; Zheng, X.F.S. mTORC1 Promotes ARID1A degradation and oncogenic chromatin remodeling in hepatocellular carcinoma. Cancer Res. 2021, 81, 5652–5665. [Google Scholar] [CrossRef] [PubMed]
- Oyama, Y.; Shigeta, S.; Tokunaga, H.; Tsuji, K.; Ishibashi, M.; Shibuya, Y.; Shimada, M.; Yasuda, J.; Yaegashi, N. CHD4 regulates platinum sensitivity through MDR1 expression in ovarian cancer: A potential role of CHD4 inhibition as a combination therapy with platinum agents. PLoS ONE 2021, 16, e0251079. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Liu, C.; Wang, L.; Sun, Y.; Jiang, Y.; Hao, T. Histone methyltransferase NSD2 regulates apoptosis and chemosensitivity in osteosarcoma. Cell Death Dis. 2019, 10, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Shi, Z.; Qian, Y.; Jiang, C.; Liu, W.; Liu, B.; Jiang, B. HDAC2- and EZH2-Mediated Histone Modifications induce PDK1 expression through miR-148a downregulation in breast cancer progression and adriamycin resistance. Cancers 2022, 14, 3600. [Google Scholar] [CrossRef] [PubMed]
- Pajares, M.J.; Alemany-Cosme, E.; Goñi, S.; Bandres, E.; Palanca-Ballester, C.; Sandoval, J. Epigenetic regulation of microRNAs in cancer: Shortening the distance from bench to bedside. Int. J. Mol. Sci. 2021, 22, 7350. [Google Scholar] [CrossRef]
- Shah, V.; Shah, J. Recent trends in targeting miRNAs for cancer therapy. J. Pharm. Pharmacol. 2020, 72, 1732–1749. [Google Scholar] [CrossRef] [PubMed]
- Lopes, R.B.; Gangeswaran, R.; McNeish, I.A.; Wang, Y.; Lemoine, N.R. Expression of the IAP protein family is dysregulated in pancreatic cancer cells and is important for resistance to chemotherapy. Int. J. Cancer 2007, 120, 2344–2352. [Google Scholar] [CrossRef]
- Frassanito, M.A.; Saltarella, I.; Vinella, A.; Muzio, L.L.; Pannone, G.; Fumarulo, R.; Vacca, A.; Mariggiò, M.A. Survivin overexpression in head and neck squamous cell carcinomas as a new therapeutic target (Review). Oncol. Rep. 2019, 41, 2615–2624. [Google Scholar] [CrossRef]
- Checcoli, A.; Pol, J.G.; Naldi, A.; Noel, V.; Barillot, E.; Kroemer, G.; Thieffry, D.; Calzone, L.; Stoll, G. Dynamical boolean modeling of immunogenic cell death. Front. Physiol. 2020, 11, 590479. [Google Scholar] [CrossRef] [PubMed]
- Ahluwalia, M.S.; Reardon, D.A.; Abad, A.P.; Curry, W.T.; Wong, E.T.; Figel, S.A.; Mechtler, L.L.; Peereboom, D.M.; Hutson, A.D.; Withers, H.G.; et al. Phase IIa study of SurVaxM plus adjuvant temozolomide for newly diagnosed glioblastoma. J. Clin. Oncol. 2022, 41, 1453–1465. [Google Scholar] [CrossRef] [PubMed]
- Burkholz, S.R.; Herst, C.V.; Carback, R.T.; Harris, P.E.; Rubsamen, R.M. Survivin (BIRC5) Peptide vaccine in the 4T1 murine mammary tumor model: A potential neoadjuvant T cell immunotherapy for triple negative breast cancer: A preliminary study. Vaccines 2023, 11, 644. [Google Scholar] [CrossRef] [PubMed]
- Tseng, J.C.; Yang, J.X.; Lee, C.Y.; Lo, C.F.; Liu, Y.L.; Zhang, M.M.; Huang, L.R.; Liu, K.J.; Wang, C.C.; Huang, C.F.; et al. Induction of immune responses and phosphatidylserine exposure by TLR9 activation results in a cooperative antitumor effect with a phosphatidylserine-targeting prodrug. Int. J. Biol. Sci. 2023, 19, 2648–2662. [Google Scholar] [CrossRef] [PubMed]
- Budhu, S.; Giese, R.; Gupta, A.; Fitzgerald, K.; Zappasodi, R.; Schad, S.; Hirschhorn, D.; Campesato, L.F.; De Henau, O.; Gigoux, M.; et al. Targeting phosphatidylserine enhances the anti-tumor response to tumor-directed radiation therapy in a preclinical model of melanoma. Cell Rep. 2021, 34, 108620. [Google Scholar] [CrossRef] [PubMed]
- Mokdad, A.A.; Zhu, H.; Beg, M.S.; Arriaga, Y.; Dowell, J.E.; Singal, A.G.; Yopp, A.C. Efficacy and safety of bavituximab in combination with sorafenib in advanced hepatocellular carcinoma: A single-arm, open-label, phase II clinical trial. Target Oncol. 2019, 14, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Fei, M.; Zhang, G.; Liang, W.C.; Lin, W.; Wu, Y.; Piskol, R.; Ridgway, J.; McNamara, E.; Huang, H.; et al. Blockade of the phagocytic receptor MerTK on tumor-associated macrophages enhances P2X7R-dependent STING activation by tumor-derived cGAMP. Immunity 2020, 52, 357–373. [Google Scholar] [CrossRef] [PubMed]
- Karam, J.A.; Msaouel, P.; Haymaker, C.L.; Matin, S.F.; Campbell, M.T.; Zurita, A.J.; Shah, A.Y.; Wistuba, I.I.; Marmonti, E.; Duose, D.Y.; et al. Phase II trial of neoadjuvant sitravatinib plus nivolumab in patients undergoing nephrectomy for locally advanced clear cell renal cell carcinoma. Nat. Commun. 2023, 14, 2684–2697. [Google Scholar] [CrossRef] [PubMed]
- Borghaei, H.; de Marinis, F.; Dumoulin, D.; Reynolds, C.; Theelen, W.S.M.E.; Percent, I.; Gutierrez Calderon, V.; Johnson, M.L.; Madroszyk-Flandin, A.; Garon, E.B.; et al. SAPPHIRE: Phase III study of sitravatinib plus nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. Ann. Oncol. 2024, 35, 66–76. [Google Scholar] [CrossRef]
- Myers Chen, K.V.; de Groot, A.E.; Mendez, S.A.; Mallin, M.M.; Amend, S.R.; Pienta, K.J. Targeting MerTK decreases efferocytosis and increases anti-tumor immune infiltrate in prostate cancer. Med. Oncol. 2023, 40, 284–295. [Google Scholar] [CrossRef]
- Li, G.; Liu, D.; Kimchi, E.T.; Kaifi, J.T.; Qi, X.; Manjunath, Y.; Liu, X.; Deering, T.; Avella, D.M.; Fox, T.; et al. Nanoliposome C6-ceramide increases the anti-tumor immune response and slows growth of liver tumors in mice. Gastroenterology 2018, 154, 1024–1036. [Google Scholar] [CrossRef] [PubMed]
- Vadevoo, S.M.P.; Gunassekaran, G.R.; Yoo, J.D.; Kwon, T.H.; Hur, K.; Chae, S.; Lee, B. Epigenetic therapy reprograms M2-type tumor-associated macrophages into an M1-like phenotype by upregulating miR-7083-5p. Front. Immunol. 2022, 13, 976196. [Google Scholar] [CrossRef] [PubMed]
- Allavena, P.; Digifico, E.; Belgiovine, C. Macrophages and cancer stem cells: A malevolent alliance. Mol. Med. 2021, 27, 121–134. [Google Scholar] [CrossRef]
- Wang, H.C.; Haung, L.Y.; Wang, C.J.; Chao, Y.J.; Hou, Y.C.; Yen, C.J.; Shan, Y.S. Tumor-associated macrophages promote resistance of hepatocellular carcinoma cells against sorafenib by activating CXCR2 signaling. J. Biomed. Sci. 2022, 29, 99–102. [Google Scholar] [CrossRef] [PubMed]
- de Carvalho, T.G.; Lara, P.; Jorquera-Cordero, C.; Aragão, C.F.S.; de Santana Oliveira, A.; Garcia, V.B.; de Paiva Souza, S.V.; Schomann, T.; Soares, L.A.L.; da Matta Guedes, P.M.; et al. Inhibition of murine colorectal cancer metastasis by targeting M2-TAM through STAT3/NF-kB/AKT signaling using macrophage 1-derived extracellular vesicles loaded with oxaliplatin, retinoic acid, and Libidibia ferrea. Biomed. Pharmacother. 2023, 168, 115663. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Bretz, A.C.; Gravemeyer, J.; Spassova, I.; Muminova, S.; Gambichler, T.; Sriram, A.; Ferrone, S.; Becker, J.C. The HDAC inhibitor domatinostat promotes cell-cycle arrest, induces apoptosis, and increases immunogenicity of merkel cell carcinoma cells. J. Investig. Dermatol. 2021, 141, 903–912. [Google Scholar] [CrossRef]
- Pérez-Romero, K.; Rodríguez, R.M.; Amedei, A.; Barceló-Coblijn, G.; Lopez, D.H. Immune landscape in tumor microenvironment: Implications for biomarker development and immunotherapy. Int. J. Mol. Sci. 2020, 21, 5521. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhang, Y. Tumor-associated macrophages: From basic research to clinical application. J. Hematol. Oncol. 2017, 10, 58–69. [Google Scholar] [CrossRef]
- Law, A.M.K.; Valdes-Mora, F.; Gallego-Ortega, D. Myeloid-derived suppressor cells as a therapeutic target for cancer. Cells 2020, 9, 561. [Google Scholar] [CrossRef]
- Camus, M.; Tosolini, M.; Mlecnik, B.; Pagès, F.; Kirilovsky, A.; Berger, A.; Costes, A.; Bindea, G.; Charoentong, P.; Bruneval, P.; et al. Coordination of intra-tumoral immune reaction and human colorectal cancer recurrence. Cancer Res. 2009, 69, 2685–2693. [Google Scholar] [CrossRef]
- Okła, K.; Farber, D.L.; Zou, W. Tissue-resident memory T cells in tumor immunity and immunotherapy. J. Exp. Med. 2021, 218, e20201605. [Google Scholar] [CrossRef] [PubMed]
- Clarke, J.; Panwar, B.; Madrigal, A.; Singh, D.; Gujar, R.; Wood, O.; Chee, S.J.; Eschweiler, S.; King, E.V.; Awad, A.S.; et al. Single-cell transcriptomic analysis of tissue-resident memory T cells in human lung cancer. J. Exp. Med. 2019, 216, 2128–2149. [Google Scholar] [CrossRef] [PubMed]
- Mackay, L.K.; Braun, A.; Macleod, B.L.; Collins, N.; Tebartz, C.; Bedoui, S.; Carbone, F.R.; Gebhardt, T. Cutting edge: CD69 interference with sphingosine-1-phosphate receptor function regulates peripheral T cell retention. J. Immunol. 2015, 194, 2059–2063. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Thommen, D.S. Tertiary lymphoid structures in cancer. Science 2022, 375, eabf9419. [Google Scholar] [CrossRef]
- Blank, C.U.; Haining, W.N.; Held, W.; Hogan, P.G.; Kallies, A.; Lugli, E.; Lynn, R.C.; Philip, M.; Rao, A.; Restifo, N.P.; et al. Defining ‘T cell exhaustion. Nat. Rev. Immunol. 2019, 19, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Van der Leun, A.M.; Thommen, D.S.; Schumacher, T.N. CD8+ T cell states in human cancer: Insights from single-cell analysis. Nat. Rev. Cancer 2020, 20, 218–232. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Xu, C. Immune checkpoint signaling and cancer immunotherapy. Cell Res. 2020, 30, 660–669. [Google Scholar] [CrossRef] [PubMed]
- Beltra, J.C.; Manne, S.; Abdel-Hakeem, M.S.; Kurachi, M.; Giles, J.R.; Chen, Z.; Casella, V.; Ngiow, S.F.; Khan, O.; Huang, Y.J.; et al. Developmental relationships of four exhausted CD8+ T cell subsets reveals underlying transcriptional and epigenetic landscape control mechanisms. Immunity 2020, 52, 825–841. [Google Scholar] [CrossRef]
- Palermo, B.; Franzese, O.; Frisullo, G.; D’Ambrosio, L.; Panetta, M.; Campo, G.; D’Andrea, D.; Sperduti, I.; De Nicola, F.; Goeman, F.; et al. CD28/PD1 co-expression: Dual impact on CD8+ T cells in peripheral blood and tumor tissue, and its significance in NSCLC patients’ survival and ICB response. J. Exp. Clin. Cancer Res. 2023, 42, 287–312. [Google Scholar] [CrossRef]
- Dąbrowska, A.; Grubba, M.; Balihodzic, A.; Szot, O.; Sobocki, B.K.; Perdyan, A. The role of regulatory T cells in cancer treatment resistance. Int. J. Mol. Sci. 2023, 24, 14114. [Google Scholar] [CrossRef]
- Franzese, O.; Mascali, A.; Capria, A.; Castagnola, V.; Paganizza, L.; Di Daniele, N. Regulatory T cells in the immunodiagnosis and outcome of kidney allograft rejection. Clin. Dev. Immunol. 2013, 2013, 852395. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, H.; Koyama, S. Mechanisms of regulatory T cell infiltration in tumors: Implications for innovative immune precision therapies. J. Immunother. Cancer 2021, 9, e002591. [Google Scholar] [CrossRef] [PubMed]
- Gabriely, G.; da Cunha, A.P.; Rezende, R.M.; Kenyon, B.; Madi, A.; Vandeventer, T.; Skillin, N.; Rubino, S.; Garo, L.; Mazzola, M.A.; et al. Targeting latency-associated peptide promotes antitumor immunity. Sci. Immunol. 2017, 2, eaaj1738. [Google Scholar] [CrossRef] [PubMed]
- Arosa, F.A.; Esgalhado, A.J.; Padrao, C.A.; Cardoso, E.M. Divide, conquer, and sense: CD8+CD28− T cells in perspective. Front. Immunol. 2016, 7, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Voss, K.; Larsen, S.E.; Snow, A.L. Metabolic reprogramming and apoptosis sensitivity: Defining the contours of a T cell response. Cancer Lett. 2017, 408, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Le Gallo, M.; Poissonnier, A.; Blanco, P.; Legembre, P. CD95/FAS, non-apoptotic signaling pathways, and kinases. Front. Immunol. 2017, 8, 1216. [Google Scholar] [CrossRef] [PubMed]
- Slomp, A.; Peperzak, V. Role and regulation of pro-survival BCL-2 proteins in multiple myeloma. Front. Oncol. 2018, 8, 533. [Google Scholar] [CrossRef] [PubMed]
- Wensveen, F.M.; Klarenbeek, P.L.; van Gisbergen, K.P.; Pascutti, M.F.; Derks, I.A.; van Schaik, B.D.; Ten Brinke, A.; de Vries, N.; Cekinovic, D.; Jonjic, S.; et al. Proapoptotic protein noxa regulates memory T cell population size and protects against lethal immunopathology. J. Immunol. 2013, 190, 1180–1191. [Google Scholar] [CrossRef]
- Fischer, S.F.; Belz, G.T.; Strasser, A. BH3-only protein Puma contributes to the death of antigen-specific T cells during shutdown of an immune response to acute viral infection. Proc. Natl. Acad. Sci. USA 2008, 105, 3035–3040. [Google Scholar] [CrossRef]
- Erlacher, M.; Labi, V.; Manzl, C.; Böck, G.; Tzankov, A.; Häcker, G.; Michalak, E.; Strasser, A.; Villunger, A. Puma cooperates with Bim, the rate-limiting BH3-only protein in cell death during lymphocyte development, in apoptosis induction. J. Exp. Med. 2006, 203, 2939–2951. [Google Scholar] [CrossRef]
- Zhang, Y.; Kurupati, R.; Liu, L.; Zhou, X.Y.; Zhang, G.; Hudaihed, A.; Filisio, F.; Giles-Davis, W.; Xu, X.; Karakousis, G.C.; et al. Enhancing CD8+ T cell fatty acid catabolism within a metabolically challenging tumor microenvironment increases the efficacy of melanoma immunotherapy. Cancer Cell 2017, 32, 377–391. [Google Scholar] [CrossRef]
- Zhang, N.; Hartig, H.; Dzhagalov, I.; Draper, D.; He, Y.W. The role of apoptosis in the development and function of T lymphocytes. Cell Res. 2005, 15, 749–769. [Google Scholar] [CrossRef]
- Russell, J.H.; White, C.L.; Loh, D.Y.; Meleedy-Rey, P. Receptor-stimulated death pathway is opened by antigen in mature T cells. Proc. Natl. Acad. Sci. USA 1991, 88, 2151–2155. [Google Scholar] [CrossRef]
- Boehme, S.A.; Lenardo, M.J. Propriocidal apoptosis of mature T lymphocytes occurs at the S phase of the cell cycle. Eur. J. Immunol. 1993, 23, 1552–1560. [Google Scholar] [CrossRef]
- Lenardo, M.J. Interleukin-2 programs mouse alpha beta T lymphocytes for apoptosis. Nature 1991, 353, 858–861. [Google Scholar] [CrossRef]
- Crawley, A.M.; Vranjkovic, A.; Faller, E.; McGuinty, M.; Busca, A.; Burke, S.C.; Cousineau, S.; Kumar, A.; Macpherson, P.A.; Angel, J.B. Jak/STAT and PI3K signaling pathways have both common and distinct roles in IL-7-mediated activities in human CD8+ T cells. J. Leukoc. Biol. 2014, 95, 117–127. [Google Scholar] [CrossRef]
- Oh, S.; Perera, L.P.; Terabe, M.; Ni, L.; Waldmann, T.A.; Berzofsky, J.A. IL-15 as a mediator of CD4+ help for CD8+ T cell longevity and avoidance of TRAIL-mediated apoptosis. Proc. Natl. Acad. Sci. USA 2008, 105, 5201–5206. [Google Scholar] [CrossRef]
- Snow, A.L.; Marsh, R.A.; Krummey, S.M.; Roehrs, P.; Young, L.R.; Zhang, K.; van Hoff, J.; Dhar, D.; Nichols, K.E.; Filipovich, A.H.; et al. Restimulation-induced apoptosis of T cells is impaired in patients with X-linked lymphoproliferative disease caused by SAP deficiency. J. Clin. Investig. 2009, 119, 2976–2989. [Google Scholar] [CrossRef]
- Strauss, L.; Bergmann, C.; Whiteside, T.L. Human circulating CD4+CD25highFoxp3+ regulatory T cells kill autologous CD8+ but not CD4+ responder cells by FAS-mediated apoptosis. J. Immunol. 2009, 182, 1469–1480. [Google Scholar] [CrossRef]
- Fritzsching, B.; Oberle, N.; Pauly, E.; Geffers, R.; Buer, J.; Poschl, J.; Krammer, P.; Linderkamp, O.; Suri-Payer, E. Naive regulatory T cells: A novel subpopulation defined by resistance toward CD95L-mediated cell death. Blood 2006, 108, 3371–3378. [Google Scholar] [CrossRef]
- Plaza-Sirvent, C.; Schuster, M.; Neumann, Y.; Heise, U.; Pils, M.C.; Schulze-Osthoff, K.; Schmitz, I. c-FLIP expression in Foxp3-expressing cells is essential for survival of regulatory T cells and prevention of autoimmunity. Cell Rep. 2017, 18, 12–22. [Google Scholar] [CrossRef]
- Yang, D.; Liu, J.; Qian, H.; Zhuang, Q. Cancer-associated fibroblasts: From basic science to anticancer therapy. Exp. Mol. Med. 2023, 55, 1322–1332. [Google Scholar] [CrossRef]
- Motz, G.T.; Santoro, S.P.; Wang, L.P.; Garrabrant, T.; Lastra, R.R.; Hagemann, I.S.; Lal, P.; Feldman, M.D.; Benencia, F.; Coukos, G. Tumor endothelium FASL establishes a selective immune barrier promoting tolerance in tumors. Nat. Med. 2014, 20, 607–615. [Google Scholar] [CrossRef]
- Krishnamoorthy, M.; Gerhardt, L.; Maleki Vareki, S. Immunosuppressive effects of myeloid-derived suppressor cells in cancer and immunotherapy. Cells 2021, 10, 1170. [Google Scholar] [CrossRef]
- Annibaldi, A.; Walczak, H. Death receptors and their ligands in inflammatory disease and cancer. Cold Spring Harb. Perspect. Biol. 2020, 12, a036384. [Google Scholar] [CrossRef]
- Lünemann, J.D.; Waiczies, S.; Ehrlich, S.; Wendling, U.; Seeger, B.; Kamradt, T.; Zipp, F. Death ligand TRAIL induces no apoptosis but inhibits activation of human (auto)antigen-specific T cells. J. Immunol. 2002, 168, 4881–4888. [Google Scholar] [CrossRef]
- Chyuan, I.T.; Tsai, H.F.; Wu, C.S.; Sung, C.C.; Hsu, P.N. TRAIL-mediated suppression of T cell receptor signaling inhibits T cell activation and inflammation in experimental autoimmune encephalomyelitis. Front. Immunol. 2018, 9, 15–28. [Google Scholar] [CrossRef]
- Franzese, O.; Palermo, B.; Di Donna, C.; Sperduti, I.; Ferraresi, V.; Stabile, H.; Gismondi, A.; Santoni, A.; Nisticò, P. Polyfunctional melan-A-specific tumor-reactive CD8+ T cells elicited by dacarbazine treatment before peptide-vaccination eepends on AKT activation sustained by ICOS. Oncoimmunology 2016, 5, e1114203. [Google Scholar] [CrossRef]
- Corthay, A. How do regulatory T cells work? Scand. J. Immunol. 2009, 70, 326–336. [Google Scholar] [CrossRef]
- Xing, J.; Zhang, J.; Wang, J. The immune regulatory role of adenosine in the tumor microenvironment. Int. J. Mol. Sci. 2023, 24, 14928. [Google Scholar] [CrossRef]
- Yang, R.; Elsaadi, S.; Misund, K.; Abdollahi, P.; Vandsemb, E.N.; Moen, S.H.; Kusnierczyk, A.; Slupphaug, G.; Standal, T.; Waage, A.; et al. Conversion of ATP to adenosine by CD39 and CD73 in multiple myeloma can be successfully targeted with adenosine receptor A2A blockade. J. Immunother. Cancer 2020, 8, e000610. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Feng, M.; Du, S.; Wei, X.; Song, H.; Xu, Y.; Song, J.; Guan, W. Adenosine generated by regulatory T cells induces CD8+ T cell exhaustion in gastric cancer through A2AR pathway. Biomed. Res. Int. 2019, 2019, 4093214. [Google Scholar] [CrossRef] [PubMed]
- Mastelic-Gavillet, B.; Navarro Rodrigo, B.; Décombaz, L.; Wang, H.; Ercolano, G.; Ahmed, R.; Lozano, L.E.; Ianaro, A.; Derré, L.; Valerio, M.; et al. Adenosine mediates functional and metabolic suppression of peripheral and tumor-infiltrating CD8+ T cells. J. Immunother. Cancer 2019, 7, 257–272. [Google Scholar] [CrossRef] [PubMed]
- Butler, J.J.; Mader, J.S.; Watson, C.L.; Zhang, H.; Blay, J.; Hoskin, D.W. Adenosine inhibits activation-induced T cell expression of CD2 and CD28 costimulatory molecules: Role of interleukin-2 and cyclic AMP signaling pathways. J. Cell. Biochem. 2003, 89, 975–991. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhao, X.; Wan, Y.Y. Intricacies of TGF-β signaling in Treg and Th17 cell biology. Cell. Mol. Immunol. 2023, 20, 1002–1022. [Google Scholar] [CrossRef] [PubMed]
- Fantini, M.C.; Becker, C.; Monteleone, G.; Pallone, F.; Galle, P.R.; Neurath, M.F. Cutting edge: TGF-beta induces a regulatory phenotype in CD4+CD25− T cells through Foxp3 induction and down-regulation of Smad7. J. Immunol. 2004, 172, 5149–5153. [Google Scholar] [CrossRef] [PubMed]
- Dahmani, A.; Delisle, J.S. TGF-β in T cell biology: Implications for cancer immunotherapy. Cancers 2018, 10, 194. [Google Scholar] [CrossRef] [PubMed]
- Sanjabi, S.; Mosaheb, M.M.; Flavell, R.A. Opposing effects of TGF-beta and IL-15 cytokines control the number of short-lived effector CD8+ T cells. Immunity 2009, 31, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Tinoco, R.; Alcalde, V.; Yang, Y.; Sauer, K.; Zuniga, E.I. Cell-intrinsic transforming growth factor-beta signaling mediates virus-specific CD8+ T cell deletion and viral persistence in vivo. Immunity 2009, 31, 145–157. [Google Scholar] [CrossRef]
- Nguyen, T.P.; Sieg, S.F. TGF-β inhibits IL-7-induced proliferation in memory but not naive human CD4+ T cells. J. Leukoc. Biol. 2017, 102, 499–506. [Google Scholar] [CrossRef]
- Weinkove, R.; George, P.; Dasyam, N.; McLellan, A.D. Selecting costimulatory domains for chimeric antigen receptors: Functional and clinical considerations. Clin. Transl. Immunol. 2019, 8, e1049. [Google Scholar] [CrossRef] [PubMed]
- Molon, B.; Liboni, C.; Viola, A. CD28 and chemokine receptors: Signalling amplifiers at the immunological synapse. Front. Immunol. 2022, 13, 938004. [Google Scholar] [CrossRef] [PubMed]
- Boise, L.H.; Minn, A.J.; Noel, P.J.; June, C.H.; Accavitti, M.A.; Lindsten, T.; Thompson, C.B. CD28 costimulation can promote T-cell survival by enhancing the expression of BCL-XL. Immunity 1995, 3, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Nagy, N.; Klein, E. Deficiency of the proapoptotic SAP function in X-linked lymphoproliferative disease aggravates Epstein-Barr virus (EBV) induced mononucleosis and promotes lymphoma development. Immunol. Lett. 2010, 4, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Shao, B. LFA-1 Activation in T-cell migration and immunological synapse formation. Cells 2023, 12, 1136. [Google Scholar] [CrossRef]
- Borthwick, N.J.; Lowdell, M.; Salmon, M.; Akbar, A.N. Loss of CD28 expression on CD8+ T cells is induced by IL-2 receptor gamma chain signaling cytokines and type I IFN and increases susceptibility to activation-induced apoptosis. Int. Immunol. 2000, 12, 1005–1013. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.Z.; Martin, P.J.; Anasetti, C. CD28 signal enhances apoptosis of CD8 T cells after strong TCR ligation. J. Immunol. 2003, 170, 3002–3006. [Google Scholar] [CrossRef]
- Hashimoto, K. CD137 as an attractive T cell co-stimulatory target in the TNFRSF for immuno-oncology drug development. Cancers 2021, 13, 2288. [Google Scholar] [CrossRef] [PubMed]
- Chester, C.; Sanmamed, M.F.; Wang, J.; Melero, I. Immunotherapy targeting 4-1BB: Mechanistic rationale, clinical results, and future strategies. Blood 2018, 131, 49–57. [Google Scholar] [CrossRef]
- Dolfi, D.V.; Boesteanu, A.C.; Petrovas, C.; Xia, D.; Butz, E.A.; Katsikis, P.D. Late signals from CD27 prevent FAS-dependent apoptosis of primary CD8+ T cells. J. Immunol. 2008, 180, 2912–2921. [Google Scholar] [CrossRef]
- Peperzak, V.; Veraar, E.A.; Keller, A.M.; Xiao, Y.; Borst, J. The Pim kinase pathway contributes to survival signaling in primed CD8+ T Cells upon CD27 costimulation. J. Immunol. 2010, 185, 6670–6678. [Google Scholar] [CrossRef]
- Linowes, B.A.; Ligons, D.L.; Nam, A.S.; Hong, C.; Keller, H.R.; Tai, X.; Luckey, M.A.; Park, J.H. Pim1 permits generation and survival of CD4+ T cells in the absence of γc cytokine receptor signaling. Eur. J. Immunol. 2013, 43, 2283–2294. [Google Scholar] [CrossRef]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef]
- Parry, R.V.; Chemnitz, J.M.; Frauwirth, K.A.; Lanfranco, A.R.; Braunstein, I.; Kobayashi, S.V.; Linsley, P.S.; Thompson, C.B.; Riley, J.L. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol. Cell. Biol. 2005, 25, 9543–9553. [Google Scholar] [CrossRef]
- Li, F.; Zhang, Z.; Xuan, Y.; Zhang, D.; Liu, J.; Li, A.; Wang, S.; Li, T.; Shi, X.; Zhang, Y. PD-1 abrogates the prolonged persistence of CD8+ CAR-T cells with 4-1BB co-stimulation. Sign. Transd. Targ. Ther. 2020, 5, 164–167. [Google Scholar] [CrossRef]
- Gibbons, R.M.; Liu, X.; Pulko, V.; Harrington, S.M.; Krco, C.J.; Kwon, E.D.; Dong, H. B7-H1 Limits the Entry of Effector CD8+ T Cells to the memory pool by upregulating Bim. Oncoimmunology 2012, 1, 1061–1073. [Google Scholar] [CrossRef]
- Sauer, N.; Janicka, N.; Szlasa, W.; Skinderowicz, B.; Kołodzińska, K.; Dwernicka, W.; Oślizło, M.; Kulbacka, J.; Novickij, V.; Karłowicz-Bodalska, K. TIM-3 as a promising target for cancer immunotherapy in a wide range of tumors. Cancer Immunol. Immunother. 2023, 72, 3405–3425. [Google Scholar] [CrossRef]
- Lv, Y.; Ma, X.; Ma, Y.; Du, Y.; Feng, J. A New Emerging target in cancer immunotherapy: Galectin-9 (LGALS9). Genes Dis. 2022, 10, 2366–2382. [Google Scholar] [CrossRef]
- Huang, Y.H.; Zhu, C.; Kondo, Y.; Anderson, A.C.; Gandhi, A.; Russell, A.; Dougan, S.K.; Petersen, B.S.; Melum, E.; Pertel, T.; et al. CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature 2015, 517, 386–390. [Google Scholar] [CrossRef]
- Lake, C.M.; Voss, K.; Bauman, B.M.; Pohida, K.; Jiang, T.; Dveksler, G.; Snow, A.L. TIM-3 drives temporal differences in restimulation-induced cell death sensitivity in effector CD8+ T cells in conjunction with CEACAM1. Cell Death Dis. 2021, 12, 400–416. [Google Scholar] [CrossRef]
- Stone, T.W.; Williams, R.O. Modulation of T cells by tryptophan metabolites in the kynurenine pathway. Trends Pharmacol. Sci. 2023, 44, 442–456. [Google Scholar] [CrossRef] [PubMed]
- Aboelella, N.S.; Brandle, C.; Kim, T.; Ding, Z.C.; Zhou, G. Oxidative stress in the tumor microenvironment and its relevance to cancer immunotherapy. Cancers 2021, 13, 986. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Song, M.; Zhang, B.; Zhang, Y. Reactive Oxygen Species Regulate T Cell Immune Response in the Tumor Microenvironment. Oxid. Med. Cell. Longev. 2016, 2016, 1580967. [Google Scholar] [CrossRef] [PubMed]
- Malmberg, K.J.; Arulampalam, V.; Ichihara, F.; Petersson, M.; Seki, K.; Andersson, T.; Lenkei, R.; Masucci, G.; Pettersson, S.; Kiessling, R. Inhibition of activated/memory (CD45RO+) T cells by oxidative stress associated with block of NF-kappaB activation. J. Immunol. 2001, 167, 2595–2601. [Google Scholar] [CrossRef] [PubMed]
- Efimova, O.; Szankasi, P.; Kelley, T.W. Ncf1 (p47phox) is essential for direct regulatory T cell mediated suppression of CD4+ effector T cells. PLoS ONE 2011, 6, e16013. [Google Scholar] [CrossRef] [PubMed]
- Mougiakakos, D.; Johansson, C.C.; Jitschin, R.; Böttcher, M.; Kiessling, R. Increased thioredoxin-1 production in human naturally occurring regulatory T cells confers enhanced tolerance to oxidative stress. Blood 2011, 117, 857–861. [Google Scholar] [CrossRef] [PubMed]
- Maj, T.; Wang, W.; Crespo, J.; Zhang, H.; Wang, W.; Wei, S.; Zhao, L.; Vatan, L.; Shao, I.; Szeliga, W.; et al. oxidative stress controls regulatory T cell apoptosis and suppressor activity and PD-L1-blockade resistance in tumor. Nat. Immunol. 2017, 18, 1332–1341. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Hsueh, P.C.; Li, Z.; Ho, P.C. Microenvironment-driven metabolic adaptations guiding CD8+ T cell anti-tumor immunity. Immunity 2023, 56, 32–42. [Google Scholar] [CrossRef]
- O’Sullivan, D.; Pearce, E.L. Targeting T cell metabolism for therapy. Trends Immunol. 2015, 36, 71–80. [Google Scholar] [CrossRef]
- Nath, S.; Balling, R. The Warburg Effect Reinterpreted 100 yr on: A First-Principles Stoichiometric Analysis and Interpretation from the Perspective of ATP Metabolism in Cancer Cells. Function 2024, 5, zqae008. [Google Scholar] [CrossRef]
- Buck, M.D.; O’Sullivan, D.; Pearce, E.L. T cell metabolism drives immunity. J. Exp. Med. 2015, 212, 1345–1360. [Google Scholar] [CrossRef]
- Salmond, R.J. mTOR regulation of glycolytic metabolism in T cells. Front. Cell Dev. Biol. 2018, 6, 122. [Google Scholar] [CrossRef]
- Shyer, J.A.; Flavell, R.A.; Bailis, W. Metabolic signaling in T cells. Cell Res. 2020, 30, 649–659. [Google Scholar] [CrossRef]
- Esensten, J.H.; Helou, Y.A.; Chopra, G.; Weiss, A.; Bluestone, J.A. CD28 costimulation: From mechanism to therapy. Immunity 2016, 44, 973–988. [Google Scholar] [CrossRef]
- Li, F.; Liu, H.; Zhang, D.; Ma, Y.; Zhu, B. Metabolic plasticity and regulation of T cell exhaustion. Immunology 2022, 167, 482–494. [Google Scholar] [CrossRef] [PubMed]
- Kishton, R.J.; Sukumar, M.; Restifo, N.P. Metabolic regulation of T cell longevity and function in tumor immunotherapy. Cell Metab. 2017, 26, 94–109. [Google Scholar] [CrossRef]
- Larsen, S.E.; Bilenkin, A.; Tarasenko, T.N.; Arjunaraja, S.; Stinson, J.R.; McGuire, P.J.; Snow, A.L. Sensitivity to restimulation-induced cell death is linked to glycolytic metabolism in human T cells. J. Immunol. 2017, 198, 147–155. [Google Scholar] [CrossRef]
- Voss, K.; Luthers, C.R.; Pohida, K.; Snow, A.L. Fatty acid synthase contributes to restimulation-induced cell death of human CD4 T cells. Front. Mol. Biosci. 2019, 6, 106–119. [Google Scholar] [CrossRef]
- Chang, C.H.; Curtis, J.D.; Maggi, L.B.; Faubert, B.; Villarino, A.V.; O’Sullivan, D.; Huang, S.C.; van der Windt, G.J.; Blagih, J.; Qiu, J.; et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 2013, 153, 1239–1251. [Google Scholar] [CrossRef]
- Ganjoo, S.; Gupta, P.; Corbali, H.I.; Nanez, S.; Riad, T.S.; Duong, L.K.; Barsoumian, H.B.; Masrorpour, F.; Jiang, H.; Welsh, J.W.; et al. The role of tumor metabolism in modulating T-cell activity and in optimizing immunotherapy. Front. Immunol. 2023, 14, 1172931. [Google Scholar] [CrossRef]
- Angelin, A.; Gil-de-Gómez, L.; Dahiya, S.; Jiao, J.; Guo, L.; Levine, M.H.; Wang, Z.; Quinn, W.J., 3rd; Kopinski, P.K.; Wang, L.; et al. Foxp3 reprograms T cell metabolism to function in low-glucose, high-lactate environments. Cell Metab. 2017, 25, 1282–1293. [Google Scholar] [CrossRef]
- Shan, T.; Chen, S.; Chen, X.; Wu, T.; Yang, Y.; Li, S.; Ma, J.; Zhao, J.; Lin, W.; Li, W.; et al. M2-TAM subsets altered by lactic acid promote T-cell apoptosis through the PD-L1/PD-1 pathway. Oncol. Rep. 2020, 44, 1885–1894. [Google Scholar] [CrossRef]
- Ma, G.; Zhang, Z.; Li, P.; Zhang, Z.; Zeng, M.; Liang, Z.; Li, D.; Wang, L.; Chen, Y.; Liang, Y.; et al. Reprogramming of glutamine metabolism and its impact on immune response in the tumor microenvironment. Cell Commun. Signal. 2022, 20, 114–128. [Google Scholar] [CrossRef]
- Blagih, J.; Coulombe, F.; Vincent, E.E.; Dupuy, F.; Galicia-Vázquez, G.; Yurchenko, E.; Raissi, T.C.; van der Windt, G.J.; Viollet, B.; Pearce, E.L.; et al. The energy sensor AMPK regulates T cell metabolic adaptation and effector responses in vivo. Immunity 2015, 42, 41–54. [Google Scholar] [CrossRef]
- Han, J.; Khatwani, N.; Searles, T.G.; Turk, M.J.; Angeles, C.V. Memory CD8+ T cell responses to cancer. Semin. Immunol. 2020, 49, 101435–101460. [Google Scholar] [CrossRef]
- Pollizzi, K.N.; Patel, C.H.; Sun, I.H.; Oh, M.H.; Waickman, A.T.; Wen, J.; Delgoffe, G.M.; Powell, J.D. mTORC1 and mTORC2 selectively regulate CD8+ T cell differentiation. J. Clin. Investig. 2015, 125, 2090–2108. [Google Scholar] [CrossRef]
- Endo, Y.; Kanno, T.; Nakajima, T. Fatty acid metabolism in T-cell function and differentiation. Int. Immunol. 2022, 34, 579–587. [Google Scholar] [CrossRef]
- Larsen, S.E.; Voss, K.; Laing, E.D.; Snow, A.L. Differential cytokine withdrawal-induced death sensitivity of effector T cells derived from distinct human CD8+ memory subsets. Cell Death Discov. 2017, 3, 17031–17038. [Google Scholar] [CrossRef]
- Patsoukis, N.; Bardhan, K.; Chatterjee, P.; Sari, D.; Liu, B.; Bell, L.N.; Karoly, E.D.; Freeman, G.J.; Petkova, V.; Seth, P.; et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat. Commun. 2015, 6, 6692–6704. [Google Scholar] [CrossRef]
- Liu, S.; Yao, S.; Yang, H.; Liu, S.; Wang, Y. Autophagy: Regulator of cell death. Cell Death Dis. 2023, 14, 648–664. [Google Scholar] [CrossRef] [PubMed]
- Angela, M.; Endo, Y.; Asou, H.K.; Yamamoto, T.; Tumes, D.J.; Tokuyama, H.; Yokote, K.; Nakayama, T. Fatty acid metabolic reprogramming via mTOR-mediated inductions of PPARγ directs early activation of T cells. Nat. Commun. 2016, 7, 13683–13697. [Google Scholar] [CrossRef]
- Xu, S.; Chaudhary, O.; Rodríguez-Morales, P.; Sun, X.; Chen, D.; Zappasodi, R.; Xu, Z.; Pinto, A.F.M.; Williams, A.; Schulze, I.; et al. Uptake of oxidized lipids by the scavenger receptor CD36 promotes lipid peroxidation and dysfunction in CD8+ T cells in tumors. Immunity 2021, 54, 1561–1577. [Google Scholar] [CrossRef]
- Cioccoloni, G.; Aquino, A.; Notarnicola, M.; Caruso, M.G.; Bonmassar, E.; Zonfrillo, M.; Caporali, S.; Faraoni, I.; Villivà, C.; Fuggetta, M.P.; et al. Fatty acid synthase inhibitor orlistat impairs cell growth and down-regulates PD-L1 expression of a human T-cell leukemia line. J. Chemother. 2020, 32, 30–40. [Google Scholar] [CrossRef]
- Geiger, R.; Rieckmann, J.C.; Wolf, T.; Basso, C.; Feng, Y.; Fuhrer, T.; Kogadeeva, M.; Picotti, P.; Meissner, F.; Mann, M.; et al. L-Arginine modulates T Cell metabolism and enhances survival and anti-tumor activity. Cell 2016, 167, 829–842. [Google Scholar] [CrossRef]
- Konjar, Š.; Veldhoen, M. Dynamic metabolic state of tissue resident CD8 T Cells. Front. Immun. 2019, 10, 1683–1692. [Google Scholar] [CrossRef]
- Sun, N.; Zhao, X. Therapeutic Implications of FABP4 in Cancer: An Emerging Target to Tackle Cancer. Front. Pharmacol. 2022, 13, 948610. [Google Scholar] [CrossRef]
- Bengsch, B.; Johnson, A.L.; Kurachi, M.; Odorizzi, P.M.; Pauken, K.E.; Attanasio, J.; Stelekati, E.; McLane, L.M.; Paley, M.A.; Delgoffe, G.M.; et al. Bioenergetic insufficiencies due to metabolic alterations regulated by the inhibitory receptor PD-1 are an early driver of CD8+ T cell exhaustion. Immunity 2016, 45, 358–373. [Google Scholar] [CrossRef]
- Alturki, N.A. Review of the immune checkpoint inhibitors in the context of cancer treatment. J. Clin. Med. 2023, 12, 4301. [Google Scholar] [CrossRef]
- Franzese, O.; Torino, F.; Fuggetta, M.P.; Aquino, A.; Roselli, M.; Bonmassar, E.; Giuliani, A.; D’Atri, S. Tumor immunotherapy: Drug-induced neoantigens (xenogenization) and immune checkpoint inhibitors. Oncotarget 2017, 8, 41641–41669. [Google Scholar] [CrossRef] [PubMed]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. Car T Cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [PubMed]
- Tschumi, B.O.; Dumauthioz, N.; Marti, B.; Zhang, L.; Lanitis, E.; Irving, M.; Schneider, P.; Mach, J.P.; Coukos, G.; Romero, P.; et al. CART cells are prone to FAS- and DR5-mediated cell death. J. Immunother. Cancer 2018, 6, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Thommen, D.S.; Schumacher, T.N. T Cell Dysfunction in Cancer. Cancer Cell 2018, 33, 547–562. [Google Scholar] [CrossRef]
- Im, S.J.; Hashimoto, M.; Gerner, M.Y.; Lee, J.; Kissick, H.T.; Burger, M.C.; Shan, Q.; Hale, J.S.; Lee, J.; Nasti, T.H.; et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 2016, 537, 417–421. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.W.; Dutta, A.; Chang, L.Y.; Mahalingam, J.; Lin, Y.C.; Chiang, J.M.; Hsu, C.Y.; Huang, C.T.; Su, W.T.; Chu, Y.Y.; et al. Apoptosis of tumor-infiltrating effector TIM-3+CD8+ T cells in colon cancer. Sci. Rep. 2015, 5, 15659–15670. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Anderson, A.C.; Schubart, A.; Xiong, H.; Imitola, J.; Khoury, S.J.; Zheng, X.X.; Strom, T.B.; Kuchroo, V.K. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat. Immunol. 2005, 6, 1245–1252. [Google Scholar] [CrossRef]
- Marin-Acevedo, J.A.; Kimbrough, E.O.; Lou, Y. Next generation of immune checkpoint inhibitors and beyond. J. Hematol. Oncol. 2021, 14, 45–73. [Google Scholar] [CrossRef] [PubMed]
- Sakuishi, K.; Apetoh, L.; Sullivan, J.M.; Blazar, B.R.; Kuchroo, V.K.; Anderson, A.C. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore antitumor immunity. J. Exp. Med. 2010, 207, 2187–2194. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.; Li, Z. Targeting Tim-3 in cancer with resistance to PD-1/PD-L1 blockade. Front. Oncol. 2021, 11, 731175. [Google Scholar] [CrossRef]
- Huo, J.L.; Wang, Y.T.; Fu, W.J.; Lu, N.; Liu, Z.S. The promising immune checkpoint LAG-3 in cancer immunotherapy: From basic research to clinical application. Front. Immunol. 2022, 13, 956090. [Google Scholar] [CrossRef]
- Andrews, L.P.; Marciscano, A.E.; Drake, C.G.; Vignali, D.A. LAG3 (CD223) as a cancer immunotherapy target. Immunol. Rev. 2017, 276, 80–96. [Google Scholar] [CrossRef]
- Lee, Y.G.; Yang, N.; Chun, I.; Porazzi, P.; Carturan, A.; Paruzzo, L.; Sauter, C.T.; Guruprasad, P.; Pajarillo, R.; Ruella, M. Apoptosis: A Janus bifrons in T-cell immunotherapy. J. Immunother. Cancer 2023, 11, e005967. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Sun, Z.; Chen, L. Memory T cells: Strategies for optimizing tumor immunotherapy. Protein Cell 2020, 11, 549–564. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Lee, K.J.; Lee, S.W. Cancer immunotherapy with T-cell targeting cytokines: IL-2 and IL-7. BMB Rep. 2021, 54, 21–30. [Google Scholar] [CrossRef]
- Berraondo, P.; Sanmamed, M.F.; Ochoa, M.C.; Etxeberria, I.; Aznar, M.A.; Pérez-Gracia, J.L.; Rodríguez-Ruiz, M.E.; Ponz-Sarvise, M.; Castañón, E.; Melero, I. Cytokines in clinical cancer immunotherapy. Br. J. Cancer 2019, 120, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Cong, Y.; Jia, M.; He, Q.; Zhong, H.; Zhao, Y.; Li, H.; Yan, M.; You, J.; Liu, J.; et al. Targeting IL-21 to tumor-reactive T cells enhances memory T cell responses and anti-PD-1 antibody therapy. Nat. Commun. 2021, 12, 951. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Liu, M.; Du, X.; Li, S.; Li, H.; Li, X.; Li, Y.; Wang, Y.; Qin, Z.; Fu, Y.X.; et al. Intratumoral delivery of IL-21 overcomes Anti-Her2/Neu resistance through shifting tumor-associated macrophages from M2 to M1 phenotype. J. Immunol. 2015, 194, 4997–5006. [Google Scholar] [CrossRef]
- Zarkavelis, G.; Kefas, A.; Pentheroudakis, G. The emerging role of interleukin-21 as an antineoplastic immunomodulatory treatment option. Transl. Cancer Res. 2017, 6, S328–S330. [Google Scholar] [CrossRef]
- Cai, X.; Li, H.; Wang, M.; Chu, E.; Wei, N.; Lin, J.; Hu, Y.; Dai, J.; Chen, A.; Zheng, H.; et al. mTOR participates in the formation, maintenance, and function of memory CD8+ T cells regulated by glycometabolism. Bioch. Pharmacol. 2022, 204, 115197. [Google Scholar] [CrossRef]
- Jung, J.W.; Veitch, M.; Bridge, J.A.; Overgaard, N.H.; Cruz, J.L.; Linedale, R.; Franklin, M.E.; Saunders, N.A.; Simpson, F.; Frazer, I.H.; et al. Clinically-relevant rapamycin treatment regimens enhance CD8+ effector memory T cell function in the skin and allow their infiltration into cutaneous squamous cell carcinoma. Oncoimmunology 2018, 7, e1479627. [Google Scholar] [CrossRef]
- Merino, D.; San Segundo, D.; Medina, J.M.; Rodrigo, E.; Asensio, E.; Irure, J.; Fernández-Fresnedo, G.; Arias, M.A.; López-Hoyos, M. Different in vitro proliferation and cytokine-production inhibition of memory T-cell subsets after calcineurin and mammalian target of rapamycin inhibitors treatment. Immunology 2016, 148, 206–215. [Google Scholar] [CrossRef]
- Crompton, J.G.; Sukumar, M.; Roychoudhuri, R.; Clever, D.; Gros, A.; Eil, R.L.; Tran, E.; Hanada, K.; Yu, Z.; Palmer, D.C.; et al. Akt inhibition enhances expansion of potent tumor-specific lymphocytes with memory cell characteristics. Cancer Res. 2015, 75, 296–305. [Google Scholar] [CrossRef] [PubMed]
- Labanieh, L.; Mackall, C.L. CAR immune cells: Design principles, resistance and the next generation. Nature 2023, 614, 635–648. [Google Scholar] [CrossRef] [PubMed]
- Orlando, E.J.; Han, X.; Tribouley, C.; Wood, P.A.; Leary, R.J.; Riester, M.; Levine, J.E.; Qayed, M.; Grupp, S.A.; Boyer, M.; et al. Genetic mechanisms of target antigen loss in CAR19 therapy of acute lymphoblastic leukemia. Nat. Med. 2018, 24, 1504–1506. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Jin, L.; Wang, F.; Zhang, Y.; Liu, B.; Zhao, T. Chimeric antigen receptor T (CAR-T) cells expanded with IL-7/IL-15 mediate superior antitumor effects. Protein Cell 2019, 10, 764–769. [Google Scholar] [CrossRef] [PubMed]
- Ugolini, A.; Nuti, M. CD137+ T-cells: Protagonists of the immunotherapy revolution. Cancers 2021, 13, 456. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.S.; Matsushita, M.; Plotkin, J.; Riviere, I.; Sadelain, M. Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/BCL-XL activation and CD8+ T cell-mediated tumor eradication. Mol. Ther. 2010, 18, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Loves, R.; Grunebaum, E. FAS signalling pathway is crucial for CAR T cell persistence. Nat. Rev. Immunol. 2024, 24, 380. [Google Scholar] [CrossRef]
- Charo, J.; Finkelstein, S.E.; Grewal, N.; Restifo, N.P.; Robbins, P.F.; Rosenberg, S.A. Bcl-2 overexpression enhances tumor-specific T-cell survival. Cancer Res. 2005, 65, 2001–2008. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Han, P.; Qi, X.; Li, F.; Li, M.; Fan, L.; Zhang, H.; Zhang, X.; Yang, X. BCL-2 enhances chimeric antigen receptor T cell persistence by reducing activation-induced apoptosis. Cancers 2021, 13, 197. [Google Scholar] [CrossRef]
- Lee, Y.G.; Guruprasad, P.; Ghilardi, G.; Pajarillo, R.; Sauter, C.T.; Patel, R.; Ballard, H.J.; Hong, S.J.; Chun, I.; Yang, N.; et al. Modulation of BCL-2 in both T cells and tumor cells to enhance chimeric antigen receptor T-cell immunotherapy against cancer. Cancer Discov. 2022, 12, 2372–2391. [Google Scholar] [CrossRef]
- Lever, J.R.; Fergason-Cantrell, E.A. Allosteric modulation of sigma receptors by BH3 mimetics ABT-737, ABT-263 (Navitoclax) and ABT-199 (Venetoclax). Pharmacol. Res. 2019, 142, 87–100. [Google Scholar] [CrossRef]
- Snajdauf, M.; Havlova, K.; Vachtenheim, J., Jr.; Ozaniak, A.; Lischke, R.; Bartunkova, J.; Smrz, D.; Strizova, Z. The TRAIL in the treatment of human cancer: An update on clinical trials. Front. Mol. Biosci. 2021, 8, 628332. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Zhang, H.; Yuan, Y.; He, Q.; Zhou, J.; Li, S.; Sun, Y.; Li, D.Y.; Qiu, H.B.; Wang, W.; et al. Fatty acid oxidation controls CD8+ tissue-resident memory T-cell survival in gastric adenocarcinoma. Cancer Immunol. Res. 2020, 8, 479–492. [Google Scholar] [CrossRef]
- Xia, C.; Yin, S.; To, K.K.W.; Fu, L. CD39/CD73/A2AR pathway and cancer immunotherapy. Mol. Cancer 2023, 22, 44–60. [Google Scholar] [CrossRef]
- Gacche, R.N. Changing landscape of anti-angiogenic therapy: Novel approaches and clinical perspectives. Biochim. Biophys. Acta Rev. Cancer 2023, 1878, 189020. [Google Scholar] [CrossRef] [PubMed]
- Blanc-Durand, F.; Clemence Wei Xian, L.; Tan, D.S.P. Targeting the immune microenvironment for ovarian cancer therapy. Front. Immunol. 2023, 14, 1328651. [Google Scholar] [CrossRef]
- Kim, B.G.; Malek, E.; Choi, S.H.; Ignatz-Hoover, J.J.; Driscoll, J.J. Novel therapies emerging in oncology to target the TGF-β pathway. J. Hematol. Oncol. 2021, 14, 55–74. [Google Scholar] [CrossRef]
- Kunisada, Y.; Eikawa, S.; Tomonobu, N.; Domae, S.; Uehara, T.; Hori, S.; Furusawa, Y.; Hase, K.; Sasaki, A.; Udono, H. Attenuation of CD4+CD25+ regulatory T cells in the tumor microenvironment by metformin, a type 2 diabetes drug. EBioMedicine 2017, 25, 154–164. [Google Scholar] [CrossRef]
- Scheffel, M.J.; Scurti, G.; Simms, P.; Garrett-Mayer, E.; Mehrotra, S.; Nishimura, M.I.; Voelkel-Johnson, C. Efficacy of adoptive T-cell therapy is improved by treatment with the antioxidant N-acetyl cysteine, which limits activation-induced T-cell death. Cancer Res. 2016, 76, 6006–6016. [Google Scholar] [CrossRef]
- Chen, C.; Wang, Z.; Ding, Y.; Qin, Y. Manipulating T-cell metabolism to enhance immunotherapy in solid tumor. Front. Immunol. 2022, 13, 1090429. [Google Scholar] [CrossRef]
- Alvanou, M.; Lysandrou, M.; Christophi, P.; Psatha, N.; Spyridonidis, A.; Papadopoulou, A.; Yannaki, E. Empowering the potential of CAR-T cell immunotherapies by epigenetic reprogramming. Cancers 2023, 15, 1935. [Google Scholar] [CrossRef] [PubMed]
- Mak, T.W.; Grusdat, M.; Duncan, G.S.; Dostert, C.; Nonnenmacher, Y.; Cox, M.; Binsfeld, C.; Hao, Z.; Brüstle, A.; Itsumi, M.; et al. Glutathione primes T cell metabolism for inflammation. Immunity 2017, 46, 1089–1090. [Google Scholar] [CrossRef] [PubMed]
- Sena, L.A.; Li, S.; Jairaman, A.; Prakriya, M.; Ezponda, T.; Hildeman, D.A.; Wang, C.R.; Schumacker, P.T.; Licht, J.D.; Perlman, H.; et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity 2013, 38, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Chamoto, K.; Chowdhury, P.S.; Kumar, A.; Sonomura, K.; Matsuda, F.; Fagarasan, S.; Honjo, T. Mitochondrial activation chemicals synergize with surface receptor PD-1 blockade for T cell-dependent anti-tumor activity. Proc. Natl. Acad. Sci. USA 2017, 114, E761–E770. [Google Scholar] [CrossRef] [PubMed]
- Buck, M.D.; O’Sullivan, D.; Klein Geltink, R.I.; Curtis, J.D.; Chang, C.H.; Sanin, D.E.; Qiu, J.; Kretz, O.; Braas, D.; van der Windt, G.J.; et al. Mitochondrial dynamics controls T cell fate through metabolic programming. Cell 2016, 166, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Mayr, T.; Muders, M.H. Competition for nutrients or cell intrinsic programming?—Metabolic mechanisms behind the tumor promoting immune microenvironment in cancer. Signal Transduct. Target. Ther. 2021, 6, 279–280. [Google Scholar] [CrossRef]
- Teijeira, A.; Garasa, S.; Etxeberria, I.; Ga-to-Cañas, M.; Melero, I.; Delgoffe, G.M. Metabolic consequences of T-cell costimulation in anticancer immunity. Cancer Immunol. Res. 2019, 7, 1564–1569. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.K.; Lee, D.Y.; Lee, D.G.; Kim, Y.H.; Kim, S.H.; Oh, H.S.; Han, C.; Kwon, B.S. 4-1BB Signaling activates glucose and fatty acid metabolism to enhance CD8+ T cell proliferation. Cell. Mol. Immunol. 2017, 14, 748–757. [Google Scholar] [CrossRef]
- van der Windt, G.J.; Everts, B.; Chang, C.H.; Curtis, J.D.; Freitas, T.C.; Amiel, E.; Pearce, E.J.; Pearce, E.L. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity 2012, 36, 68–78. [Google Scholar] [CrossRef]
- Cui, G.; Staron, M.M.; Gray, S.M.; Ho, P.C.; Amezquita, R.A.; Wu, J.; Kaech, S.M. IL-7-induced glycerol transport and TAG synthesis promotes memory CD8+ T cell longevity. Cell 2015, 161, 750–761. [Google Scholar] [CrossRef]
- Pearce, E.L.; Poffenberger, M.C.; Chang, C.H.; Jones, R.G. Fueling immunity: Insights into metabolism and lymphocyte function. Science 2013, 342, 1242454. [Google Scholar] [CrossRef]
- Wang, T.; Gnanaprakasam, J.N.R.; Chen, X.; Kang, S.; Xu, X.; Sun, H.; Liu, L.; Rodgers, H.; Miller, E.; Cassel, T.A.; et al. Inosine is an alternative carbon source for CD8+-T-cell function under glucose restriction. Nat. Metab. 2020, 2, 635–647. [Google Scholar] [CrossRef]
- Gao, Z.W.; Yang, L.; Liu, C.; Wang, X.; Guo, W.T.; Zhang, H.Z.; Dong, K. Distinct roles of adenosine deaminase isoenzymes ADA1 and ADA2: A pan-cancer analysis. Front Immunol. 2022, 13, 903461. [Google Scholar] [CrossRef]
- Qu, Y.; Dunn, Z.S.; Chen, X.; MacMullan, M.; Cinay, G.; Wang, H.Y.; Liu, J.; Hu, F.; Wang, P. Adenosine deaminase 1 overexpression enhances the antitumor efficacy of chimeric antigen receptor-engineered T cells. Hum. Gene Ther. 2022, 33, 223–236. [Google Scholar] [CrossRef]
- Chi, H. Immunometabolism at the intersection of metabolic signaling, cell fate, and systems immunology. Cell. Mol. Immunol. 2022, 19, 299–302. [Google Scholar] [CrossRef]
- Abdelmoneim, M.; Aboalela, M.A.; Naoe, Y.; Matsumura, S.; Eissa, I.R.; Bustos-Villalobos, I.; Sibal, P.A.; Takido, Y.; Kodera, Y.; Kasuya, H. The impact of metformin on tumor-infiltrated immune cells: Preclinical and clinical studies. Int. J. Mol. Sci. 2023, 24, 13353. [Google Scholar] [CrossRef]
- Worsley, C.M.; Veale, R.B.; Mayne, E.S. The acidic tumour microenvironment: Manipulating the immune response to elicit escape. Hum. Immunol. 2022, 83, 399–408. [Google Scholar] [CrossRef]
- Halama, A.; Riesen, N.; Möller, G.; Hrabě de Angelis, M.; Adamski, J.J. Identification of biomarkers for apoptosis in cancer cell lines using metabolomics: Tools for individualized medicine. Intern. Med. 2013, 274, 425–439. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.E.; Pazdur, R.J. Docetaxel. Clin. Oncol. 1995, 13, 2643–2655. [Google Scholar] [CrossRef]
- Lyseng-Williamson, K.A.; Fenton, C. Docetaxel: A review of its use in metastatic breast cancer. Drugs 2005, 65, 2513–2531. [Google Scholar] [CrossRef]
- Mhaidat, N.M.; Wang, Y.; Kiejda, K.A.; Zhang, X.D.; Hersey, P. Docetaxel-induced apoptosis in melanoma cells is dependent on activation of caspase-2. Mol. Cancer Ther. 2007, 6, 752–761. [Google Scholar] [CrossRef]
- Alken, S.; Kelly, C.M. Benefit risk assessment and update on the use of docetaxel in the management of breast cancer. Cancer Manag. Res. 2013, 5, 357–365. [Google Scholar] [CrossRef]
- Wang, D.; Tang, Y.; Feng, F.; Qi, M.; Fang, J.; Zhang, Y.; Chai, Y.; Cao, Y.; Lv, D. Investigation of the apoptosis-inducing effect of docetaxel by comprehensive LC-MS-based metabolomics and network pharmacology approaches. Biomed. Chromatogr. 2022, 36, e5417. [Google Scholar] [CrossRef]
- Chen, S.; Sutiman, N.; Zhang, C.Z.; Yu, Y.; Lam, S.; Khor, C.C.; Chowbay, B. Pharmacogenetics of irinotecan, doxorubicin and docetaxel transporters in asian and caucasian cancer patients: A comparative review. Drug Metab. Rev. 2016, 48, 502–540. [Google Scholar] [CrossRef]
- Hu, K.; Li, K.; Lv, J.; Feng, J.; Chen, J.; Wu, H.; Cheng, F.; Jiang, W.; Wang, J.; Pei, H.; et al. Suppression of the SLC7A11/glutathione axis causes synthetic lethality in KRAS-mutant lung adenocarcinoma. J. Clin. Investig. 2020, 130, 1752–1766. [Google Scholar] [CrossRef]
- Ogiwara, H.; Takahashi, K.; Sasaki, M.; Kuroda, T.; Yoshida, H.; Watanabe, R.; Maruyama, A.; Makinoshima, H.; Chiwaki, F.; Sasaki, H.; et al. Targeting the vulnerability of glutathione metabolism in ARID1A-deficient cancers. Cancer Cell 2019, 35, 177–190. [Google Scholar] [CrossRef]
- Ursini, F.; Maiorino, M. Lipid peroxidation and ferroptosis: The role of GSH and GPx4. Free Radic. Biol. Med. 2020, 152, 175–185. [Google Scholar] [CrossRef]
- Liu, X.; Peng, S.; Tang, G.; Xu, G.; Xie, Y.; Shen, D.; Zhu, M.; Huang, Y.; Wang, X.; Yu, H.; et al. Fasting-mimicking diet synergizes with ferroptosis against quiescent, chemotherapy-resistant cells. EBioMedicine 2023, 90, 104496. [Google Scholar] [CrossRef]
- Darwiche, W.; Gomila, C.; Ouled-Haddou, H.; Naudot, M.; Doualle, C.; Morel, P.; Nguyen-Khac, F.; Garçon, L.; Marolleau, J.P.; Ghamlouch, H. Ascorbic acid (vitamin C) synergistically enhances the therapeutic effect of targeted therapy in chronic lymphocytic leukemia. J. Exp. Clin. Cancer Res. 2020, 39, 228–244. [Google Scholar] [CrossRef]
- Gao, J.; Zhao, Y.; Tao, T.; Gan, X.; Yu, H. The role of PKM2 in the regulation of mitochondrial function: Focus on mitochondrial metabolism, oxidative stress, dynamic, and apoptosis. PKM2 in mitochondrial function. Oxid. Med. Cell. Longev. 2022, 2022, 7702681. [Google Scholar] [CrossRef]
- Asgharzadeh, M.R.; Barar, J.; Pourseif, M.M.; Eskandani, M.; Jafari Niya, M.; Mashayekhi, M.R.; Omidi, Y. Molecular machineries of pH dysregulation in tumor microenvironment: Potential targets for cancer therapy. Bioimpacts 2017, 7, 115–133. [Google Scholar] [CrossRef]
- Yeo, M.; Kim, D.K.; Kim, Y.B.; Oh, T.Y.; Lee, J.E.; Cho, S.W.; Kim, H.C.; Hahm, K.B. Selective induction of apoptosis with proton pump inhibitor in gastric cancer cells. Clin. Cancer Res. 2004, 10, 8687–8696. [Google Scholar] [CrossRef]
- Lee, J.; Choi, M.K.; Song, I.S. Recent advances in doxorubicin formulation to enhance pharmacokinetics and tumor targeting. Pharmaceuticals 2023, 16, 802. [Google Scholar] [CrossRef]
- Kalinin, S.; Malkova, A.; Sharonova, T.; Sharoyko, V.; Bunev, A.; Supuran, C.T.; Krasavin, M. Carbonic anhydrase IX inhibitors as candidates for combination therapy of solid tumors. Int. J. Mol. Sci. 2021, 22, 13405. [Google Scholar] [CrossRef]
- Zhou, D.; Duan, Z.; Li, Z.; Ge, F.; Wei, R.; Kong, L. The significance of glycolysis in tumor progression and its relationship with the tumor microenvironment. Front. Pharmacol. 2022, 13, 1091779. [Google Scholar] [CrossRef]
- Dubyak, G.R. Luciferase-assisted detection of extracellular ATP and ATP metabolites during immunogenic death of cancer cells. Methods Enzymol. 2019, 629, 81–102. [Google Scholar] [CrossRef]
- Chong, W.P.; Yusufi, F.N.; Lee, D.Y.; Reddy, S.G.; Wong, N.S.; Heng, C.K.; Yap, M.G.; Ho, Y.S. Metabolomics-based identification of apoptosis-inducing metabolites in recombinant fed-batch CHO culture media. J. Biotechnol. 2010, 151, 218–224. [Google Scholar] [CrossRef]
- Zhao, H.; Achreja, A.; Iessi, E.; Logozzi, M.; Mizzoni, D.; Di Raimo, R.; Nagrath, D.; Fais, S. The key role of extracellular vesicles in the metastatic process. Biochim. Biophys. Acta Rev. Cancer 2018, 1869, 64–77. [Google Scholar] [CrossRef]
- Jiramongkol, Y.; Lam, E.W. Multifaceted oncogenic role of adipocytes in the tumour microenvironment. Adv. Exp. Med. Biol. 2020, 1219, 125–142. [Google Scholar] [CrossRef]
- Delort, L.; Cholet, J.; Decombat, C.; Vermerie, M.; Dumontet, C.; Castelli, F.A.; Fenaille, F.; Auxenfans, C.; Rossary, A.; Caldefie-Chezet, F. The adipose microenvironment dysregulates the mammary myoepithelial cells and could participate to the progression of breast cancer. Front. Cell Dev. Biol. 2021, 8, 571948. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug/ Compound | Metabolite/ Enzyme/Pathway | Cancer/Model | Reference |
|---|---|---|---|
| staurosporine 5-fluorouracil etoposide | increase of alanine, arginine, glutamate, and acetyl carnitine (taurine metabolism) | HEK293 and HepG2 cells | [238] |
| docetaxel | glycolysis/gluconeogenesis, cysteine, and methionine metabolism, and arginine biosynthesis | cervical cancer | [243] |
| sulfasalazine HG106 inhibitor | increase in intracellular cysteine and glutamate secretion (GSH biosynthesis) | patients affected by KRAS-mutated lung adenocarcinoma | [245] |
| fasting combined with 5-fluorouracil oxaliplatin | low levels of adenosine and deoxyadenosine monophosphate, high levels of lipidic and organic compounds | colorectal cancer cells | [248] |
| ascorbic acid combined with ibrutinib, idelalisib, and venetoclax | transport of H+ ions and generation of ROS | chronic lymphocytic leukemia | [249] |
| inhibitors of NHE: amiloride, benzyolguanidinium, cimetidine, clonidine, harmaline cariporide, compound 9T, 2-aminophenoxazine inhibitor of NHE-3-one, ethylisopropylamiloride, hexamethylamiloride, dimethylamiloride | HCO3− and H+-based transporting systems | human cancer cells | [251] |
| V-ATPase inhibitors: bafilomycin, concanamycin, salicylihalamide A, apicularen A, lobatamide A, oximidine I, cruentaren, NiK12192, PPI SB 242784, and FR202126 | H+ gradient produced by H+-ATPase | breast cancer, esophageal carcinoma, lung carcinoma, hepatocellular and pancreatic carcinoma, oral squamous cell carcinoma, sarcoma and other solid tumors | [251] |
| pantoprazole | H+/K+-ATPase | xenograft model of nude mice with gastric cancer | [252] |
| vinblastine, doxorubicin, vincristine, mitoxantrone, paclitaxel in combination with CA IX inhibitors | H+ in the extracellular space | many cancer cell lines; xenograft model; patients | [254] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franzese, O.; Ancona, P.; Bianchi, N.; Aguiari, G. Apoptosis, a Metabolic “Head-to-Head” between Tumor and T Cells: Implications for Immunotherapy. Cells 2024, 13, 924. https://doi.org/10.3390/cells13110924
Franzese O, Ancona P, Bianchi N, Aguiari G. Apoptosis, a Metabolic “Head-to-Head” between Tumor and T Cells: Implications for Immunotherapy. Cells. 2024; 13(11):924. https://doi.org/10.3390/cells13110924
Chicago/Turabian StyleFranzese, Ornella, Pietro Ancona, Nicoletta Bianchi, and Gianluca Aguiari. 2024. "Apoptosis, a Metabolic “Head-to-Head” between Tumor and T Cells: Implications for Immunotherapy" Cells 13, no. 11: 924. https://doi.org/10.3390/cells13110924
APA StyleFranzese, O., Ancona, P., Bianchi, N., & Aguiari, G. (2024). Apoptosis, a Metabolic “Head-to-Head” between Tumor and T Cells: Implications for Immunotherapy. Cells, 13(11), 924. https://doi.org/10.3390/cells13110924