
The Impact of Natriuretic Peptides on Heart Development, Homeostasis, and Disease


Abstract
1. Introduction of Natriuretic Peptides
2. Regulation of NPPA and NPPB Expression during Development and Disease
3. Natriuretic Peptide Production and Receptor Interactions
4. The Role of NPs in CM Cell Cycle Activity during Development, Maturation, and Stress
5. The Role of NPs in CM Hypertrophy
6. Role of NPs in Cardiac Contractility
7. Role of NPs in Cardiac Fibrosis and Inflammation
8. NPs in Cardiac Energy Metabolism and Mitochondrial Function
9. Role of NPs in Cardiac Rhythm
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Abbreviation
| ANP | Atrial natriuretic peptide |
| ANF | Atrial natriuretic factor |
| BNP | B-type/Brain natriuretic peptide |
| CNP | C-type natriuretic peptide |
| NPR-A | Natriuretic peptide receptor-A |
| NRP-B | Natriuretic peptide receptor-B |
| NRP-C | Natriuretic peptide receptor-C |
| Nppa | Natriuretic peptide A |
| Nppb | Natriuretic peptide B |
| Nppc | Natriuretic peptide C |
| Npr1 | Natriuretic peptide receptor-1 |
| Npr2 | Natriuretic peptide receptor-2 |
| Npr3 | Natriuretic peptide receptor-3 |
References
- De Bold, A.J.; Borenstein, H.B.; Veress, A.T.; Sonnenberg, H. A rapid and potent natriuretic response to intravenous injection of atrial myocardial extract in rats. Life Sci. 1981, 28, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Sudoh, T.; Kangawa, K.; Minamino, N.; Matsuo, H. A new natriuretic peptide in porcine brain. Nature 1988, 332, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Mukoyama, M.; Nakao, K.; Saito, Y.; Ogawa, Y.; Hosoda, K.; Suga, S.; Shirakami, G.; Jougasaki, M.; Imura, H. Increased Human Brain Natriuretic Peptide in Congestive Heart Failure. New Engl. J. Med. 1990, 323, 757–758. [Google Scholar] [CrossRef]
- Mukoyama, M.; Nakao, K.; Hosoda, K.; Suga, S.; Saito, Y.; Ogawa, Y.; Shirakami, G.; Jougasaki, M.; Obata, K.; Yasue, H.; et al. Brain natriuretic peptide as a novel cardiac hormone in humans. Evidence for an exquisite dual natriuretic peptide system, atrial natriuretic peptide and brain natriuretic peptide. J. Clin. Investig. 1991, 87, 1402–1412. [Google Scholar] [CrossRef] [PubMed]
- Sudoh, T.; Minamino, N.; Kangawa, K.; Matsuo, H. C-Type natriuretic peptide (CNP): A new member of natriuretic peptide family identified in porcine brain. Biochem. Biophys. Res. Commun. 1990, 168, 863–870. [Google Scholar] [CrossRef] [PubMed]
- Vollmar, A.M.; Gerbes, A.L.; Nemer, M.; Schulz, R. Detection of C-type natriuretic peptide (CNP) transcript in the rat heart and immune organs. Endocrinology 1993, 132, 1872–1874. [Google Scholar] [CrossRef]
- Del Ry, S.; Cabiati, M.; Vozzi, F.; Battolla, B.; Caselli, C.; Forini, F.; Segnani, C.; Prescimone, T.; Giannessi, D.; Mattii, L. Expression of C-type natriuretic peptide and its receptor NPR-B in cardiomyocytes. Peptides 2011, 32, 1713–1718. [Google Scholar] [CrossRef] [PubMed]
- Del Ry, S.; Maltinti, M.; Piacenti, M.; Passino, C.; Emdin, M.; Giannessi, D. Cardiac production of C-type natriuretic peptide in heart failure. J. Cardiovasc. Med. 2006, 7, 397–399. [Google Scholar] [CrossRef] [PubMed]
- Tarazón, E.; Roselló-Lletí, E.; Ortega, A.; Molina-Navarro, M.M.; Sánchez-Lázaro, I.; Lago, F.; González-Juanatey, J.R.; Rivera, M.; Portolés, M. Differential gene expression of C-type natriuretic peptide and its related molecules in dilated and ischemic cardiomyopathy. A new option for the management of heart failure. Int. J. Cardiol. 2014, 174, e84–e86. [Google Scholar] [CrossRef]
- Del Ry, S.; Passino, C.; Maltinti, M.; Emdin, M.; Giannessi, D. C-type natriuretic peptide plasma levels increase in patients with chronic heart failure as a function of clinical severity. Eur. J. Hear. Fail. 2005, 7, 1145–1148. [Google Scholar] [CrossRef]
- Goetze, J.P.; Bruneau, B.G.; Ramos, H.R.; Ogawa, T.; de Bold, M.K.; de Bold, A.J. Cardiac natriuretic peptides. Nat. Rev. Cardiol. 2020, 17, 698–717. [Google Scholar] [CrossRef] [PubMed]
- Scott, N.J.; Ellmers, L.J.; Lainchbury, J.G.; Maeda, N.; Smithies, O.; Richards, A.M.; Cameron, V.A. Influence of natriuretic peptide receptor-1 on survival and cardiac hypertrophy during development. Biochim. et Biophys. Acta (BBA) - Mol. Basis Dis. 2009, 1792, 1175–1184. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, M.; Holtwick, R.; A Baba, H.; Perriard, J.C.; Schmitz, W.; Ehler, E. Progressive cardiac hypertrophy and dysfunction in atrial natriuretic peptide receptor (GC-A) deficient mice. Heart 2002, 87, 368–374. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, H.; Kuwahara, K.; Nishida, M.; Jian, Z.; Rong, X.; Kiyonaka, S.; Kuwabara, Y.; Kurose, H.; Inoue, R.; Mori, Y.; et al. Inhibition of TRPC6 Channel Activity Contributes to the Antihypertrophic Effects of Natriuretic Peptides-Guanylyl Cyclase-A Signaling in the Heart. Circ. Res. 2010, 106, 1849–1860. [Google Scholar] [CrossRef]
- Tokudome, T.; Kishimoto, I.; Horio, T.; Arai, Y.; Schwenke, D.O.; Hino, J.; Okano, I.; Kawano, Y.; Kohno, M.; Miyazato, M.; et al. Regulator of G-Protein Signaling Subtype 4 Mediates Antihypertrophic Effect of Locally Secreted Natriuretic Peptides in the Heart. Circulation 2008, 117, 2329–2339. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Kishimoto, I.; Saito, Y.; Harada, M.; Kuwahara, K.; Izumi, T.; Takahashi, N.; Kawakami, R.; Tanimoto, K.; Nakagawa, Y.; et al. Guanylyl cyclase-A inhibits angiotensin II type 1A receptor-mediated cardiac remodeling, an endogenous protective mechanism in the heart. Circulation 2002, 106, 1722–1728. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, M.; Saito, Y.; Kishimoto, I.; Harada, M.; Kuwahara, K.; Takahashi, N.; Kawakami, R.; Nakagawa, Y.; Tanimoto, K.; Yasuno, S.; et al. Role of Natriuretic Peptide Receptor Guanylyl Cyclase-A in Myocardial Infarction Evaluated Using Genetically Engineered Mice. Hypertension 2005, 46, 441–447. [Google Scholar] [CrossRef]
- Tokudome, T.; Horio, T.; Kishimoto, I.; Soeki, T.; Mori, K.; Kawano, Y.; Kohno, M.; Garbers, D.L.; Nakao, K.; Kangawa, K. Calcineurin-nuclear factor of activated T cells pathway-dependent cardiac remodeling in mice deficient in guanylyl cyclase A, a receptor for atrial and brain natriuretic peptides. Circulation 2005, 111, 3095–3104. [Google Scholar] [CrossRef] [PubMed]
- Vellaichamy, E.; Khurana, M.L.; Fink, J.; Pandey, K.N. Involvement of the NF-kappa B/matrix metalloproteinase pathway in cardiac fibrosis of mice lacking guanylyl cyclase/natriuretic peptide receptor A. J. Biol. Chem. 2005, 280, 19230–19242. [Google Scholar] [CrossRef]
- Knowles, J.W.; Esposito, G.; Mao, L.; Hagaman, J.R.; Fox, J.E.; Smithies, O.; Rockman, H.A.; Maeda, N. Pressure-independent enhancement of cardiac hypertrophy in natriuretic peptide receptor A-deficient mice. J. Clin. Investig. 2001, 107, 975–984. [Google Scholar] [CrossRef]
- Kishimoto, I.; Rossi, K.; Garbers, D.L. A genetic model provides evidence that the receptor for atrial natriuretic peptide (guanylyl cyclase-A) inhibits cardiac ventricular myocyte hypertrophy. Proc. Natl. Acad. Sci. USA 2001, 98, 2703–2706. [Google Scholar] [CrossRef]
- Otani, K.; Tokudome, T.; Kamiya, C.A.; Mao, Y.; Nishimura, H.; Hasegawa, T.; Arai, Y.; Kaneko, M.; Shioi, G.; Ishida, J.; et al. Deficiency of Cardiac Natriuretic Peptide Signaling Promotes Peripartum Cardiomyopathy-Like Remodeling in the Mouse Heart. Circulation 2020, 141, 571–588. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H.; Oberwinkler, H.; Nikolaev, V.O.; Gaßner, B.; Umbenhauer, S.; Wagner, H.; Saito, Y.; Baba, H.A.; Frantz, S.; Kuhn, M. Atrial Natriuretic Peptide Locally Counteracts the Deleterious Effects of Cardiomyocyte Mineralocorticoid Receptor Activation. Circ. Hear. Fail. 2014, 7, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Klaiber, M.; Kruse, M.; Volker, K.; Schroter, J.; Feil, R.; Freichel, M.; Gerling, A.; Feil, S.; Dietrich, A.; Londono, J.E.; et al. Novel insights into the mechanisms mediating the local antihypertrophic effects of cardiac atrial natriuretic peptide: Role of cGMP-dependent protein kinase and RGS. Basic Res. Cardiol. 2010, 105, 583–595. [Google Scholar] [CrossRef] [PubMed]
- Zahabi, A.; Picard, S.; Fortin, N.; Reudelhuber, T.L.; Deschepper, C.F. Expression of Constitutively Active Guanylate Cyclase in Cardiomyocytes Inhibits the Hypertrophic Effects of Isoproterenol and Aortic Constriction on Mouse Hearts. J. Biol. Chem. 2003, 278, 47694–47699. [Google Scholar] [CrossRef]
- Wagner, B.M.; Robinson, J.W.; Healy, C.L.; Gauthier, M.; Dickey, D.M.; Yee, S.P.; Osborn, J.W.; O’Connell, T.D.; Potter, L.R. Guanylyl cyclase-A phosphorylation decreases cardiac hypertrophy and improves systolic function in male, but not female, mice. FASEB J. 2022, 36, e22069. [Google Scholar] [CrossRef] [PubMed]
- Vellaichamy, E.; Das, S.; Subramanian, U.; Maeda, N.; Pandey, K.N. Genetically Altered Mutant Mouse Models of Guanylyl Cyclase/Natriuretic Peptide Receptor-A Exhibit the Cardiac Expression of Proinflammatory Mediators in a Gene-Dose-Dependent Manner. Endocrinology 2014, 155, 1045–1056. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Oparil, S.; Feng, J.A.; Li, P.; Perry, G.; Chen, L.B.; Dai, M.; John, S.W.; Chen, Y.-F. Effects of Pressure Overload on Extracellular Matrix Expression in the Heart of the Atrial Natriuretic Peptide–Null Mouse. Hypertension 2003, 42, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Franco, V.; Chen, Y.F.; Oparil, S.; Feng, J.A.; Wang, D.; Hage, F.; Perry, G. Atrial natriuretic peptide dose-dependently inhibits pressure overload-induced cardiac remodeling. Hypertension 2004, 44, 746–750. [Google Scholar] [CrossRef]
- Mori, T.; Chen, Y.-F.; Feng, J.A.; Hayashi, T.; Oparil, S.; Perry, G.J. Volume overload results in exaggerated cardiac hypertrophy in the atrial natriuretic peptide knockout mouse. Cardiovasc. Res. 2004, 61, 771–779. [Google Scholar] [CrossRef]
- Feng, J.A.; Perry, G.; Mori, T.; Hayashi, T.; Oparil, S.; Chen, Y. Pressure-independent enhancement of cardiac hypertrophy in atrial natriuretic peptide–deficient mice. Clin. Exp. Pharmacol. Physiol. 2003, 30, 343–349. [Google Scholar] [CrossRef]
- Houng, A.K.; McNamee, R.A.; Kerner, A.; Sharma, P.; Mohamad, A.; Tronolone, J.; Reed, G.L. Atrial natriuretic peptide increases inflammation, infarct size, and mortality after experimental coronary occlusion. Am. J. Physiol. Circ. Physiol. 2009, 296, H655–H661. [Google Scholar] [CrossRef] [PubMed]
- Holditch, S.J.; Schreiber, C.A.; Nini, R.; Tonne, J.M.; Peng, K.-W.; Geurts, A.; Jacob, H.J.; Burnett, J.C.; Cataliotti, A.; Ikeda, Y.; et al. B-Type Natriuretic Peptide Deletion Leads to Progressive Hypertension, Associated Organ Damage, and Reduced Survival. Hypertension 2015, 66, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Cataliotti, A.; Tonne, J.M.; Bellavia, D.; Martin, F.L.; Oehler, E.A.; Harders, G.E.; Campbell, J.M.; Peng, K.-W.; Russell, S.J.; Malatino, L.S.; et al. Long-Term Cardiac pro-B-Type Natriuretic Peptide Gene Delivery Prevents the Development of Hypertensive Heart Disease in Spontaneously Hypertensive Rats. Circulation 2011, 123, 1297–1305. [Google Scholar] [CrossRef] [PubMed]
- Moyes, A.J.; Chu, S.M.; Aubdool, A.A.; Dukinfield, M.S.; Margulies, K.B.; Bedi, K.C.; Hodivala-Dilke, K.; Baliga, R.S.; Hobbs, A.J. C-type natriuretic peptide co-ordinates cardiac structure and function. Eur. Hear. J. 2020, 41, 1006–1020. [Google Scholar] [CrossRef] [PubMed]
- Izumi, T.; Saito, Y.; Kishimoto, I.; Harada, M.; Kuwahara, K.; Hamanaka, I.; Takahashi, N.; Kawakami, R.; Li, Y.; Takemura, G.; et al. Blockade of the natriuretic peptide receptor guanylyl cyclase-A inhibits NF-kappaB activation and alleviates myocardial ischemia/reperfusion injury. J. Clin. Investig. 2001, 108, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Lu, Y.; Wang, X.; Cheng, C.; Xue, F.; Xie, L.; Zhang, Y.; Sui, W.; Zhang, M.; Zhang, Y.; et al. NPRC deletion attenuates cardiac fibrosis in diabetic mice by activating PKA/PKG and inhibiting TGF-beta1/Smad pathways. Sci. Adv. 2023, 9, eadd4222. [Google Scholar] [CrossRef] [PubMed]
- Forte, M.; Marchitti, S.; Di Nonno, F.; Stanzione, R.; Schirone, L.; Cotugno, M.; Bianchi, F.; Schiavon, S.; Raffa, S.; Ranieri, D.; et al. NPPA/atrial natriuretic peptide is an extracellular modulator of autophagy in the heart. Autophagy 2022, 19, 1087–1099. [Google Scholar] [CrossRef] [PubMed]
- Moilanen, A.-M.; Rysä, J.; Mustonen, E.; Serpi, R.; Aro, J.; Tokola, H.; Leskinen, H.; Manninen, A.; Levijoki, J.; Vuolteenaho, O.; et al. Intramyocardial BNP Gene Delivery Improves Cardiac Function Through Distinct Context-Dependent Mechanisms. Circ. Hear. Fail. 2011, 4, 483–495. [Google Scholar] [CrossRef]
- Tamura, N.; Ogawa, Y.; Chusho, H.; Nakamura, K.; Nakao, K.; Suda, M.; Kasahara, M.; Hashimoto, R.; Katsuura, G.; Mukoyama, M.; et al. Cardiac fibrosis in mice lacking brain natriuretic peptide. Proc. Natl. Acad. Sci. USA 2000, 97, 4239–4244. [Google Scholar] [CrossRef]
- Ogawa, Y.; Tamura, N.; Chusho, H.; Nakao, K. Brain natriuretic peptide appears to act locally as an antifibrotic factor in the heart. Can. J. Physiol. Pharmacol. 2001, 79, 723–729. [Google Scholar] [CrossRef]
- Kawakami, R.; Saito, Y.; Kishimoto, I.; Harada, M.; Kuwahara, K.; Takahashi, N.; Nakagawa, Y.; Nakanishi, M.; Tanimoto, K.; Usami, S.; et al. Overexpression of Brain Natriuretic Peptide Facilitates Neutrophil Infiltration and Cardiac Matrix Metalloproteinase-9 Expression After Acute Myocardial Infarction. Circulation 2004, 110, 3306–3312. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Ma, M.; Cao, D.; Zheng, A.; Zhang, Y.; Su, Y.; Wang, J.; Xu, Y.; Zhou, M.; Tang, Y.; et al. Inhibition of Fap Promotes Cardiac Repair by Stabilizing BNP. Circ. Res. 2023, 132, 586–600. [Google Scholar] [CrossRef] [PubMed]
- Schipke, J.; Roloff, K.; Kuhn, M.; Mühlfeld, C. Systemic, but not cardiomyocyte-specific, deletion of the natriuretic peptide receptor guanylyl cyclase A increases cardiomyocyte number in neonatal mice. Histochem. Cell. Biol. 2015, 144, 365–375. [Google Scholar] [CrossRef]
- Ali, M.; Liccardo, D.; Cao, T.; Tian, Y. Natriuretic peptides and Forkhead O transcription factors act in a cooperative manner to promote cardiomyocyte cell cycle re-entry in the postnatal mouse heart. BMC Dev. Biol. 2021, 21, 1–13. [Google Scholar] [CrossRef]
- Bon-Mathier, A.-C.; Déglise, T.; Rignault-Clerc, S.; Bielmann, C.; Mazzolai, L.; Rosenblatt-Velin, N. Brain Natriuretic Peptide Protects Cardiomyocytes from Apoptosis and Stimulates Their Cell Cycle Re-Entry in Mouse Infarcted Hearts. Cells 2022, 12, 7. [Google Scholar] [CrossRef]
- Chen, B.; Chang, P.; Shen, X.; Zhang, X.; Zhang, J.; Wang, X.; Yu, J. Cardiac-specific deletion of natriuretic peptide receptor A induces differential myocardial expression of circular RNA and mRNA molecules involved in metabolism in mice. Mol. Med. Rep. 2021, 23, 50. [Google Scholar] [CrossRef]
- Chang, P.; Niu, Y.; Zhang, X.; Zhang, J.; Wang, X.; Shen, X.; Chen, B.; Yu, J. Integrative Proteomic and Metabolomic Analysis Reveals Metabolic Phenotype in Mice With Cardiac-Specific Deletion of Natriuretic Peptide Receptor, A. Mol. Cell. Proteom. 2021, 20, 100072. [Google Scholar] [CrossRef]
- Mishra, A.; Tavasoli, M.; Sokolenko, S.; McMaster, C.R.; Pasumarthi, K.B. Atrial natriuretic peptide signaling co-regulates lipid metabolism and ventricular conduction system gene expression in the embryonic heart. iScience 2024, 27, 108748. [Google Scholar] [CrossRef] [PubMed]
- Calamera, G.; Ndongson-Dongmo, B.; Arunthavarajah, D.; Ovesen, M.; Choel, K.; Levy, F.O.; Andressen, K.W.; Moltzau, L.R. Natriuretic peptides increase cGMP around cardiomyocyte mitochondria and protect against apoptosis. bioRxiv 2022. [CrossRef]
- Chang, P.; Zhang, X.; Zhang, J.; Wang, J.; Wang, X.; Li, M.; Wang, R.; Yu, J.; Fu, F. BNP protects against diabetic cardiomyopathy by promoting Opa1-mediated mitochondrial fusion via activating the PKG-STAT3 pathway. Redox Biol. 2023, 62, 102702. [Google Scholar] [CrossRef] [PubMed]
- Dorey, T.W.; Mackasey, M.; Jansen, H.J.; McRae, M.D.; Bohne, L.J.; Liu, Y.; Belke, D.D.; Atkinson, L.; Rose, R.A. Natriuretic peptide receptor B maintains heart rate and sinoatrial node function via cyclic GMP-mediated signalling. Cardiovasc. Res. 2021, 118, 1917–1931. [Google Scholar] [CrossRef] [PubMed]
- Hall, E.J.; Pal, S.; Glennon, M.S.; Shridhar, P.; Satterfield, S.L.; Weber, B.; Zhang, Q.; Salama, G.; Lal, H.; Becker, J.R. Cardiac natriuretic peptide deficiency sensitizes the heart to stress-induced ventricular arrhythmias via impaired CREB signalling. Cardiovasc. Res. 2021, 118, 2124–2138. [Google Scholar] [CrossRef] [PubMed]
- Egom, E.E.; Vella, K.; Hua, R.; Jansen, H.J.; Moghtadaei, M.; Polina, I.; Bogachev, O.; Hurnik, R.; Mackasey, M.; Rafferty, S.; et al. Impaired sinoatrial node function and increased susceptibility to atrial fibrillation in mice lacking natriuretic peptide receptor C. J. Physiol. 2015, 593, 1127–1146. [Google Scholar] [CrossRef]
- Mackasey, M.; Egom, E.E.; Jansen, H.J.; Hua, R.; Moghtadaei, M.; Liu, Y.; Kaur, J.; McRae, M.D.; Bogachev, O.; Rafferty, S.A.; et al. Natriuretic Peptide Receptor-C Protects Against Angiotensin II-Mediated Sinoatrial Node Disease in Mice. JACC Basic Transl. Sci. 2018, 3, 824–843. [Google Scholar] [CrossRef] [PubMed]
- Jansen, H.J.; Mackasey, M.; Moghtadaei, M.; Liu, Y.; Kaur, J.; Egom, E.E.; Tuomi, J.M.; Rafferty, S.A.; Kirkby, A.W.; Rose, R.A. NPR-C (Natriuretic Peptide Receptor-C) Modulates the Progression of Angiotensin II–Mediated Atrial Fibrillation and Atrial Remodeling in Mice. Circ. Arrhythmia Electrophysiol. 2019, 12, e006863. [Google Scholar] [CrossRef] [PubMed]
- Michel, K.; Herwig, M.; Werner, F.; Spes, K.; Abeßer, M.; Schuh, K.; Dabral, S.; Mügge, A.; Baba, H.A.; Skryabin, B.V.; et al. C-type natriuretic peptide moderates titin-based cardiomyocyte stiffness. J. Clin. Investig. 2020, 5, e139910. [Google Scholar] [CrossRef] [PubMed]
- Manfra, O.; Calamera, G.; Froese, A.; Arunthavarajah, D.; Surdo, N.C.; Meier, S.; Melleby, A.O.; Aasrum, M.; Aronsen, J.M.; Nikolaev, V.O.; et al. CNP regulates cardiac contractility and increases cGMP near both SERCA and TnI: Difference from BNP visualized by targeted cGMP biosensors. Cardiovasc. Res. 2021, 118, 1506–1519. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Naruse, K.; Yamagami, S.; Mitani, H.; Suzuki, N.; Takei, Y. Four functionally distinct C-type natriuretic peptides found in fish reveal evolutionary history of the natriuretic peptide system. Proc. Natl. Acad. Sci. USA 2003, 100, 10079–10084. [Google Scholar] [CrossRef]
- Zeller, R.; Bloch, K.D.; Williams, B.S.; Arceci, R.J.; Seidman, C.E. Localized expression of the atrial natriuretic factor gene during cardiac embryogenesis. Genes Dev. 1987, 1, 693–698. [Google Scholar] [CrossRef]
- Houweling, A.C.; van Borren, M.M.; Moorman, A.F.; Christoffels, V.M. Expression and regulation of the atrial natriuretic factor encoding gene Nppa during development and disease. Cardiovasc. Res. 2005, 67, 583–593. [Google Scholar] [CrossRef]
- Sergeeva, I.A.; Hooijkaas, I.B.; Van Der Made, I.; Jong, W.M.; Creemers, E.E.; Christoffels, V.M. A transgenic mouse model for the simultaneous monitoring of ANF and BNP gene activity during heart development and disease. Cardiovasc. Res. 2013, 101, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Casserly, B.; Klinger, J.R. Brain natriuretic peptide in pulmonary arterial hypertension: Biomarker and potential therapeutic agent. Drug Des. Devel Ther. 2009, 3, 269–287. [Google Scholar]
- Van Duijvenboden, K.; de Bakker, D.E.M.; Man, J.C.K.; Janssen, R.; Gunthel, M.; Hill, M.C.; Hooijkaas, I.B.; van der Made, I.; van der Kraak, P.H.; Vink, A.; et al. Conserved NPPB+ Border Zone Switches From MEF2- to AP-1-Driven Gene Program. Circulation 2019, 140, 864–879. [Google Scholar] [CrossRef]
- Field, L.J. Atrial Natriuretic Factor-SV40 T Antigen Transgenes Produce Tumors and Cardiac Arrhythmias in Mice. Science 1988, 239, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Sergeeva, I.A.; Christoffels, V.M. Regulation of expression of atrial and brain natriuretic peptide, biomarkers for heart development and disease. Biochim. et Biophys. Acta (BBA) - Mol. Basis Dis. 2013, 1832, 2403–2413. [Google Scholar] [CrossRef] [PubMed]
- Stefanovic, S.; Barnett, P.; van Duijvenboden, K.; Weber, D.; Gessler, M.; Christoffels, V.M. GATA-dependent regulatory switches establish atrioventricular canal specificity during heart development. Nat. Commun. 2014, 5, 3680. [Google Scholar] [CrossRef] [PubMed]
- Luna-Zurita, L.; Stirnimann, C.U.; Glatt, S.; Kaynak, B.L.; Thomas, S.; Baudin, F.; Samee, A.H.; He, D.; Small, E.M.; Mileikovsky, M.; et al. Complex Interdependence Regulates Heterotypic Transcription Factor Distribution and Coordinates Cardiogenesis. Cell 2016, 164, 999–1014. [Google Scholar] [CrossRef] [PubMed]
- Sergeeva, I.A.; Hooijkaas, I.B.; Ruijter, J.M.; van der Made, I.; de Groot, N.E.; van de Werken, H.J.G.; Creemers, E.E.; Christoffels, V.M. Identification of a regulatory domain controlling the Nppa-Nppb gene cluster during heart development and stress. Development 2016, 143, 2135–2146. [Google Scholar] [CrossRef]
- Man, J.; Barnett, P.; Christoffels, V.M. Structure and function of the Nppa-Nppb cluster locus during heart development and disease. Cell Mol. Life Sci. 2018, 75, 1435–1444. [Google Scholar] [CrossRef]
- Knowlton, K.U.; Rockman, H.A.; Itani, M.; Vovan, A.; Seidman, C.E.; Chien, K.R. Divergent pathways mediate the induction of ANF transgenes in neonatal and hypertrophic ventricular myocardium. J. Clin. Investig. 1995, 96, 1311–1318. [Google Scholar] [CrossRef] [PubMed]
- Man, J.C.; van Duijvenboden, K.; Krijger, P.H.; Hooijkaas, I.B.; van der Made, I.; Vries, C.d.G.-D.; Wakker, V.; Creemers, E.E.; de Laat, W.; Boukens, B.J.; et al. Genetic Dissection of a Super Enhancer Controlling the Nppa-Nppb Cluster in the Heart. Circ. Res. 2021, 128, 115–129. [Google Scholar] [CrossRef] [PubMed]
- Gilsbach, R.; Schwaderer, M.; Preissl, S.; Grüning, B.A.; Kranzhöfer, D.; Schneider, P.; Nührenberg, T.G.; Mulero-Navarro, S.; Weichenhan, D.; Braun, C.; et al. Distinct epigenetic programs regulate cardiac myocyte development and disease in the human heart in vivo. Nat. Commun. 2018, 9, 391. [Google Scholar] [CrossRef] [PubMed]
- Annilo, T.; Kepp, K.; Laan, M. Natural antisense transcript of natriuretic peptide precursor A (NPPA): Structural organization and modulation of NPPA expression. BMC Mol. Biol. 2009, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Celik, S.; Sadegh, M.K.; Morley, M.; Roselli, C.; Ellinor, P.T.; Cappola, T.; Smith, J.G.; Gidlöf, O. Antisense regulation of atrial natriuretic peptide expression. J. Clin. Investig. 2019, 4, e130978. [Google Scholar] [CrossRef]
- Fu, W.; Ren, H.; Shou, J.; Liao, Q.; Li, L.; Shi, Y.; Jose, P.A.; Zeng, C.; Wang, W.E. Loss of NPPA-AS1 promotes heart regeneration by stabilizing SFPQ–NONO heteromer-induced DNA repair. Basic Res. Cardiol. 2022, 117, 1–19. [Google Scholar] [CrossRef]
- Feng, Y.; Cai, L.; Hong, W.; Zhang, C.; Tan, N.; Wang, M.; Wang, C.; Liu, F.; Wang, X.; Ma, J.; et al. Rewiring of 3D Chromatin Topology Orchestrates Transcriptional Reprogramming and the Development of Human Dilated Cardiomyopathy. Circulation 2022, 145, 1663–1683. [Google Scholar] [CrossRef] [PubMed]
- Semenov, A.G.; Postnikov, A.B.; Tamm, N.N.; Seferian, K.R.; Karpova, N.S.; Bloshchitsyna, M.N.; Koshkina, E.V.; Krasnoselsky, M.I.; Serebryanaya, D.V.; Katrukha, A.G. Processing of Pro–Brain Natriuretic Peptide Is Suppressed by O-Glycosylation in the Region Close to the Cleavage Site. Clin. Chem. 2009, 55, 489–498. [Google Scholar] [CrossRef]
- Moyes, A.J.; Hobbs, A.J. C-Type Natriuretic Peptide: A Multifaceted Paracrine Regulator in the Heart and Vasculature. Int. J. Mol. Sci. 2019, 20, 2281. [Google Scholar] [CrossRef]
- Cody, R.J.; Atlas, S.A.; Laragh, J.H.; Kubo, S.H.; Covit, A.B.; Ryman, K.S.; Shaknovich, A.; Pondolfino, K.; Clark, M.; Camargo, M.J.; et al. Atrial natriuretic factor in normal subjects and heart failure patients. Plasma levels and renal, hormonal, and hemodynamic responses to peptide infusion. J. Clin. Investig. 1986, 78, 1362–1374. [Google Scholar] [CrossRef]
- Burnett, J.C.; Kao, P.C.; Hu, D.C.; Heser, D.W.; Heublein, D.; Granger, J.P.; Opgenorth, T.J.; Reeder, G.S. Atrial Natriuretic Peptide Elevation in Congestive Heart Failure in the Human. Science 1986, 231, 1145–1147. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Platt, M.J.; Shibasaki, T.; Quaggin, S.E.; Backx, P.H.; Seino, S.; Simpson, J.A.; Drucker, D.J. GLP-1 receptor activation and Epac2 link atrial natriuretic peptide secretion to control of blood pressure. Nat. Med. 2013, 19, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Liang, F.; O’rear, J.; Schellenberger, U.; Tai, L.; Lasecki, M.; Schreiner, G.F.; Apple, F.S.; Maisel, A.S.; Pollitt, N.S.; Protter, A.A. Evidence for Functional Heterogeneity of Circulating B-Type Natriuretic Peptide. J. Am. Coll. Cardiol. 2007, 49, 1071–1078. [Google Scholar] [CrossRef]
- Nakao, K.; Sugawara, A.; Morii, N.; Sakamoto, M.; Yamada, T.; Itoh, H.; Shiono, S.; Saito, Y.; Nishimura, K.; Ban, T.; et al. The pharmacokinetics of alpha-human atrial natriuretic polypeptide in healthy subjects. Eur. J. Clin. Pharmacol. 1986, 31, 101–103. [Google Scholar] [CrossRef] [PubMed]
- Potter, L.R.; Yoder, A.R.; Flora, D.R.; Antos, L.K.; Dickey, D.M. Natriuretic peptides: Their structures, receptors, physiologic functions and therapeutic applications. In Handbook of Experimental Pharmacology; Schmidt, H.H.H.W., Hofmann, F., Stasch, J.-P., Eds.; Springer: Berlin/Heidelberg, Germany, 2009; Volume 191, pp. 341–366. ISBN 9783540689607. [Google Scholar]
- Dickey, D.M.; Flora, D.R.; Bryan, P.M.; Xu, X.; Chen, Y.; Potter, L.R. Differential Regulation of Membrane Guanylyl Cyclases in Congestive Heart Failure: Natriuretic Peptide Receptor (NPR)-B, Not NPR-A, Is the Predominant Natriuretic Peptide Receptor in the Failing Heart. Endocrinology 2007, 148, 3518–3522. [Google Scholar] [CrossRef] [PubMed]
- Rose, R.A.; Giles, W.R. Natriuretic peptide C receptor signalling in the heart and vasculature. J. Physiol. 2008, 586, 353–366. [Google Scholar] [CrossRef] [PubMed]
- Moffatt, P.; Thomas, G.; Sellin, K.; Bessette, M.-C.; Lafrenière, F.; Akhouayri, O.; St-Arnaud, R.; Lanctôt, C. Osteocrin Is a Specific Ligand of the Natriuretic Peptide Clearance Receptor That Modulates Bone Growth. J. Biol. Chem. 2007, 282, 36454–36462. [Google Scholar] [CrossRef]
- Kenny, A.J.; Bourne, A.; Ingram, J. Hydrolysis of human and pig brain natriuretic peptides, urodilatin, C-type natriuretic peptide and some C-receptor ligands by endopeptidase-24.11. Biochem. J. 1993, 291, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Keane, F.M.; Nadvi, N.A.; Yao, T.W.; Gorrell, M.D. Neuropeptide Y, B-type natriuretic peptide, substance P and peptide YY are novel substrates of fibroblast activation protein-alpha. FEBS J. 2011, 278, 1316–1332. [Google Scholar] [CrossRef]
- John, S.W.; Krege, J.H.; Oliver, P.M.; Hagaman, J.R.; Hodgin, J.B.; Pang, S.C.; Flynn, T.G.; Smithies, O. Genetic decreases in atrial natriuretic peptide and salt-sensitive hypertension. Science 1995, 267, 679–681. [Google Scholar] [CrossRef]
- Rubattu, S.; Bigatti, G.; Evangelista, A.; Lanzani, C.; Stanzione, R.; Zagato, L.; Manunta, P.; Marchitti, S.; Venturelli, V.; Bianchi, G.; et al. Association of Atrial Natriuretic Peptide and Type A Natriuretic Peptide Receptor Gene Polymorphisms With Left Ventricular Mass in Human Essential Hypertension. J. Am. Coll. Cardiol. 2006, 48, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Tamura, N.; Doolittle, L.K.; Hammer, R.E.; Shelton, J.M.; Richardson, J.A.; Garbers, D.L. Critical roles of the guanylyl cyclase B receptor in endochondral ossification and development of female reproductive organs. Proc. Natl. Acad. Sci. USA 2004, 101, 17300–17305. [Google Scholar] [CrossRef] [PubMed]
- Langenickel, T.H.; Buttgereit, J.; Pagel-Langenickel, I.; Lindner, M.; Monti, J.; Beuerlein, K.; Al-Saadi, N.; Plehm, R.; Popova, E.; Tank, J.; et al. Cardiac hypertrophy in transgenic rats expressing a dominant-negative mutant of the natriuretic peptide receptor B. Proc. Natl. Acad. Sci. USA 2006, 103, 4735–4740. [Google Scholar] [CrossRef]
- Jaubert, J.; Jaubert, F.; Martin, N.; Washburn, L.L.; Lee, B.K.; Eicher, E.M.; Guénet, J.-L. Three new allelic mouse mutations that cause skeletal overgrowth involve the natriuretic peptide receptor C gene (Npr3). Proc. Natl. Acad. Sci. USA 1999, 96, 10278–10283. [Google Scholar] [CrossRef]
- Rahmutula, D.; Zhang, H.; Wilson, E.E.; Olgin, J.E. Absence of natriuretic peptide clearance receptor attenuates TGF-beta1-induced selective atrial fibrosis and atrial fibrillation. Cardiovasc. Res. 2019, 115, 357–372. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.R.; Chatterjee, S.; Robinson, T.Y.; Bennett, J.S.; Panakova, D.; Galindo, C.L.; Zhong. L.; Shin, J.T.; Coy, S.M.; Kelly, A.E.; et al. Differential activation of natriuretic peptide receptors modulates cardiomyocyte proliferation during development. Development 2014, 141, 335–345. [Google Scholar] [CrossRef]
- Hotchkiss, A.; Feridooni, T.; Baguma-Nibasheka, M.; McNeil, K.; Chinni, S.; Pasumarthi, K.B.S. Atrial natriuretic peptide inhibits cell cycle activity of embryonic cardiac progenitor cells via its NPRA receptor signaling axis. Am. J. Physiol. Physiol. 2015, 308, C557–C569. [Google Scholar] [CrossRef]
- Koide, M.; Akins, R.E.; Harayama, H.; Yasui, K.; Yokota, M.; Tuan, R.S. Atrial natriuretic peptide accelerates proliferation of chick embryonic cardiomyocytes in vitro. Differentiation 1996, 61, 1–11. [Google Scholar] [CrossRef]
- Honkoop, H.; de Bakker, D.E.; Aharonov, A.; Kruse, F.; Shakked, A.; Nguyen, P.D.; de Heus, C.; Garric, L.; Muraro, M.J.; Shoffner, A.; et al. Single-cell analysis uncovers that metabolic reprogramming by ErbB2 signaling is essential for cardiomyocyte proliferation in the regenerating heart. eLife 2019, 8, e50163. [Google Scholar] [CrossRef] [PubMed]
- Travers, J.G.; Kamal, F.A.; Robbins, J.; Yutzey, K.E.; Blaxall, B.C. Cardiac Fibrosis: The Fibroblast Awakens. Circ. Res. 2016, 118, 1021–1040. [Google Scholar] [CrossRef]
- Chu, L.; Xie, D.; Xu, D. Epigenetic Regulation of Fibroblasts and Crosstalk between Cardiomyocytes and Non-Myocyte Cells in Cardiac Fibrosis. Biomolecules 2023, 13, 1382. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Wang, D.; Lucas, J.; Oparil, S.; Xing, D.; Cao, X.; Novak, L.; Renfrow, M.B.; Chen, Y.F. Atrial natriuretic peptide inhibits transforming growth factor beta-induced Smad signaling and myofibroblast transformation in mouse cardiac fibroblasts. Circ. Res. 2008, 102, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Huntley, B.K.; Sandberg, S.M.; Noser, J.A.; Cataliotti, A.; Redfield, M.M.; Matsuda, Y.; Burnett, J.C., Jr. BNP-induced activation of cGMP in human cardiac fibroblasts: Interactions with fibronectin and natriuretic peptide receptors. J. Cell Physiol. 2006, 209, 943–949. [Google Scholar] [CrossRef] [PubMed]
- Kapoun, A.M.; Liang, F.; O’Young, G.; Damm, D.L.; Quon, D.; White, R.T.; Munson, K.; Lam, A.; Schreiner, G.F.; Protter, A.A. B-type natriuretic peptide exerts broad functional opposition to transforming growth factor-beta in primary human cardiac fibroblasts: Fibrosis, myofibroblast conversion, proliferation, and inflammation. Circ. Res. 2004, 94, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Glezeva, N.; Collier, P.; Voon, V.; Ledwidge, M.; McDonald, K.; Watson, C.; Baugh, J. Attenuation of Monocyte Chemotaxis—A Novel Anti-inflammatory Mechanism of Action for the Cardio-protective Hormone B-Type Natriuretic Peptide. J. Cardiovasc. Transl. Res. 2013, 6, 545–557. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.Q.; Liu, Y.L.; Li, G.; Li, B.; Liu, Y.; Li, X.F.; Liu, A.J. Inhibitory effects of C-type natriuretic peptide on the differentiation of cardiac fibroblasts, and secretion of monocyte chemoattractant protein-1 and plasminogen activator inhibitor-1. Mol. Med. Rep. 2015, 11, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Eisner, V.; Csordás, G.; Hajnóczky, G. Interactions between sarco-endoplasmic reticulum and mitochondria in cardiac and skeletal muscle—Pivotal roles in Ca2+ and reactive oxygen species signaling. J. Cell Sci. 2013, 126, 2965–2978. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Tian, R. Mitochondrial dysfunction in pathophysiology of heart failure. J. Clin. Investig. 2018, 128, 3716–3726. [Google Scholar] [CrossRef] [PubMed]
- Zois, N.E.; Bartels, E.D.; Hunter, I.; Kousholt, B.S.; Olsen, L.H.; Goetze, J.P. Natriuretic peptides in cardiometabolic regulation and disease. Nat. Rev. Cardiol. 2014, 11, 403–412. [Google Scholar] [CrossRef]
- Arjamaa, O. The Endocrine Heart: Natriuretic Peptides and Oxygen Metabolism in Cardiac Diseases. CJC Open 2021, 3, 1149–1152. [Google Scholar] [CrossRef]
- Suffee, N.; Moore-Morris, T.; Farahmand, P.; Rücker-Martin, C.; Dilanian, G.; Fradet, M.; Sawaki, D.; Derumeaux, G.; LePrince, P.; Clément, K.; et al. Atrial natriuretic peptide regulates adipose tissue accumulation in adult atria. Proc. Natl. Acad. Sci. USA 2017, 114, E771–E780. [Google Scholar] [CrossRef]
- Suffee, N.; Moore-Morris, T.; Jagla, B.; Mougenot, N.; Dilanian, G.; Berthet, M.; Proukhnitzky, J.; Le Prince, P.; Tregouet, D.A.; Pucéat, M.; et al. Reactivation of the Epicardium at the Origin of Myocardial Fibro-Fatty Infiltration During the Atrial Cardiomyopathy. Circ. Res. 2020, 126, 1330–1342. [Google Scholar] [CrossRef]
- Govindapillai, A.; Hotchkiss, A.; Baguma-Nibasheka, M.; Rose, R.A.; Miquerol, L.; Smithies, O.; Maeda, N.; Pasumarthi, K.B.S. Characterizing the role of atrial natriuretic peptide signaling in the development of embryonic ventricular conduction system. Sci. Rep. 2018, 8, 6939. [Google Scholar] [CrossRef]
- Sankar, S.; Jayabalan, M.; Venkatesh, S.; Ibrahim, M. Effect of hyperglycemia on tbx5a and nppa gene expression and its correlation to structural and functional changes in developing zebrafish heart. Cell Biol. Int. 2022, 46, 2173–2184. [Google Scholar] [CrossRef]
- Hamano, M.; Nomura, S.; Iida, M.; Komuro, I.; Yamanishi, Y. Prediction of single-cell mechanisms for disease progression in hypertrophic remodelling by a trans-omics approach. Sci. Rep. 2021, 11, 1–17. [Google Scholar] [CrossRef]
- Guo, Y.; Cen, X.-F.; Li, D.; Qiu, H.-L.; Chen, Y.-J.; Zhang, M.; Huang, S.-H.; Xia, H.; Xu, M. Identify Tcea3 as a novel anti-cardiomyocyte hypertrophy gene involved in fatty acid oxidation and oxidative stress. Front. Cardiovasc. Med. 2023, 10, 1137429. [Google Scholar] [CrossRef] [PubMed]
- Hong, L.; Xi, J.; Zhang, Y.; Tian, W.; Xu, J.; Cui, X.; Xu, Z. Atrial natriuretic peptide prevents the mitochondrial permeability transition pore opening by inactivating glycogen synthase kinase 3beta via PKG and PI3K in cardiac H9c2 cells. Eur. J. Pharmacol. 2012, 695, 13–19. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, Y.; Yan, M.; Wu, Y.; Zheng, X. B-Type Natriuretic Peptide-Induced Cardioprotection against Reperfusion Is Associated with Attenuation of Mitochondrial Permeability Transition. Biol. Pharm. Bull. 2009, 32, 1545–1551. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Wang, L.-L.; Liu, Z.-B.; Chen, C.; Ren, X.; Luo, A.-T.; Ma, J.-H.; Antzelevitch, C.; Barajas-Martínez, H.; Hu, D. Underlying mechanism of atrial fibrillation associated Nppa-I137T mutation and cardiac effect of potential drug therapy. Hear. Rhythm. 2023, 21, 184–196. [Google Scholar] [CrossRef] [PubMed]
- Cunha, P.S.; Antunes, D.O.; Laranjo, S.; Coutinho, A.; Abecasis, J.; Oliveira, M.M. Case report: Mutation in NPPA gene as a cause of fibrotic atrial myopathy. Front. Cardiovasc. Med. 2023, 10, 1149717. [Google Scholar] [CrossRef]
- Hodgson-Zingman, D.M.; Karst, M.L.; Zingman, L.V.; Heublein, D.M.; Darbar, D.; Herron, K.J.; Ballew, J.D.; de Andrade, M.; Burnett, J.C.; Olson, T.M. Atrial Natriuretic Peptide Frameshift Mutation in Familial Atrial Fibrillation. New Engl. J. Med. 2008, 359, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Cachorro, E.; Gunscht, M.; Schubert, M.; Sadek, M.S.; Siegert, J.; Dutt, F.; Bauermeister, C.; Quickert, S.; Berning, H.; Nowakowski, F.; et al. CNP Promotes Antiarrhythmic Effects via Phosphodiesterase 2. Circ. Res. 2023, 132, 400–414. [Google Scholar] [CrossRef] [PubMed]
- Azer, J.; Hua, R.; Vella, K.; Rose, R.A. Natriuretic peptides regulate heart rate and sinoatrial node function by activating multiple natriuretic peptide receptors. J. Mol. Cell. Cardiol. 2012, 53, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Meijers, W.C.; Bayes-Genis, A.; Mebazaa, A.; Bauersachs, J.; Cleland, J.G.F.; Coats, A.J.S.; Januzzi, J.L.; Maisel, A.S.; McDonald, K.; Mueller, T.; et al. Circulating heart failure biomarkers beyond natriuretic peptides: Review from the Biomarker Study Group of the Heart Failure Association (HFA), European Society of Cardiology (ESC). Eur. J. Hear. Fail. 2021, 23, 1610–1632. [Google Scholar] [CrossRef] [PubMed]
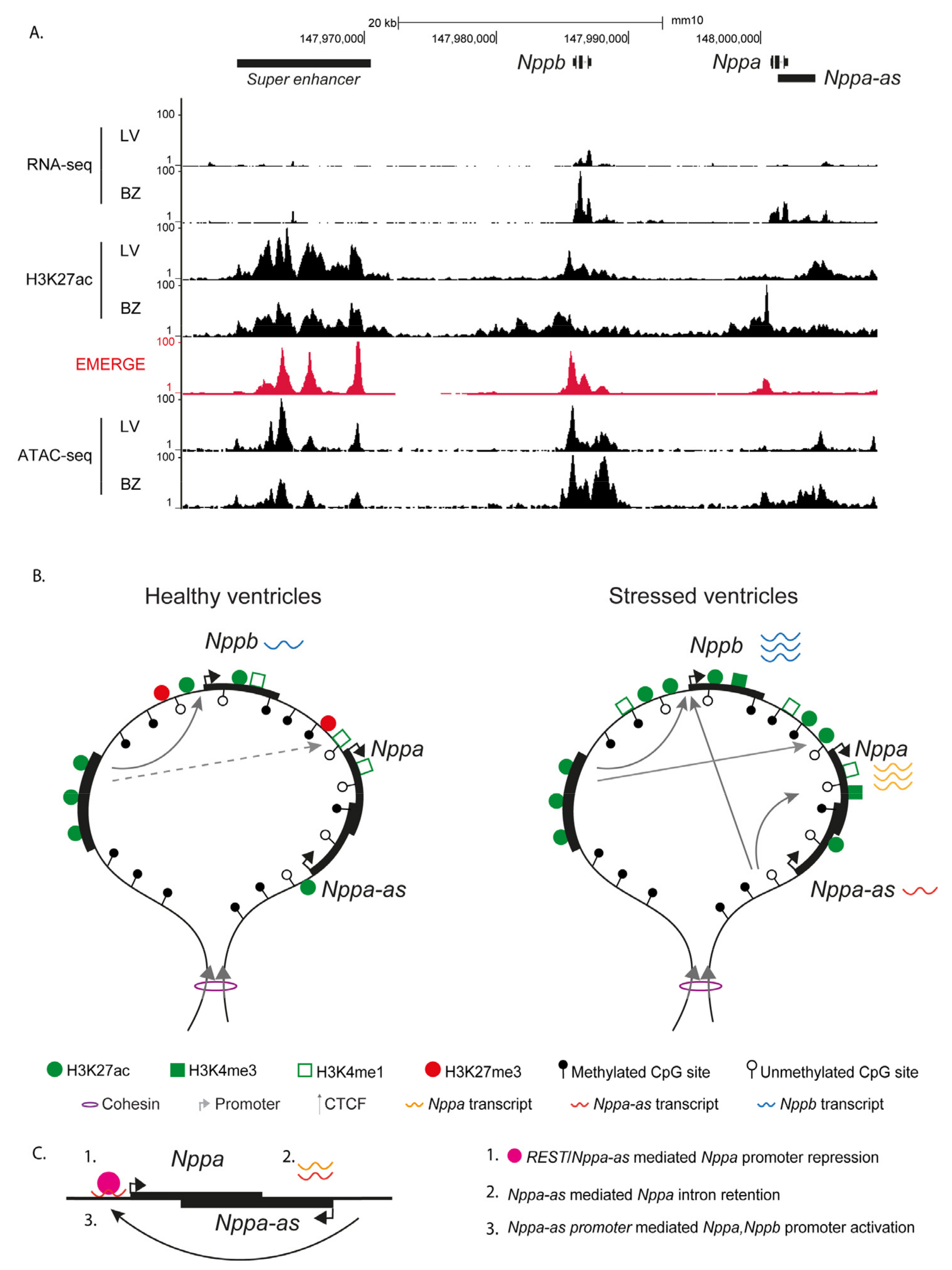
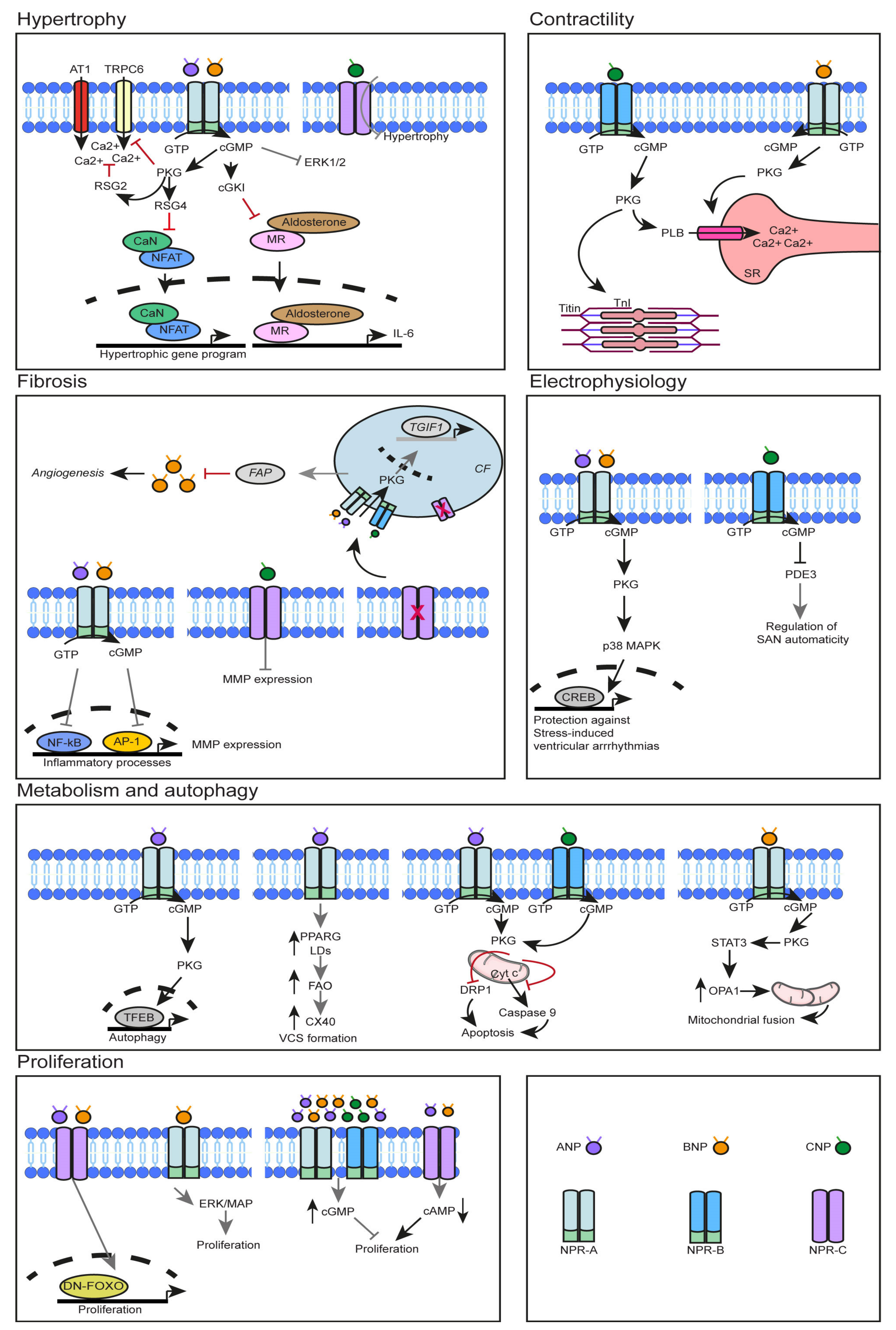

{kind=link}
{kind=link}
| Hypertrophy | ||||||
|---|---|---|---|---|---|---|
| NPs or NP receptors | Experimental model | Manipulation | Age/species | Intracardiac observations | Proposed pathways | Ref. |
| Npr1 | Global KO | None | E15.5, ND1 | Increased heart size, reduced number of nuclei | N/A | [12] |
| Npr1 | Global KO | None | 4 months, 12 months/mice | Increased CM size, increased fibrosis | N/A | [13] |
| Npr1 | Global KO | TRPC channel inhibition+ Ang II treatment | 12 weeks/mice | Reduction in hypertrophy | ANP/cGMP/PKG inhibition of the Ca2+ channel TRPC6 activity | [14] |
| Npr1 | Global KO | NPRA−/− x RGS4 CM-specific OE | 16 weeks/mice | Reduction in hypertrophy | NPRA/RGS4-mediated suppression of the calcineurin–NFAT pathway | [15] |
| Npr1 | Global KO | NPRA−/− NPRA−/− x AT1−/− DKO | 16 weeks/mice | CM hypertrophy Attenuated hypertrophic response | N/A | [16] |
| Npr1 | Global KO | NPRA−/− + MI NPRA−/− x AT1−/− DKO + MI | 8–10 weeks/mice | CM hypertrophy Attenuated hypertrophic response | N/A | [17] |
| Npr1 | Global KO | Calcineurin inhibitor (FK506) | 14 weeks/mice | Reduction in hypertrophy, decreased fibrosis | ANP pathway antagonizes the calcineurin–NFAT pathway to regulate the hypertrophic response | [18] |
| Npr1 | Global KO | None | Neonatal (P2), young (4 weeks), adult mice (22 weeks) | Increased CM hypertrophy, increased fibrosis | NF-kB/AP1-mediated MPP activation (fibrosis) disruption of sarcoplasmic reticulum Ca2+ handling (hypertrophy) | [19] |
| MMP-inhibition | adult mice (22 weeks) | Increased hypertrophy, decreased fibrosis | ||||
| Npr1 | Global KO | TAC | 3–6 months/mice | Cardiac hypertrophy | N/A | [20] |
| Npr1 | Global KO | None | 8–12 weeks/mice | CM hypertrophy | N/A | [21] |
| CM-specific NPRA OE | None | CM cell size reduction | ||||
| Global KO x CM-specific CM NPRA OE | None | Reduced hypertrophic response | ||||
| Npr1 | Global KO | Consecutive pregnancy–lactation cycles | 8 weeks/mice | Lactation-induced cardiac hypertrophy | NP/NPRA suppressed aldosterone/MR signaling and cardiac IL-6 expression | [22] |
| CM-specific KO | ||||||
| Npr1 | CM-specific KO | TAC | 8–12 weeks/mice | CM hypertrophy | ANP/cGMP/cGKI-mediated inhibition of MR nuclear translocation | [23] |
| TAC+ MR agonist (eplerenone) | Attenuation of CM hypertrophy | |||||
| Npr1 | CM-specific KO | Treatment with AngII/ISO | 4–6 weeks/mice | CM hypertrophy | ANP/cGMP/PKG I/RGS2-mediated suppression of Ang II-stimulated Ca2+ handling | [24] |
| Npr1 | CM-specific constitutively active NPRA | ISO infusion/AAC | 8–12 weeks/mice | Prevention of CM hypertrophy | N/A | [25] |
| Npr1 | Global constitutively active NPRA | None | 12 weeks/mice | Male-specific CM size reduction | NP/NPRA-mediated reduction of ERK1/2 activity | [26] |
| Npr1 | Global Npr1++/++ | None | 24–26 weeks/mice | Decreased CM area, reduced inflammatory cytokines, | NPRA/cGMP/mediated suppression of Nf-KB/AP1 | [27] |
| Nppa | Global KO | TAC | 10 weeks | Increased hypertrophy, increased ECM gene expression | N/A | [28] |
| Nppa | Global KO | Nppa+/− + TAC Nppa−/− + TAC | 9–12 weeks/mice | Dose-dependent cardiac hypertrophy in untreated and TAC mice | N/A | [29] |
| Nppa | Global KO | Aorto-caval fistula (ACF) for volume overload and low-salt diet | 8–10 weeks/mice | Increased hypertrophy | N/A | [30] |
| Nppa | Global KO | Low-salt diet | 8–9 weeks/mice | Increased CM hypertrophy | N/A | [31] |
| Nppa | Global KO | MI MI + ANP infusion | 6–24 weeks/mice | Increased hypertrophy | N/A | [32] |
| Nppb | Global KO | none | 4–8 weeks/rats | Progressive hypertrophy, hypertension fibrosis | N/A | [33] |
| AAV9-rBNP for 9 months | 9 months/rats | Reduced hypertrophy | ||||
| BNP | AAV9-rBNP for 9 months | Normotensive | 3-4 weeks/rats | Prevention of age-associated hypertrophy and fibrosis | N/A | [34] |
| Spontaneously hypertensive rat model | Prevention of hypertension-associated hypertrophy and fibrosis | |||||
| CNP/Npr3 | CM-specific deletion | none | Adult/mice | Preserved cardiac functionality, no increase in hypertrophy or fibrosis | N/A | [35] |
| Abdominal aortic constriction | Functional decline, increased hypertrophic response, fibrosis | |||||
| Isoproterenol | ||||||
| Angiotensin II | Increased hypertrophic response | |||||
| Angiotensin II + CNP | Decreased hypertrophic response | |||||
| IR | Increased infarct size, prolonged impairment in LV function | |||||
| Npr3 global deletion | none | Preserved cardiac functionality, no increase in hypertrophy or fibrosis | ||||
| Abdominal aortic constriction | Functional decline, increased hypertrophic response, fibrosis | |||||
| Abdominal aortic constriction + CNP | No reversal of pathologic remodeling | |||||
| IR | Increased infarct size, prolonged impairment in LV function | |||||
| Fibrosis | ||||||
| NPs or NP receptors | Experimental model | Manipulation | Age/species | Intracardiac observations | Proposed pathways | Ref. |
| Npr1 | Global KO | Consecutive pregnancy–lactation cycles | 8 weeks/mice | Lactation-induced cardiac hypertrophy+ fibrosis | NP/NPRA suppressed aldosterone/MR signaling and cardiac IL-6 expression | [22] |
| CM-specific KO | ||||||
| Npr1 | Global KO | None | Neonatal (P2), young (4 weeks), adult mice (22 weeks) | Increased CM hypertrophy, increased fibrosis | NF-kB/AP-1-mediated MPP activation (fibrosis) disruption of sarcoplasmic reticulum Ca2+ handling (hypertrophy) | [19] |
| MMP-inhibition | Adult mice (22 weeks) | Increased hypertrophy, decreased fibrosis | ||||
| Npr1 | Global KO | 30 min I/R | 10–14 weeks/mice | N/A | NPRA-mediated suppression of NF-kB-mediated inflammatory processes | [36] |
| Npr1 | Global Npr1++/++ | None | 24–26 weeks/mice | Decreased CM area, reduced inflammatory cytokines, | NPRA/cGMP-mediated suppression of Nf-KB/AP1 | [27] |
| Npr3 | Global KO | Npr3−/− | 20 weeks/mice | No effect on left ventricular and left atrial fibrosis | NPRC deletion results in cAMP/PKA- and cGMP/PKG-mediated TGIF1 upregulation | [37] |
| Npr3−/− with STZ-induced diabetic cardiomyopathy | Attenuation of diabetes-induced cardiac fibrosis, no difference in atrial fibrosis | |||||
| AAV9-shNPRC | AAV9-shNPRC with STZ-induced diabetic cardiomyopathy | Attenuation of diabetes-induced cardiac fibrosis | ||||
| Nppa | Global KO | Nppa+/− + TAC Nppa−/− + TAC | 9–12 weeks/mice | Dose-dependent cardiac hypertrophy and fibrosis both in untreated and TAC mice | N/A | [29] |
| Nppa | Global KO | Nppa−/− + I/R | 8–10 weeks/mice | Increased infarct size and reduced autophagy | ANP/NPR1/PRKG-mediated stimulation of autophagy through activation of TFEB | [38] |
| BNP | hAAV5-BNP intracardiac injection | Unmanipulated | rats | Reduced fibrosis, increase capillary density | N/A | [39] |
| MI | Improved cardiac function | Normalization of SERCA2 expression and PLN phosphorylation | ||||
| Ang II infusion | Decreased fibrosis | N/A | ||||
| Nppb | Global KO | None | 20 weeks/mice | Increased fibrosis | N/A | [40] |
| AAC | ||||||
| Nppb | Global KO | None | 20 weeks/mice | Increased fibrosis, Increased sarcomere contraction and disorganized myofibrils | N/A | [41] |
| AAC | ||||||
| Nppb | Liver-specific human serum amyloid P component promoter-mediated BNP plasma OE | MI | 8–12 weeks/mice | Increased neutrophile infiltration in the infracted area and increased cardiac MMP-9 expression | N/A | [42] |
| Nppb, Npr1 | Global KO Nppb Global KO Npr1 | TAC + FAP inhibition | 8–10 weeks | Decreased fibrosis, improved cardiac function, increased angiogenesis in BNP+/+ and NPRA+/+ | Fap inhibition results in BNP/NPRA-mediated cardioprotection | [43] |
| MI + FAP inhibition | ||||||
| Proliferation | ||||||
| NPs or NP receptors | Experimental model | Manipulation | Age/species | Intracardiac observations | Proposed pathways | Ref. |
| Npr1 | Global KO | None | P2/mice | Increase in CM number | N/A | [44] |
| CM-specific KO | None | No difference | ||||
| ANP | None | Intracardiac ANP + MI | Neonatal (ND7)/mice | No difference observed compared to control littermates | ANP, BNP/NPRC signaling and FOXO nuclear activity cooperatively regulate CM cell cycle activity | [45] |
| Intracardiac ANP + DN-FOXO + MI | Reactivation of CM cell cycle, reduced scar formation | |||||
| BNP | None | Bidaily BNP IP injection | Neonatal mice | Increased number of CMs, increase in CM cell cycle re-entry | BNP activation of the MAPK/ERK pathway | [46] |
| None | Bidaily BNP IP injection+MI | 8 weeks/mice | Increased number of CMs, increase in CM cell cycle re-entry, decreased apoptosis | |||
| Myh6 MerCreMer (tamoxifen -D14) | Bidaily BNP IP injection+MI | Increase in TNTI+/GFP+ cells, increase in CM cell cycle re-entry | ||||
| Npr1−/− | Bidaily BNP IP injection | Unchanged number of CMs | ||||
| None | Inhibitor of BNP degradation (LCZ696) + MI | Increased number of CMs, increase in CM cell cycle re-entry | ||||
| Metabolism and autophagy | ||||||
| NPs or NP receptors | Experimental model | Manipulation | Age/species | Intracardiac observations | Proposed pathways | Ref. |
| Npr1 | CM specific KO | None | Not described/mice | Increase in metabolic processes | circRNA and microRNA | [47] |
| Npr1 | CM specific KO | None | 8 weeks/mice | Metabolic deregulation | Positive enrichment in nucleotide synthesis and histidine metabolism, negative enrichment of mitochondrial proteins | [48] |
| Nppa | Global KO | Nppa−/− + I/R | 8–10 weeks/mice | Increased infarct size and reduced autophagy | ANP/NPR1/PRKG-mediated stimulation of autophagy through activation of TFEB | [38] |
| ANP | Embryonic ventricular cell cultures | ANP treatment | E11.5/mice | Increased VCS cell proliferation | ANP/NPRA-mediated increase in PPARG and FAO promotes VCS formation | [49] |
| ANP | Primary cardiomyocyte cultures | ANP treatment | Adult/rats | Reduced cardiomyocyte apoptosis | ANP- and CNP-mediated DrpI phosphorylation and caspase 9 decrease | [50] |
| CNP | CNP treatment | |||||
| BNP | No genetic modification | 4 weeks of BNP administration 7 days after STZ-induced diabetic cardiomyopathy | 8 weeks/mice | Preservation of mitochondrial function and prevention of DMC onset | BNP/NPRA/PKG/STAT3-OPA1-mediated mitochondrial fusion activation | [51] |
| AAV9-shBNP after 5 days of STZ-induced diabetic cardiomyopathy | Impaired mitochondrial function, exaggerated cardiac dysfunction | |||||
| Electrophysiology | ||||||
| NPs or NP receptors | Experimental model | Manipulation | Age/species | Intracardiac observations | Proposed pathways | Ref. |
| Npr2 | Global KO (Npr2−/+) | none | 20 weeks/mice | Increased cSNRT, spontaneous AP frequency, slower HR, reduced If and Ica, L currents | NPR-B/cGMP-mediated inhibition of PDE3 | [52] |
| Nppa, Nppb | Global KO Nppa−/− or Nppb−/− | none | 1–12 months/mice | Mild ventricular remodeling, no cardiac functional dysfunction | ANP, BNP/cGMP/PKG1/p38MAPK-mediated phophorylation of CREB | [53] |
| TAC | ||||||
| Isoproterenol treatment | Increased incidence of ventricular arrythmias | |||||
| Npr3 | Global KO | None | 10–15 weeks/mice | SAN dysfunction, atrial suspeptibility and increased fibrosis | N/A | [54] |
| Npr3 | Global KO | none | 10–15 weeks/mice | SAN dysfunction and increased SAN fibrosis | N/A | [55] |
| Ang-II treatment | ||||||
| Npr3 | Global KO | none | 10–15 weeks/mice | Atrial suspeptibility and increased atrial fibrosis | N/A | [56] |
| Ang-II treatment | ||||||
| Contractility | ||||||
| NPs or NP receptors | Experimental model | Manipulation | Age/species | Intracardiac observations | Proposed pathways | Ref. |
| Npr2 | CM specific Npr2 KO | TAC | 2 months/mice | Increased ventricular stiffness, LV diastolic and systolic dysfunction | CNP/NPRB/cGMP/PKGI-mediated Ser4080 phosphorylationof Tintin | [57] |
| CNP, BNP | cGMP biosensor | Isolated cardiomyocytes | Neonatal-adult/rats | Increased lucitropic and negative inotropic effects by CNP | CNP increases cGMP near TnI and PLB, regulated by PDE2 and PDE3 BNP increases cGMP only near PLB | [58] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giovou, A.E.; Gladka, M.M.; Christoffels, V.M. The Impact of Natriuretic Peptides on Heart Development, Homeostasis, and Disease. Cells 2024, 13, 931. https://doi.org/10.3390/cells13110931
Giovou AE, Gladka MM, Christoffels VM. The Impact of Natriuretic Peptides on Heart Development, Homeostasis, and Disease. Cells. 2024; 13(11):931. https://doi.org/10.3390/cells13110931
Chicago/Turabian StyleGiovou, Alexandra E., Monika M. Gladka, and Vincent M. Christoffels. 2024. "The Impact of Natriuretic Peptides on Heart Development, Homeostasis, and Disease" Cells 13, no. 11: 931. https://doi.org/10.3390/cells13110931
APA StyleGiovou, A. E., Gladka, M. M., & Christoffels, V. M. (2024). The Impact of Natriuretic Peptides on Heart Development, Homeostasis, and Disease. Cells, 13(11), 931. https://doi.org/10.3390/cells13110931