

Mitochondrial Aldehyde Dehydrogenase 2 (ALDH2) Protects against Binge Alcohol-Mediated Gut and Brain Injury

 , ,
, ,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Histological H&E and TUNEL Staining
2.2. Measurements of Serum LPS, IL-6, TNF-α, and EtOH Contents and Caspase-3 Activity
2.3. FITC-Dextran 4 kDa Analysis to Determine the Rate of Intestinal Permeability
2.4. Fluoro-Jade-C Staining to Determine the Rate of Neurodegeneration
2.5. Cell Culture, ALDH2 Activity Measurement, and Immunoblot Analysis
2.6. MTT Assay for Measurement of Neural Cell Viability
2.7. Immunoblot Analysis of Gut Enterocytes and Hippocampal Extracts
2.8. Statistical Analysis
3. Results
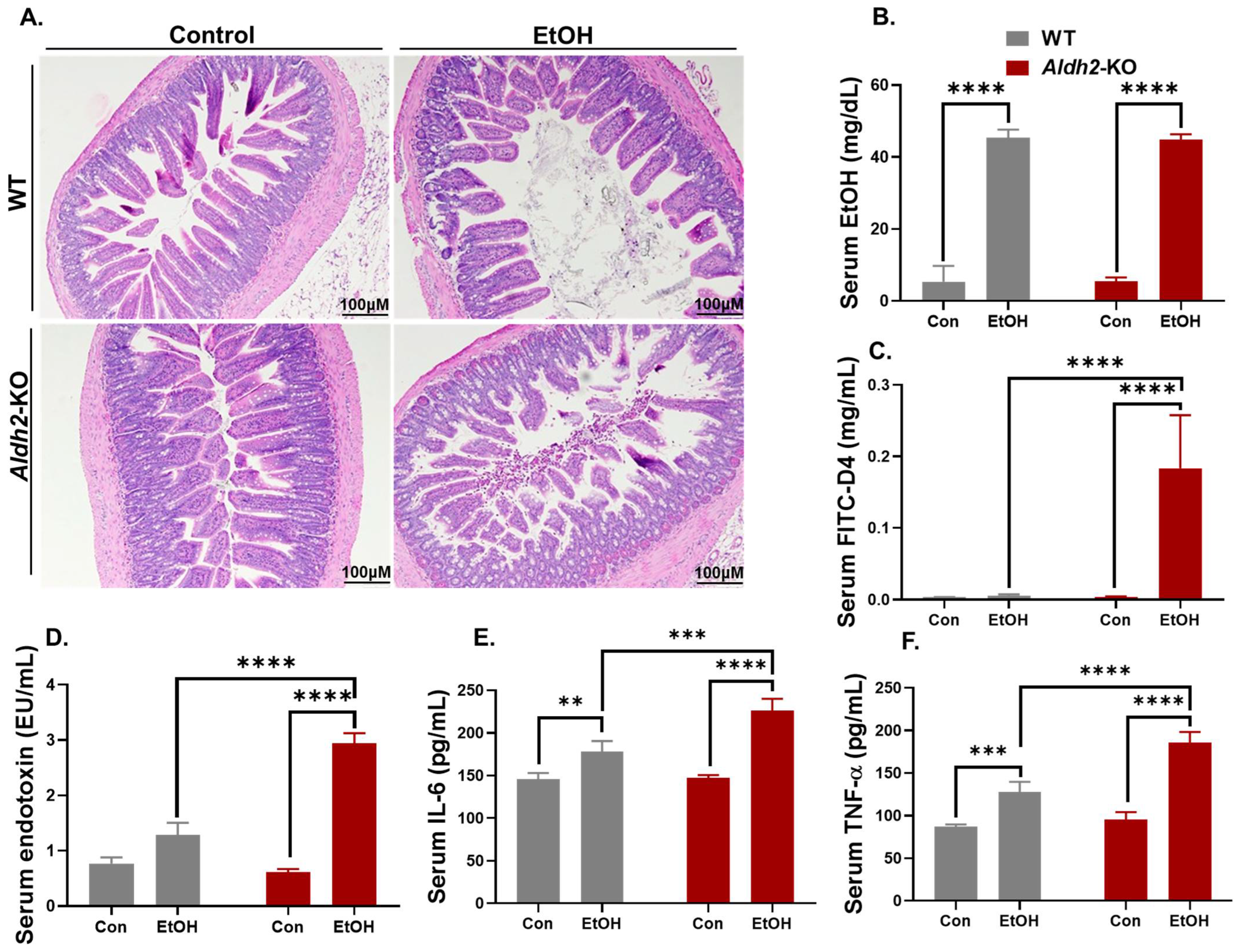
3.1. Binge Alcohol Exposure Caused Intestinal Disintegration and Increased Serum FITC-D4 and Endotoxin (LPS) Levels in Aldh2-KO Mice
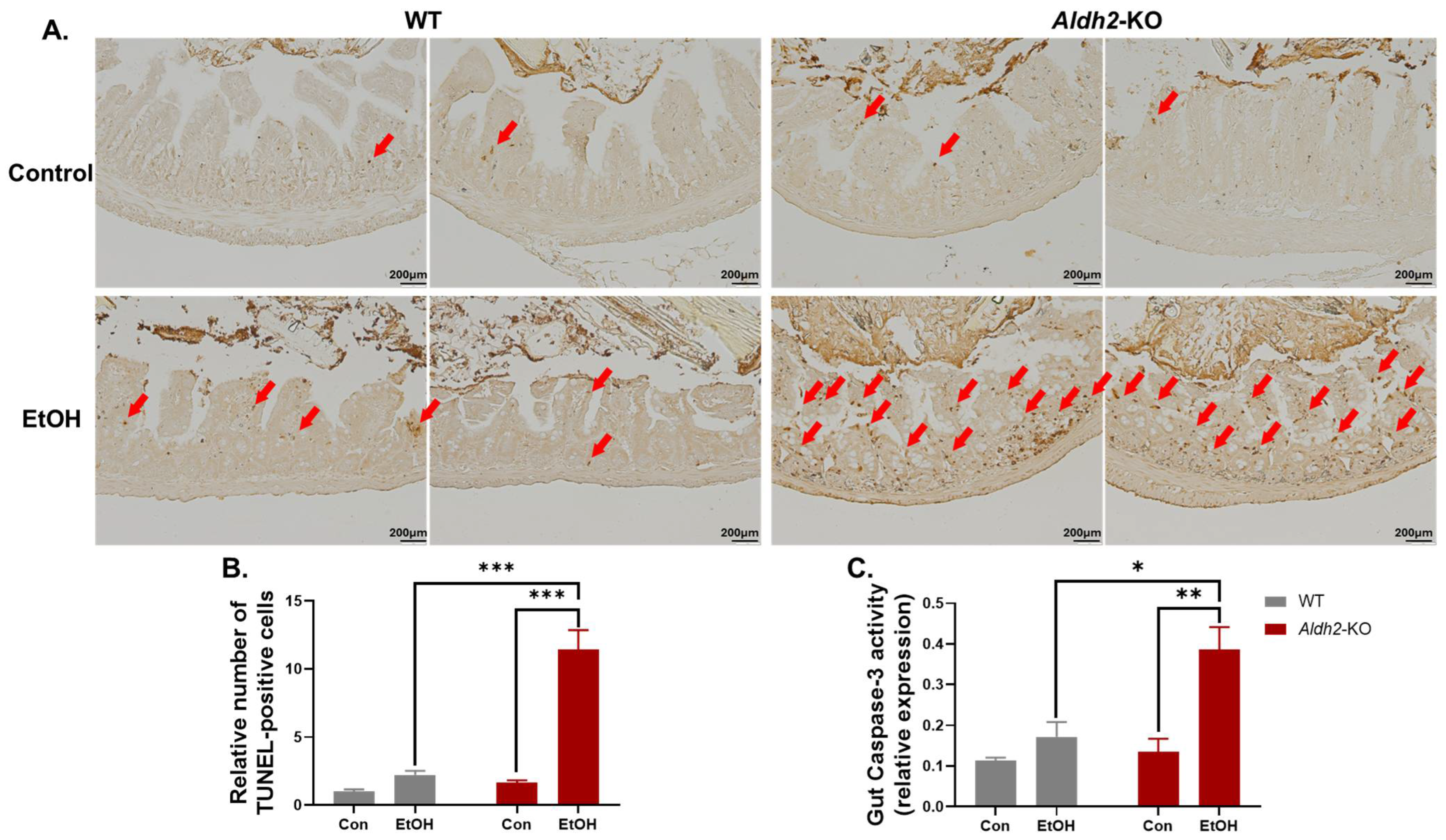
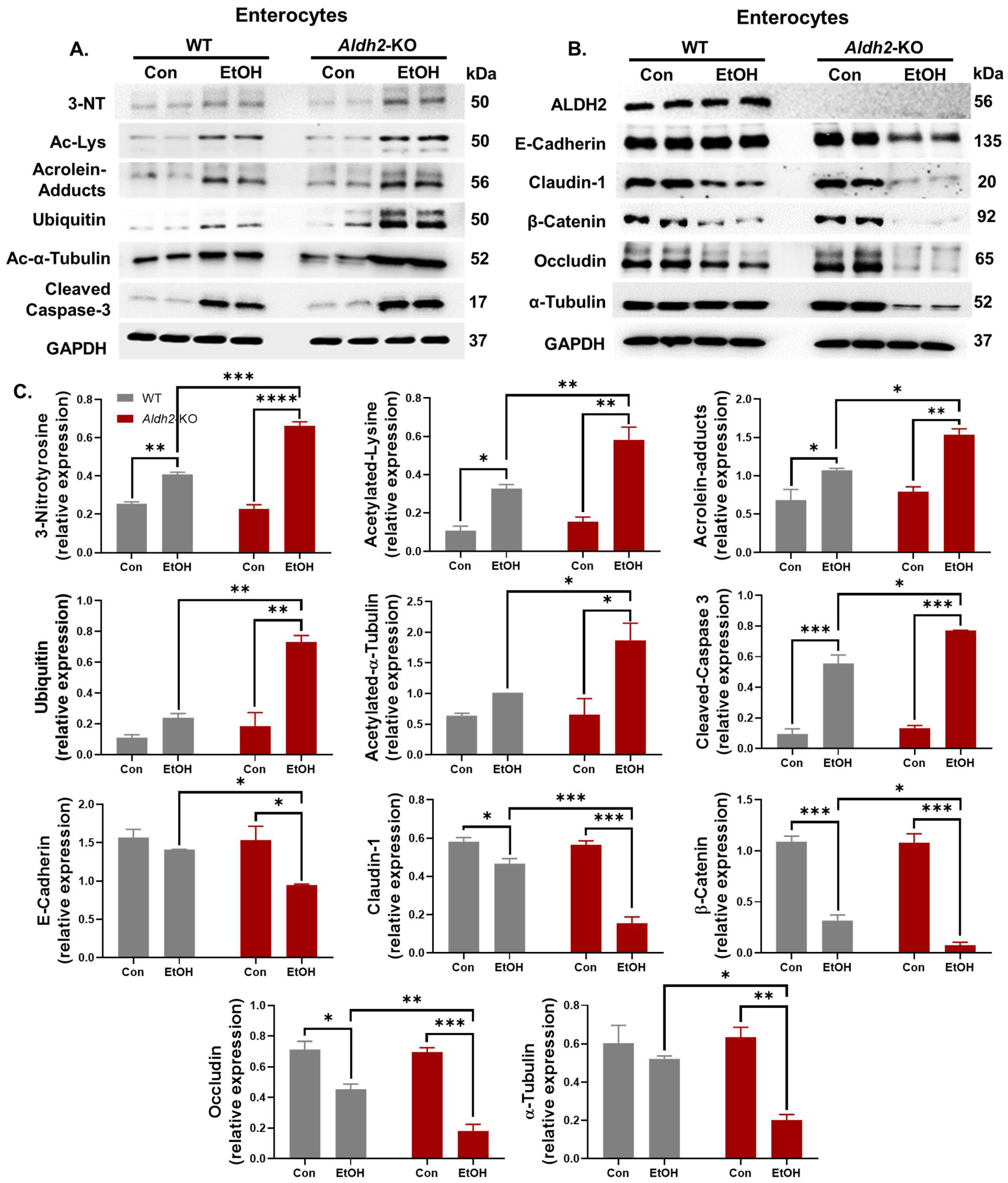
3.2. Binge Alcohol Exposure Elevated Intestinal Apoptosis, Oxidative Stress-Related PTMs, and Degradation of Gut TJ/AJ Proteins in Aldh2-KO Mice
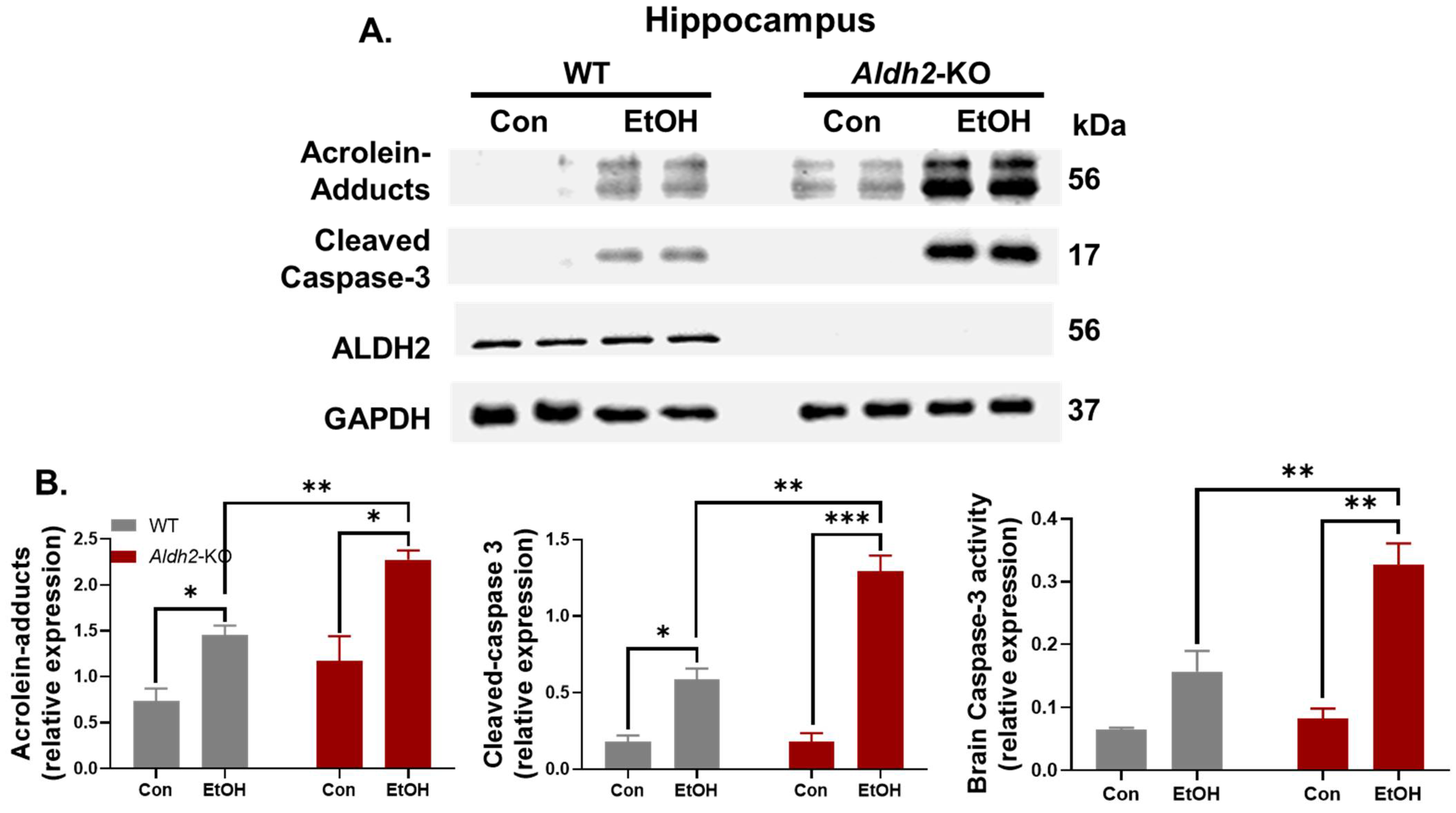
3.3. Binge Alcohol Exposure Induced Neurodegeneration, Oxidative Stress, and Neuronal Apoptosis in Aldh2-KO Mice
3.4. ALDH2 Suppression Enhanced Oxidative Stress-Mediated PTMs and Apoptosis of Neuronal Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zakhari, S. Overview: How is alcohol metabolized by the body? Alcohol Res. Health 2006, 29, 245–254. [Google Scholar] [PubMed]
- Lieber, C.S. Microsomal ethanol-oxidizing system (MEOS): The first 30 years (1968–1998)—A review. Alcohol. Clin. Exp. Res. 1999, 23, 991–1007. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.U.; Van Wassenhove, L.D.; Logas, K.R.; Minhas, P.S.; Andreasson, K.I.; Weinberg, K.I.; Chen, C.H.; Mochly-Rosen, D. Aldehyde dehydrogenase 2 activity and aldehydic load contribute to neuroinflammation and Alzheimer’s disease related pathology. Acta Neuropathol. Commun. 2019, 7, 190. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Borinskaya, S.; Yoshimura, K.; Kal’ina, N.; Marusin, A.; Stepanov, V.A.; Qin, Z.; Khaliq, S.; Lee, M.Y.; Yang, Y.; et al. Refined geographic distribution of the oriental ALDH2*504Lys (nee 487Lys) variant. Ann. Hum. Genet. 2009, 73, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Goldman, D. Aldehyde Dehydrogenase Deficiency as Cause of Facial Flushing Reaction to Alcohol in Japanese. Alcohol Health Res. World 1995, 19, 48–49. [Google Scholar] [PubMed]
- Masaki, T.; Mochizuki, H.; Matsushita, S.; Yokoyama, A.; Kamakura, K.; Higuchi, S. Association of aldehyde dehydrogenase-2 polymorphism with alcoholic polyneuropathy in humans. Neurosci. Lett. 2004, 363, 288–290. [Google Scholar] [CrossRef]
- D’Souza, Y.; Elharram, A.; Soon-Shiong, R.; Andrew, R.D.; Bennett, B.M. Characterization of Aldh2 (-/-) mice as an age-related model of cognitive impairment and Alzheimer’s disease. Mol. Brain 2015, 8, 27. [Google Scholar] [CrossRef]
- Kamino, K.; Nagasaka, K.; Imagawa, M.; Yamamoto, H.; Yoneda, H.; Ueki, A.; Kitamura, S.; Namekata, K.; Miki, T.; Ohta, S. Deficiency in mitochondrial aldehyde dehydrogenase increases the risk for late-onset Alzheimer’s disease in the Japanese population. Biochem. Biophys. Res. Commun. 2000, 273, 192–196. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.M.; Stewart, R.; Shin, I.S.; Jung, J.S.; Yoon, J.S. Assessment of association between mitochondrial aldehyde dehydrogenase polymorphism and Alzheimer’s disease in an older Korean population. Neurobiol. Aging 2004, 25, 295–301. [Google Scholar] [CrossRef]
- Wang, B.; Wang, J.; Zhou, S.; Tan, S.; He, X.; Yang, Z.; Xie, Y.C.; Li, S.; Zheng, C.; Ma, X. The association of mitochondrial aldehyde dehydrogenase gene (ALDH2) polymorphism with susceptibility to late-onset Alzheimer’s disease in Chinese. J. Neurol. Sci. 2008, 268, 172–175. [Google Scholar] [CrossRef]
- Ueno, M.; Yoshino, Y.; Mori, H.; Funahashi, Y.; Kumon, H.; Ochi, S.; Ozaki, T.; Tachibana, A.; Yoshida, T.; Shimizu, H.; et al. Association Study and Meta-Analysis of Polymorphisms and Blood mRNA Expression of the ALDH2 Gene in Patients with Alzheimer’s Disease. J. Alzheimer’s Dis. JAD 2022, 87, 863–871. [Google Scholar] [CrossRef]
- Yokoyama, A.; Muramatsu, T.; Ohmori, T.; Yokoyama, T.; Okuyama, K.; Takahashi, H.; Hasegawa, Y.; Higuchi, S.; Maruyama, K.; Shirakura, K.; et al. Alcohol-related cancers and aldehyde dehydrogenase-2 in Japanese alcoholics. Carcinogenesis 1998, 19, 1383–1387. [Google Scholar] [CrossRef]
- Brooks, P.J.; Enoch, M.A.; Goldman, D.; Li, T.K.; Yokoyama, A. The alcohol flushing response: An unrecognized risk factor for esophageal cancer from alcohol consumption. PLoS Med. 2009, 6, e50. [Google Scholar] [CrossRef]
- Chen, W.Y.; Zhang, J.; Ghare, S.; Barve, S.; McClain, C.; Joshi-Barve, S. Acrolein Is a Pathogenic Mediator of Alcoholic Liver Disease and the Scavenger Hydralazine Is Protective in Mice. Cell. Mol. Gastroenterol. Hepatol. 2016, 2, 685–700. [Google Scholar] [CrossRef] [PubMed]
- Vatsalya, V.; Kong, M.; Gobejishvili, L.; Chen, W.Y.; Srivastava, S.; Barve, S.; McClain, C.; Joshi-Barve, S. Urinary acrolein metabolite levels in severe acute alcoholic hepatitis patients. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 316, G115–G122. [Google Scholar] [CrossRef] [PubMed]
- Keshavarzian, A.; Farhadi, A.; Forsyth, C.B.; Rangan, J.; Jakate, S.; Shaikh, M.; Banan, A.; Fields, J.Z. Evidence that chronic alcohol exposure promotes intestinal oxidative stress, intestinal hyperpermeability and endotoxemia prior to development of alcoholic steatohepatitis in rats. J. Hepatol. 2009, 50, 538–547. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.E.; Song, B.J. Pomegranate prevents binge alcohol-induced gut leakiness and hepatic inflammation by suppressing oxidative and nitrative stress. Redox Biol. 2018, 18, 266–278. [Google Scholar] [CrossRef]
- Lowe, P.P.; Morel, C.; Ambade, A.; Iracheta-Vellve, A.; Kwiatkowski, E.; Satishchandran, A.; Furi, I.; Cho, Y.; Gyongyosi, B.; Catalano, D.; et al. Chronic alcohol-induced neuroinflammation involves CCR2/5-dependent peripheral macrophage infiltration and microglia alterations. J. Neuroinflamm. 2020, 17, 296. [Google Scholar] [CrossRef]
- Leclercq, S.; Cani, P.D.; Neyrinck, A.M.; Stärkel, P.; Jamar, F.; Mikolajczak, M.; Delzenne, N.M.; de Timary, P. Role of intestinal permeability and inflammation in the biological and behavioral control of alcohol-dependent subjects. Brain Behav. Immun. 2012, 26, 911–918. [Google Scholar] [CrossRef]
- Engen, P.A.; Green, S.J.; Voigt, R.M.; Forsyth, C.B.; Keshavarzian, A. The Gastrointestinal Microbiome: Alcohol Effects on the Composition of Intestinal Microbiota. Alcohol Res. Curr. Rev. 2015, 37, 223–236. [Google Scholar]
- Stärkel, P.; Leclercq, S.; de Timary, P.; Schnabl, B. Intestinal dysbiosis and permeability: The yin and yang in alcohol dependence and alcoholic liver disease. Clin. Sci. 2018, 132, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Obrenovich, M.E.M. Leaky Gut, Leaky Brain? Microorganisms 2018, 6, 107. [Google Scholar] [CrossRef]
- Carbia, C.; Bastiaanssen, T.F.S.; Iannone, L.F.; García-Cabrerizo, R.; Boscaini, S.; Berding, K.; Strain, C.R.; Clarke, G.; Stanton, C.; Dinan, T.G.; et al. The Microbiome-Gut-Brain axis regulates social cognition & craving in young binge drinkers. EBioMedicine 2023, 89, 104442. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, B.; Astarita, C.; Boffo, S.; Massaro-Giordano, M.; Antonella Ianuzzi, C.; Caporaso, A.; Macaluso, M.; Giordano, A. LPS-induced inflammatory response triggers cell cycle reactivation in murine neuronal cells through retinoblastoma proteins induction. Cell Cycle 2017, 16, 2330–2336. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Xue, Z.; Zheng, Y.; Li, S.; Zhou, L.; Cao, L.; Zou, Y. Selenium supplementation enhanced the expression of selenoproteins in hippocampus and played a neuroprotective role in LPS-induced neuroinflammation. Int. J. Biol. Macromol. 2023, 234, 123740. [Google Scholar] [CrossRef] [PubMed]
- Kalyan, M.; Tousif, A.H.; Sonali, S.; Vichitra, C.; Sunanda, T.; Praveenraj, S.S.; Ray, B.; Gorantla, V.R.; Rungratanawanich, W.; Mahalakshmi, A.M.; et al. Role of Endogenous Lipopolysaccharides in Neurological Disorders. Cells 2022, 11, 4038. [Google Scholar] [CrossRef] [PubMed]
- Chidambaram, S.B.; Essa, M.M.; Rathipriya, A.G.; Bishir, M.; Ray, B.; Mahalakshmi, A.M.; Tousif, A.H.; Sakharkar, M.K.; Kashyap, R.S.; Friedland, R.P.; et al. Gut dysbiosis, defective autophagy and altered immune responses in neurodegenerative diseases: Tales of a vicious cycle. Pharmacol. Ther. 2022, 231, 107988. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Q.; Su, Z.; Dai, C.G.; Song, J.L.; Qian, B. Multi-omics analysis reveals BDE47 induces depression-like behaviors in mice by interfering with the 2-arachidonoyl glycerol-associated microbiota-gut-brain axis. Ecotoxicol. Environ. Saf. 2023, 259, 115041. [Google Scholar] [CrossRef]
- Ghosh, S.S.; Wang, J.; Yannie, P.J.; Ghosh, S. Intestinal Barrier Dysfunction, LPS Translocation, and Disease Development. J. Endocr. Soc. 2020, 4, bvz039. [Google Scholar] [CrossRef]
- Alnouti, Y.; Klaassen, C.D. Tissue distribution, ontogeny, and regulation of aldehyde dehydrogenase (Aldh) enzymes mRNA by prototypical microsomal enzyme inducers in mice. Toxicol. Sci. Off. J. Soc. Toxicol. 2008, 101, 51–64. [Google Scholar] [CrossRef]
- Picklo, M.J.; Olson, S.J.; Markesbery, W.R.; Montine, T.J. Expression and activities of aldo-keto oxidoreductases in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2001, 60, 686–695. [Google Scholar] [CrossRef] [PubMed]
- Song, K.; Kim, S.; Na, J.Y.; Park, J.H.; Kim, J.K.; Kim, J.H.; Kwon, J. Rutin attenuates ethanol-induced neurotoxicity in hippocampal neuronal cells by increasing aldehyde dehydrogenase 2. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2014, 72, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Deza-Ponzio, R.; Herrera, M.L.; Bellini, M.J.; Virgolini, M.B.; Hereñú, C.B. Aldehyde dehydrogenase 2 in the spotlight: The link between mitochondria and neurodegeneration. Neurotoxicology 2018, 68, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Xia, P.; Zhang, F.; Yuan, Y.; Chen, C.; Huang, Y.; Li, L.; Wang, E.; Guo, Q.; Ye, Z. ALDH 2 conferred neuroprotection on cerebral ischemic injury by alleviating mitochondria-related apoptosis through JNK/caspase-3 signing pathway. Int. J. Biol. Sci. 2020, 16, 1303–1323. [Google Scholar] [CrossRef] [PubMed]
- Rungratanawanich, W.; Lin, Y.; Wang, X.; Kawamoto, T.; Chidambaram, S.B.; Song, B.J. ALDH2 deficiency increases susceptibility to binge alcohol-induced gut leakiness, endotoxemia, and acute liver injury in mice through the gut-liver axis. Redox Biol. 2023, 59, 102577. [Google Scholar] [CrossRef] [PubMed]
- Gow, A.J.; Duran, D.; Malcolm, S.; Ischiropoulos, H. Effects of peroxynitrite-induced protein modifications on tyrosine phosphorylation and degradation. FEBS Lett. 1996, 385, 63–66. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.T.; Mun, S.H.; Lee, C.S.; Hwang, C.S. Control of protein degradation by N-terminal acetylation and the N-end rule pathway. Exp. Mol. Med. 2018, 50, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.E.; Yu, L.R.; Abdelmegeed, M.A.; Yoo, S.H.; Song, B.J. Apoptosis of enterocytes and nitration of junctional complex proteins promote alcohol-induced gut leakiness and liver injury. J. Hepatol. 2018, 69, 142–153. [Google Scholar] [CrossRef] [PubMed]
- Ikenari, T.; Kurata, H.; Satoh, T.; Hata, Y.; Mori, T. Evaluation of Fluoro-Jade C Staining: Specificity and Application to Damaged Immature Neuronal Cells in the Normal and Injured Mouse Brain. Neuroscience 2020, 425, 146–156. [Google Scholar] [CrossRef]
- Ikenari, T.; Kawaguchi, T.; Ota, R.; Matsui, M.; Yoshida, R.; Mori, T. Improvement in Double Staining with Fluoro-Jade C and Fluorescent Immunostaining: FJC Staining Is Not Specific to Degenerating Mature Neurons. J. Histochem. Cytochem. Off. J. Histochem. Soc. 2021, 69, 597–610. [Google Scholar] [CrossRef]
- Garcia, J.; Chang, R.; Steinberg, R.A.; Arce, A.; Yang, J.; Van Der Eb, P.; Abdullah, T.; Chandrashekar, D.V.; Eck, S.M.; Meza, P.; et al. Modulation of hepatic amyloid precursor protein and lipoprotein receptor-related protein 1 by chronic alcohol intake: Potential link between liver steatosis and amyloid-β. Front. Physiol. 2022, 13, 930402. [Google Scholar] [CrossRef]
- İlhan, A.O.; Can, B.; Kar, F.; Gündoğdu, A.; Söğüt, İ.; Kanbak, G. An Investigation into the Protective Effects of Various Doses of Boric Acid on Liver, Kidney, and Brain Tissue Damage Caused by High Levels of Acute Alcohol Consumption. Biol. Trace Elem. Res. 2023, 201, 5346–5357. [Google Scholar] [CrossRef]
- Wei, H.; Yu, C.; Zhang, C.; Ren, Y.; Guo, L.; Wang, T.; Chen, F.; Li, Y.; Zhang, X.; Wang, H.; et al. Butyrate ameliorates chronic alcoholic central nervous damage by suppressing microglia-mediated neuroinflammation and modulating the microbiome-gut-brain axis. Biomed. Pharmacother. Biomed. Pharmacother. 2023, 160, 114308. [Google Scholar] [CrossRef] [PubMed]
- Hamada, K.; Ferguson, L.B.; Mayfield, R.D.; Krishnan, H.R.; Maienschein-Cline, M.; Lasek, A.W. Binge-like ethanol drinking activates anaplastic lymphoma kinase signaling and increases the expression of STAT3 target genes in the mouse hippocampus and prefrontal cortex. Genes Brain Behav. 2021, 20, e12729. [Google Scholar] [CrossRef]
- Rapp, C.; Hamilton, J.; Richer, K.; Sajjad, M.; Yao, R.; Thanos, P.K. Alcohol binge drinking decreases brain glucose metabolism and functional connectivity in adolescent rats. Metab. Brain Dis. 2022, 37, 1901–1908. [Google Scholar] [CrossRef]
- Sun, J.K.; Wu, D.; Wong, G.C.; Lau, T.M.; Yang, M.; Hart, R.P.; Kwan, K.M.; Chan, H.Y.E.; Chow, H.M. Chronic alcohol metabolism results in DNA repair infidelity and cell cycle-induced senescence in neurons. Aging Cell 2023, 22, e13772. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.C. The endotoxin hypothesis of neurodegeneration. J. Neuroinflamm. 2019, 16, 180. [Google Scholar] [CrossRef]
- Choi, Y.; Abdelmegeed, M.A.; Song, B.J. Preventive effects of indole-3-carbinol against alcohol-induced liver injury in mice via antioxidant, anti-inflammatory, and anti-apoptotic mechanisms: Role of gut-liver-adipose tissue axis. J. Nutr. Biochem. 2018, 55, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.M.; Liu, A.J.; Zang, P.; Dong, W.Z.; Ying, L.; Wang, W.; Xu, P.; Song, X.R.; Cai, J.; Zhang, S.Q.; et al. ALDH2 protects against stroke by clearing 4-HNE. Cell Res. 2013, 23, 915–930. [Google Scholar] [CrossRef]
- Fu, S.H.; Zhang, H.F.; Yang, Z.B.; Li, T.B.; Liu, B.; Lou, Z.; Ma, Q.L.; Luo, X.J.; Peng, J. Alda-1 reduces cerebral ischemia/reperfusion injury in rat through clearance of reactive aldehydes. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2014, 387, 87–94. [Google Scholar] [CrossRef]
- Chen, W.Y.; Wang, M.; Zhang, J.; Barve, S.S.; McClain, C.J.; Joshi-Barve, S. Acrolein Disrupts Tight Junction Proteins and Causes Endoplasmic Reticulum Stress-Mediated Epithelial Cell Death Leading to Intestinal Barrier Dysfunction and Permeability. Am. J. Pathol. 2017, 187, 2686–2697. [Google Scholar] [CrossRef] [PubMed]
- Rungratanawanich, W.; Qu, Y.; Wang, X.; Essa, M.M.; Song, B.J. Advanced glycation end products (AGEs) and other adducts in aging-related diseases and alcohol-mediated tissue injury. Exp. Mol. Med. 2021, 53, 168–188. [Google Scholar] [CrossRef] [PubMed]
- Simon, L.; Souza-Smith, F.M.; Molina, P.E. Alcohol-Associated Tissue Injury: Current Views on Pathophysiological Mechanisms. Annu. Rev. Physiol. 2022, 84, 87–112. [Google Scholar] [CrossRef] [PubMed]
- Isse, T.; Matsuno, K.; Oyama, T.; Kitagawa, K.; Kawamoto, T. Aldehyde dehydrogenase 2 gene targeting mouse lacking enzyme activity shows high acetaldehyde level in blood, brain, and liver after ethanol gavages. Alcohol. Clin. Exp. Res. 2005, 29, 1959–1964. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, K.K.; Samak, G.; Shukla, P.K.; Mir, H.; Gangwar, R.; Manda, B.; Isse, T.; Kawamoto, T.; Salaspuro, M.; Kaihovaara, P.; et al. ALDH2 Deficiency Promotes Ethanol-Induced Gut Barrier Dysfunction and Fatty Liver in Mice. Alcohol. Clin. Exp. Res. 2015, 39, 1465–1475. [Google Scholar] [CrossRef] [PubMed]
- Knopp, R.C.; Lee, S.H.; Hollas, M.; Nepomuceno, E.; Gonzalez, D.; Tam, K.; Aamir, D.; Wang, Y.; Pierce, E.; BenAissa, M.; et al. Interaction of oxidative stress and neurotrauma in ALDH2(-/-) mice causes significant and persistent behavioral and pro-inflammatory effects in a tractable model of mild traumatic brain injury. Redox Biol. 2020, 32, 101486. [Google Scholar] [CrossRef] [PubMed]
- Herr, S.A.; Shi, L.; Gianaris, T.; Jiao, Y.; Sun, S.; Race, N.; Shapiro, S.; Shi, R. Critical role of mitochondrial aldehyde dehydrogenase 2 in acrolein sequestering in rat spinal cord injury. Neural Regen. Res. 2022, 17, 1505–1511. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.S.; Chen, Y.H.; Hu, J.T.; Chiu, C.F.; Hung, S.W.; Chang, Y.C.; Chiu, C.C.; Chuang, H.L. Aldehyde Dehydrogenase Mutation Exacerbated High-Fat-Diet-Induced Nonalcoholic Fatty Liver Disease with Gut Microbiota Remodeling in Male Mice. Biology 2021, 10, 737. [Google Scholar] [CrossRef]
- Yoval-Sánchez, B.; Rodríguez-Zavala, J.S. Differences in susceptibility to inactivation of human aldehyde dehydrogenases by lipid peroxidation byproducts. Chem. Res. Toxicol. 2012, 25, 722–729. [Google Scholar] [CrossRef]
- Osna, N.A.; Donohue, T.M., Jr.; Kharbanda, K.K. Alcoholic Liver Disease: Pathogenesis and Current Management. Alcohol Res. Curr. Rev. 2017, 38, 147–161. [Google Scholar]
- Schnabl, B.; Brenner, D.A. Interactions between the intestinal microbiome and liver diseases. Gastroenterology 2014, 146, 1513–1524. [Google Scholar] [CrossRef] [PubMed]
- Mutlu, E.A.; Gillevet, P.M.; Rangwala, H.; Sikaroodi, M.; Naqvi, A.; Engen, P.A.; Kwasny, M.; Lau, C.K.; Keshavarzian, A. Colonic microbiome is altered in alcoholism. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G966–G978. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Wu, X.; Block, M.L.; Liu, Y.; Breese, G.R.; Hong, J.S.; Knapp, D.J.; Crews, F.T. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 2007, 55, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Zimatkin, S.M.; Rout, U.K.; Koivusalo, M.; Bühler, R.; Lindros, K.O. Regional distribution of low-Km mitochondrial aldehyde dehydrogenase in the rat central nervous system. Alcohol. Clin. Exp. Res. 1992, 16, 1162–1167. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M.J.; Malek, K.; Crabb, D.W. Distribution of messenger RNAs for aldehyde dehydrogenase 1, aldehyde dehydrogenase 2, and aldehyde dehydrogenase 5 in human tissues. J. Investig. Med. Off. Publ. Am. Fed. Clin. Res. 1996, 44, 42–46. [Google Scholar]
- Palmer, K.R.; Jenkins, W.J. Aldehyde dehydrogenase in alcoholic subjects. Hepatology 1985, 5, 260–263. [Google Scholar] [CrossRef] [PubMed]
- Tomita, Y.; Haseba, T.; Kurosu, M.; Watanabe, T. Effects of chronic ethanol intoxication on aldehyde dehydrogenase in mouse liver. Alcohol Alcohol. 1992, 27, 171–180. [Google Scholar] [PubMed]
- Moon, K.H.; Hood, B.L.; Kim, B.J.; Hardwick, J.P.; Conrads, T.P.; Veenstra, T.D.; Song, B.J. Inactivation of oxidized and S-nitrosylated mitochondrial proteins in alcoholic fatty liver of rats. Hepatology 2006, 44, 1218–1230. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.; Yu, L.R.; Abdelmegeed, M.A.; Gao, Y.; Banerjee, A.; Song, B.J. Critical role of c-jun N-terminal protein kinase in promoting mitochondrial dysfunction and acute liver injury. Redox Biol. 2015, 6, 552–564. [Google Scholar] [CrossRef]
- Abdelmegeed, M.A.; Jang, S.; Banerjee, A.; Hardwick, J.P.; Song, B.J. Robust protein nitration contributes to acetaminophen-induced mitochondrial dysfunction and acute liver injury. Free Radic. Biol. Med. 2013, 60, 211–222. [Google Scholar] [CrossRef]
- Moon, K.H.; Upreti, V.V.; Yu, L.R.; Lee, I.J.; Ye, X.; Eddington, N.D.; Veenstra, T.D.; Song, B.J. Mechanism of 3,4-methylenedioxymethamphetamine (MDMA, ecstasy)-mediated mitochondrial dysfunction in rat liver. Proteomics 2008, 8, 3906–3918. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ray, B.; Rungratanawanich, W.; LeFort, K.R.; Chidambaram, S.B.; Song, B.-J. Mitochondrial Aldehyde Dehydrogenase 2 (ALDH2) Protects against Binge Alcohol-Mediated Gut and Brain Injury. Cells 2024, 13, 927. https://doi.org/10.3390/cells13110927
Ray B, Rungratanawanich W, LeFort KR, Chidambaram SB, Song B-J. Mitochondrial Aldehyde Dehydrogenase 2 (ALDH2) Protects against Binge Alcohol-Mediated Gut and Brain Injury. Cells. 2024; 13(11):927. https://doi.org/10.3390/cells13110927
Chicago/Turabian StyleRay, Bipul, Wiramon Rungratanawanich, Karli R. LeFort, Saravana Babu Chidambaram, and Byoung-Joon Song. 2024. "Mitochondrial Aldehyde Dehydrogenase 2 (ALDH2) Protects against Binge Alcohol-Mediated Gut and Brain Injury" Cells 13, no. 11: 927. https://doi.org/10.3390/cells13110927
APA StyleRay, B., Rungratanawanich, W., LeFort, K. R., Chidambaram, S. B., & Song, B.-J. (2024). Mitochondrial Aldehyde Dehydrogenase 2 (ALDH2) Protects against Binge Alcohol-Mediated Gut and Brain Injury. Cells, 13(11), 927. https://doi.org/10.3390/cells13110927