
The Role of Mesenchymal Reprogramming in Malignant Clonal Evolution and Intra-Tumoral Heterogeneity in Glioblastoma

Abstract
:1. Introduction
2. GBM Stem Cells in the GBM Microenvironment
3. Heterogeneity in GBM
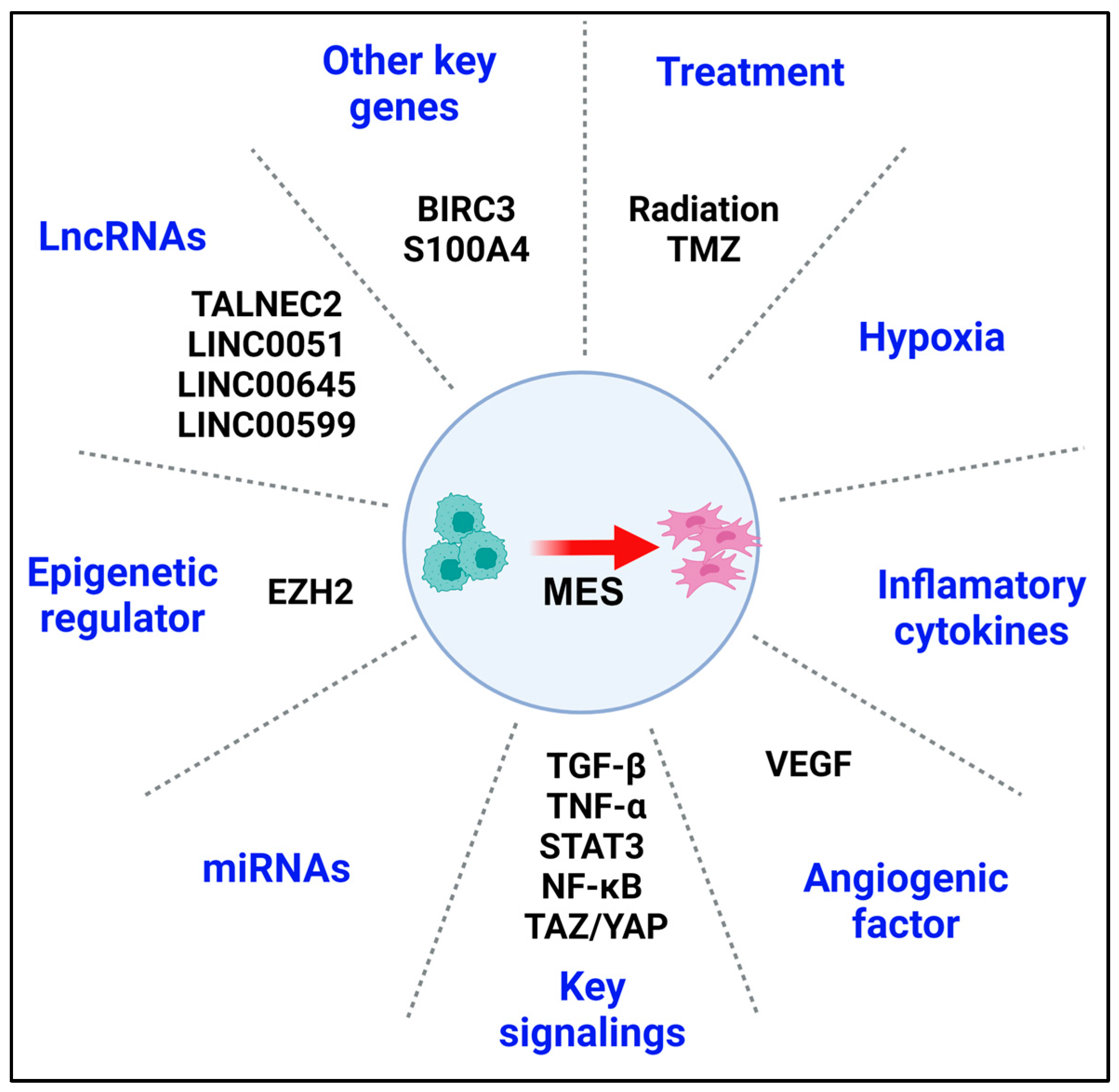
4. MES Reprogramming
4.1. Treatment-Induced MES Reprogramming and Clinical Relevance
4.2. Heterogenous Tumor Microenvironment
4.3. Key Regulators, Pathways, and Clinical Targets in MES Reprogramming
4.4. Recent and Potential Therapies Targeting MES Reprogramming
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| BIRC3 | Baculoviral IAP repeat containing 3 |
| CXCL2 | CXC-chemokine ligand 2 |
| DNMT | DNA methyltransferase |
| EMT | Epithelial–mesenchymal transition |
| EZH2 | Enhancer of zeste homolog |
| FAK | Focal adhesion kinase |
| FOXO1 | Forkhead box protein O1 |
| GBM | Glioblastoma |
| GSC | GBM stem cell |
| HDAC | Histone deacetylase |
| IDH | Isocitrate dehydrogenase |
| lncRNA | Long Non-Coding RNAs |
| MM9 | Matrix metalloproteinase 9 |
| MES | Mesenchymal |
| MGMT | O-6-methylguanine-DNA methyltransferase |
| PN | Proneural |
| RANKL | Receptor activator of nuclear factor kappa beta |
| RTKs | Receptor tyrosine kinases |
| scRNA-Seq | Single-cell RNA sequencing |
| STAT3 | Signal transducer and activator of transcription 3 |
| TAMs | Tumor-associated macrophages |
| TAZ | Transcriptional activator with PDZ-binding motif |
| TGM2 | The enzyme transglutaminase 2 |
| TME | Tumor microenvironment |
| TMZ | Temozolomide |
| YAP | Yes-associated protein |
References
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ostrom, Q.T.; Gittleman, H.; Farah, P.; Ondracek, A.; Chen, Y.; Wolinsky, Y.; Stroup, N.E.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS statistical report: Primary brain and central nervous system tumors diagnosed in the United States in 2006–2010. Neuro-Oncology 2013, 15 (Suppl. S2), ii1–ii56. [Google Scholar] [CrossRef] [PubMed]
- Tran, B.; Rosenthal, M.A. Survival comparison between glioblastoma multiforme and other incurable cancers. J. Clin. Neurosci. 2010, 17, 417–421. [Google Scholar] [CrossRef] [PubMed]
- Thakkar, J.P.; Dolecek, T.A.; Horbinski, C.; Ostrom, Q.T.; Lightner, D.D.; Barnholtz-Sloan, J.S.; Villano, J.L. Epidemiologic and molecular prognostic review of glioblastoma. Cancer Epidemiol. Biomark. Prev. 2014, 23, 1985–1996. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bleeker, F.E.; Atai, N.A.; Lamba, S.; Jonker, A.; Rijkeboer, D.; Bosch, K.S.; Tigchelaar, W.; Troost, D.; Vandertop, W.P.; Bardelli, A.; et al. The prognostic IDH1R132 mutation is associated with reduced NADP+-dependent IDH activity in glioblastoma. Acta Neuropathol. 2010, 119, 487–494. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Guo, J.; Wang, W.; Wang, D. Relationship between MGMT gene expression and treatment effectiveness and prognosis in glioma. Oncol. Lett. 2017, 14, 229–233. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rivera, A.L.; Pelloski, C.E.; Gilbert, M.R.; Colman, H.; De La Cruz, C.; Sulman, E.P.; Bekele, B.N.; Aldape, K.D. MGMT promoter methylation is predictive of response to radiotherapy and prognostic in the absence of adjuvant alkylating chemotherapy for glioblastoma. Neuro-Oncology 2010, 12, 116–121. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Taylor, O.G.; Brzozowski, J.S.; Skelding, K.A. Glioblastoma Multiforme: An Overview of Emerging Therapeutic Targets. Front. Oncol. 2019, 9, 963. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Omuro, A.; Vlahovic, G.; Lim, M.; Sahebjam, S.; Baehring, J.; Cloughesy, T.; Voloschin, A.; Ramkissoon, S.H.; Ligon, K.L.; Latek, R.; et al. Nivolumab with or without ipilimumab in patients with recurrent glioblastoma: Results from exploratory phase I cohorts of CheckMate 143. Neuro-Oncology 2018, 20, 674–686. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.; Lim, M.; Wick, A.; Baehring, J.; Ahluwalia, M.S.; Roth, P.; Bähr, O.; et al. Effect of Nivolumab vs Bevacizumab in Patients With Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1003–1010. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sampson, J.H.; Aldape, K.D.; Archer, G.E.; Coan, A.; Desjardins, A.; Friedman, A.H.; Friedman, H.S.; Gilbert, M.R.; Herndon, J.E.; McLendon, R.E.; et al. Greater chemotherapy-induced lymphopenia enhances tumor-specific immune responses that eliminate EGFRvIII-expressing tumor cells in patients with glioblastoma. Neuro-Oncology 2011, 13, 324–333. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Schuster, J.; Lai, R.K.; Recht, L.D.; Reardon, D.A.; Paleologos, N.A.; Groves, M.D.; Mrugala, M.M.; Jensen, R.; Baehring, J.M.; Sloan, A.; et al. A phase II, multicenter trial of rindopepimut (CDX-110) in newly diagnosed glioblastoma: The ACT III study. Neuro-Oncology 2015, 17, 854–861. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Weller, M.; Butowski, N.; Tran, D.D.; Recht, L.D.; Lim, M.; Hirte, H.; Ashby, L.; Mechtler, L.; Goldlust, S.A.; Iwamoto, F.; et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): A randomised, double-blind, international phase 3 trial. Lancet. Oncol. 2017, 18, 1373–1385. [Google Scholar] [CrossRef] [PubMed]
- Chuntova, P.; Chow, F.; Watchmaker, P.B.; Galvez, M.; Heimberger, A.B.; Newell, E.W.; Diaz, A.; DePinho, R.A.; Li, M.O.; Wherry, E.J.; et al. Unique challenges for glioblastoma immunotherapy-discussions across neuro-oncology and non-neuro-oncology experts in cancer immunology. Meeting Report from the 2019 SNO Immuno-Oncology Think Tank. Neuro-Oncology 2021, 23, 356–375. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.R.; Hugo, W.; Lee, A.H.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M.; et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat. Med. 2019, 25, 477–486. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Corso, C.D.; Bindra, R.S. Success and Failures of Combined Modalities in Glioblastoma Multiforme: Old Problems and New Directions. Semin. Radiat. Oncol. 2016, 26, 281–298. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, T.; Therkelsen, K.E.; Ahmad, S.; Nagpal, S. Current State of Immunotherapy for Treatment of Glioblastoma. Curr. Treat. Options Oncol. 2019, 20, 24. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Westphal, M.; Maire, C.L.; Lamszus, K. EGFR as a Target for Glioblastoma Treatment: An Unfulfilled Promise. CNS Drugs 2017, 31, 723–735. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chongsathidkiet, P.; Jackson, C.; Koyama, S.; Loebel, F.; Cui, X.; Farber, S.H.; Woroniecka, K.; Elsamadicy, A.A.; Dechant, C.A.; Kemeny, H.R.; et al. Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat. Med. 2018, 24, 1459–1468. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hodges, T.R.; Ott, M.; Xiu, J.; Gatalica, Z.; Swensen, J.; Zhou, S.; Huse, J.T.; de Groot, J.; Li, S.; Overwijk, W.W.; et al. Mutational burden, immune checkpoint expression, and mismatch repair in glioma: Implications for immune checkpoint immunotherapy. Neuro-Oncology 2017, 19, 1047–1057. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Indraccolo, S.; Lombardi, G.; Fassan, M.; Pasqualini, L.; Giunco, S.; Marcato, R.; Gasparini, A.; Candiotto, C.; Nalio, S.; Fiduccia, P.; et al. Genetic, Epigenetic, and Immunologic Profiling of MMR-Deficient Relapsed Glioblastoma. Clin. Cancer Res. 2019, 25, 1828–1837. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.M.; Choi, J.; Lim, M. Mechanisms of immunotherapy resistance: Lessons from glioblastoma. Nat. Immunol. 2019, 20, 1100–1109. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, B.C.; Maier, L.M.; D’Amico, R.; Mandigo, C.E.; Fontana, E.J.; Waziri, A.; Assanah, M.C.; Canoll, P.; Anderson, R.C.; Anderson, D.E.; et al. Dynamics of central and peripheral immunomodulation in a murine glioma model. BMC Immunol. 2009, 10, 11. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cifarelli, C.P.; Jacques, A.; Bobko, A. Heterogeneity of radiation response in mesenchymal subtype glioblastoma: Molecular profiling and reactive oxygen species generation. J. Neurooncol. 2021, 152, 245–255. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kim, Y.; Varn, F.S.; Park, S.H.; Yoon, B.W.; Park, H.R.; Lee, C.; Verhaak, R.G.W.; Paek, S.H. Perspective of mesenchymal transformation in glioblastoma. Acta Neuropathol. Commun. 2021, 9, 50. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Thomas, J.G.; Parker Kerrigan, B.C.; Hossain, A.; Gumin, J.; Shinojima, N.; Nwajei, F.; Ezhilarasan, R.; Love, P.; Sulman, E.P.; Lang, F.F. Ionizing radiation augments glioma tropism of mesenchymal stem cells. J. Neurosurg. 2018, 128, 287–295. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bradshaw, A.; Wickremsekera, A.; Tan, S.T.; Peng, L.; Davis, P.F.; Itinteang, T. Cancer Stem Cell Hierarchy in Glioblastoma Multiforme. Front. Surg. 2016, 3, 21. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mitchell, K.; Troike, K.; Silver, D.J.; Lathia, J.D. The evolution of the cancer stem cell state in glioblastoma: Emerging insights into the next generation of functional interactions. Neuro-Oncology 2021, 23, 199–213. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lee, J.H.; Lee, J.E.; Kahng, J.Y.; Kim, S.H.; Park, J.S.; Yoon, S.J.; Um, J.Y.; Kim, W.K.; Lee, J.K.; Park, J.; et al. Human glioblastoma arises from subventricular zone cells with low-level driver mutations. Nature 2018, 560, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Vescovi, A.L.; Galli, R.; Reynolds, B.A. Brain tumour stem cells. Nat. Rev. Cancer 2006, 6, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Iacopino, F.; Angelucci, C.; Piacentini, R.; Biamonte, F.; Mangiola, A.; Maira, G.; Grassi, C.; Sica, G. Isolation of cancer stem cells from three human glioblastoma cell lines: Characterization of two selected clones. PLoS ONE 2014, 9, e105166. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Pointer, K.B.; Clark, P.A.; Zorniak, M.; Alrfaei, B.M.; Kuo, J.S. Glioblastoma cancer stem cells: Biomarker and therapeutic advances. Neurochem. Int. 2014, 71, 1–7. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Singh, S.K.; Clarke, I.D.; Hide, T.; Dirks, P.B. Cancer stem cells in nervous system tumors. Oncogene 2004, 23, 7267–7273. [Google Scholar] [CrossRef] [PubMed]
- Hambardzumyan, D.; Bergers, G. Glioblastoma: Defining Tumor Niches. Trends Cancer 2015, 1, 252–265. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chen, J.; Li, Y.; Yu, T.S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Dey, M.; Ulasov, I.V.; Lesniak, M.S. Virotherapy against malignant glioma stem cells. Cancer Lett. 2010, 289, 1–10. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hombach-Klonisch, S.; Mehrpour, M.; Shojaei, S.; Harlos, C.; Pitz, M.; Hamai, A.; Siemianowicz, K.; Likus, W.; Wiechec, E.; Toyota, B.D.; et al. Glioblastoma and chemoresistance to alkylating agents: Involvement of apoptosis, autophagy, and unfolded protein response. Pharmacol. Ther. 2018, 184, 13–41. [Google Scholar] [CrossRef] [PubMed]
- Farahani, E.; Patra, H.K.; Jangamreddy, J.R.; Rashedi, I.; Kawalec, M.; Rao Pariti, R.K.; Batakis, P.; Wiechec, E. Cell adhesion molecules and their relation to (cancer) cell stemness. Carcinogenesis 2014, 35, 747–759. [Google Scholar] [CrossRef] [PubMed]
- Plaks, V.; Kong, N.; Werb, Z. The cancer stem cell niche: How essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liang, J.; Piao, Y.; Holmes, L.; Fuller, G.N.; Henry, V.; Tiao, N.; de Groot, J.F. Neutrophils promote the malignant glioma phenotype through S100A4. Clin. Cancer Res. 2014, 20, 187–198. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Badie, B.; Schartner, J. Role of microglia in glioma biology. Microsc. Res. Tech. 2001, 54, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 2016, 19, 20–27. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Becher, O.J.; Hambardzumyan, D.; Fomchenko, E.I.; Momota, H.; Mainwaring, L.; Bleau, A.M.; Katz, A.M.; Edgar, M.; Kenney, A.M.; Cordon-Cardo, C.; et al. Gli activity correlates with tumor grade in platelet-derived growth factor-induced gliomas. Cancer Res. 2008, 68, 2241–2249. [Google Scholar] [CrossRef] [PubMed]
- Edwards, L.A.; Woolard, K.; Son, M.J.; Li, A.; Lee, J.; Ene, C.; Mantey, S.A.; Maric, D.; Song, H.; Belova, G.; et al. Effect of brain- and tumor-derived connective tissue growth factor on glioma invasion. J. Natl. Cancer Inst. 2011, 103, 1162–1178. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; deCarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56.e6. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kolla, V.; Zhuang, T.; Higashi, M.; Naraparaju, K.; Brodeur, G.M. Role of CHD5 in human cancers: 10 years later. Cancer Res. 2014, 74, 652–658. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Reifenberger, G.; Liu, L.; Ichimura, K.; Schmidt, E.E.; Collins, V.P. Amplification and overexpression of the MDM2 gene in a subset of human malignant gliomas without p53 mutations. Cancer Res. 1993, 53, 2736–2739. [Google Scholar] [PubMed]
- Toledo, F.; Wahl, G.M. Regulating the p53 pathway: In vitro hypotheses, in vivo veritas. Nat. Rev. Cancer 2006, 6, 909–923. [Google Scholar] [CrossRef] [PubMed]
- Classon, M.; Harlow, E. The retinoblastoma tumour suppressor in development and cancer. Nat. Rev. Cancer 2002, 2, 910–917. [Google Scholar] [CrossRef] [PubMed]
- Esteban-Villarrubia, J.; Soto-Castillo, J.J.; Pozas, J.; San Román-Gil, M.; Orejana-Martín, I.; Torres-Jiménez, J.; Carrato, A.; Alonso-Gordoa, T.; Molina-Cerrillo, J. Tyrosine Kinase Receptors in Oncology. Int. J. Mol. Sci. 2020, 21, 20201112. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Li, E.; Hristova, K. Role of receptor tyrosine kinase transmembrane domains in cell signaling and human pathologies. Biochemistry 2006, 45, 6241–6251. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Frederick, L.; Wang, X.Y.; Eley, G.; James, C.D. Diversity and frequency of epidermal growth factor receptor mutations in human glioblastomas. Cancer Res. 2000, 60, 1383–1387. [Google Scholar] [PubMed]
- Lee, J.C.; Vivanco, I.; Beroukhim, R.; Huang, J.H.; Feng, W.L.; DeBiasi, R.M.; Yoshimoto, K.; King, J.C.; Nghiemphu, P.; Yuza, Y.; et al. Epidermal growth factor receptor activation in glioblastoma through novel missense mutations in the extracellular domain. PLoS Med. 2006, 3, e485. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [CrossRef] [PubMed] [PubMed Central]
- Ozawa, T.; Brennan, C.W.; Wang, L.; Squatrito, M.; Sasayama, T.; Nakada, M.; Huse, J.T.; Pedraza, A.; Utsuki, S.; Yasui, Y.; et al. PDGFRA gene rearrangements are frequent genetic events in PDGFRA-amplified glioblastomas. Genes Dev. 2010, 24, 2205–2218. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhang, Y.; Zhang, B.; Lv, C.; Zhang, N.; Xing, K.; Wang, Z.; Lv, R.; Yu, M.; Xu, C.; Wang, Y. Single-cell RNA sequencing identifies critical transcription factors of tumor cell invasion induced by hypoxia microenvironment in glioblastoma. Theranostics 2023, 13, 3744–3760. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Alban, T.J.; Alvarado, A.G.; Sorensen, M.D.; Bayik, D.; Volovetz, J.; Serbinowski, E.; Mulkearns-Hubert, E.E.; Sinyuk, M.; Hale, J.S.; Onzi, G.R.; et al. Global immune fingerprinting in glioblastoma patient peripheral blood reveals immune-suppression signatures associated with prognosis. JCI Insight 2018, 3, e122264. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Miska, J.; Rashidi, A.; Lee-Chang, C.; Gao, P.; Lopez-Rosas, A.; Zhang, P.; Burga, R.; Castro, B.; Xiao, T.; Han, Y.; et al. Polyamines drive myeloid cell survival by buffering intracellular pH to promote immunosuppression in glioblastoma. Sci. Adv. 2021, 7, eabc8929. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chanoch-Myers, R.; Wider, A.; Suva, M.L.; Tirosh, I. Elucidating the diversity of malignant mesenchymal states in glioblastoma by integrative analysis. Genome Med. 2022, 14, 106. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, L.; Jung, J.; Babikir, H.; Shamardani, K.; Jain, S.; Feng, X.; Gupta, N.; Rosi, S.; Chang, S.; Raleigh, D.; et al. A single-cell atlas of glioblastoma evolution under therapy reveals cell-intrinsic and cell-extrinsic therapeutic targets. Nat. Cancer 2022, 3, 1534–1552. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Perrault, E.N.; Shireman, J.M.; Ali, E.S.; Lin, P.; Preddy, I.; Park, C.; Budhiraja, S.; Baisiwala, S.; Dixit, K.; James, C.D.; et al. Ribonucleotide reductase regulatory subunit M2 drives glioblastoma TMZ resistance through modulation of dNTP production. Sci. Adv. 2023, 9, eade7236. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Shireman, J.M.; Atashi, F.; Lee, G.; Ali, E.S.; Saathoff, M.R.; Park, C.H.; Savchuk, S.; Baisiwala, S.; Miska, J.; Lesniak, M.S.; et al. De novo purine biosynthesis is a major driver of chemoresistance in glioblastoma. Brain 2021, 144, 1230–1246. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ravi, V.M.; Neidert, N.; Will, P.; Joseph, K.; Maier, J.P.; Kückelhaus, J.; Vollmer, L.; Goeldner, J.M.; Behringer, S.P.; Scherer, F.; et al. T-cell dysfunction in the glioblastoma microenvironment is mediated by myeloid cells releasing interleukin-10. Nat. Commun. 2022, 13, 925. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ravi, V.M.; Will, P.; Kueckelhaus, J.; Sun, N.; Joseph, K.; Salié, H.; Vollmer, L.; Kuliesiute, U.; von Ehr, J.; Benotmane, J.K.; et al. Spatially resolved multi-omics deciphers bidirectional tumor-host interdependence in glioblastoma. Cancer Cell 2022, 40, 639–655.e13. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Huang, Z.; Zhou, L.; Xiao, P.; Song, J.; He, P.; Xie, C.; Zhou, R.; Li, M.; Dong, X.; et al. Spatial transcriptomics reveals niche-specific enrichment and vulnerabilities of radial glial stem-like cells in malignant gliomas. Nat. Commun. 2023, 14, 1028. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Greenwald, A.C.; Darnell, N.G.; Hoefflin, R.; Simkin, D.; Mount, C.W.; Gonzalez Castro, L.N.; Harnik, Y.; Dumont, S.; Hirsch, D.; Nomura, M.; et al. Integrative spatial analysis reveals a multi-layered organization of glioblastoma. Cell 2024, 187, 2485–2501.e26. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- De Silva, M.I.; Stringer, B.W.; Bardy, C. Neuronal and tumourigenic boundaries of glioblastoma plasticity. Trends Cancer 2023, 9, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849.e21. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Suvà, M.L.; Tirosh, I. The Glioma Stem Cell Model in the Era of Single-Cell Genomics. Cancer Cell 2020, 37, 630–636. [Google Scholar] [CrossRef] [PubMed]
- Couturier, C.P.; Ayyadhury, S.; Le, P.U.; Nadaf, J.; Monlong, J.; Riva, G.; Allache, R.; Baig, S.; Yan, X.; Bourgey, M.; et al. Single-cell RNA-seq reveals that glioblastoma recapitulates a normal neurodevelopmental hierarchy. Nat. Commun. 2020, 11, 3406. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef] [PubMed]
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavare, S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Behnan, J.; Finocchiaro, G.; Hanna, G. The landscape of the mesenchymal signature in brain tumours. Brain 2019, 142, 847–866. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bhat, K.P.L.; Balasubramaniyan, V.; Vaillant, B.; Ezhilarasan, R.; Hummelink, K.; Hollingsworth, F.; Wani, K.; Heathcock, L.; James, J.D.; Goodman, L.D.; et al. Mesenchymal differentiation mediated by NF-κB promotes radiation resistance in glioblastoma. Cancer Cell 2013, 24, 331–346. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Singh, S.K.; Fiorelli, R.; Kupp, R.; Rajan, S.; Szeto, E.; Lo Cascio, C.; Maire, C.L.; Sun, Y.; Alberta, J.A.; Eschbacher, J.M.; et al. Post-translational Modifications of OLIG2 Regulate Glioma Invasion through the TGF-β Pathway. Cell Rep. 2016, 16, 950–966. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sciacovelli, M.; Frezza, C. Metabolic reprogramming and epithelial-to-mesenchymal transition in cancer. FEBS J. 2017, 284, 3132–3144. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liu, T.; Ma, W.; Xu, H.; Huang, M.; Zhang, D.; He, Z.; Zhang, L.; Brem, S.; O’Rourke, D.M.; Gong, Y.; et al. PDGF-mediated mesenchymal transformation renders endothelial resistance to anti-VEGF treatment in glioblastoma. Nat. Commun. 2018, 9, 3439. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Piao, Y.; Liang, J.; Holmes, L.; Henry, V.; Sulman, E.; de Groot, J.F. Acquired resistance to anti-VEGF therapy in glioblastoma is associated with a mesenchymal transition. Clin. Cancer Res. 2013, 19, 4392–4403. [Google Scholar] [CrossRef] [PubMed]
- Segerman, A.; Niklasson, M.; Haglund, C.; Bergström, T.; Jarvius, M.; Xie, Y.; Westermark, A.; Sönmez, D.; Hermansson, A.; Kastemar, M.; et al. Clonal Variation in Drug and Radiation Response among Glioma-Initiating Cells Is Linked to Proneural-Mesenchymal Transition. Cell Rep. 2016, 17, 2994–3009. [Google Scholar] [CrossRef] [PubMed]
- Halliday, J.; Helmy, K.; Pattwell, S.S.; Pitter, K.L.; LaPlant, Q.; Ozawa, T.; Holland, E.C. In vivo radiation response of proneural glioma characterized by protective p53 transcriptional program and proneural-mesenchymal shift. Proc. Natl. Acad. Sci. USA 2014, 111, 5248–5253. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Martinez, R.; Rohde, V.; Schackert, G. Different molecular patterns in glioblastoma multiforme subtypes upon recurrence. J. Neuro-Oncol. 2010, 96, 321–329. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Takashima, Y.; Kawaguchi, A.; Yamanaka, R. Promising Prognosis Marker Candidates on the Status of Epithelial-Mesenchymal Transition and Glioma Stem Cells in Glioblastoma. Cells 2019, 8, 1312. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Fedele, M.; Cerchia, L.; Pegoraro, S.; Sgarra, R.; Manfioletti, G. Proneural-Mesenchymal Transition: Phenotypic Plasticity to Acquire Multitherapy Resistance in Glioblastoma. Int. J. Mol. Sci. 2019, 20, 2746. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kim, J.; Lee, I.H.; Cho, H.J.; Park, C.K.; Jung, Y.S.; Kim, Y.; Nam, S.H.; Kim, B.S.; Johnson, M.D.; Kong, D.S.; et al. Spatiotemporal Evolution of the Primary Glioblastoma Genome. Cancer Cell 2015, 28, 318–328. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.; Lu, X.; Liao, Y.; Ouyang, P.; Wang, H.; Zhang, X.; Huang, G.; Qi, S.; Li, Y. Crosstalk between glioblastoma and tumor microenvironment drives proneural-mesenchymal transition through ligand-receptor interactions. Genes Dis. 2024, 11, 874–889. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Herting, C.J.; Chen, Z.; Pitter, K.L.; Szulzewsky, F.; Kaffes, I.; Kaluzova, M.; Park, J.C.; Cimino, P.J.; Brennan, C.; Wang, B.; et al. Genetic driver mutations define the expression signature and microenvironmental composition of high-grade gliomas. Glia 2017, 65, 1914–1926. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Su, X.; Yang, Y.; Guo, C.; Zhang, R.; Sun, S.; Wang, Y.; Qiao, Q.; Fu, Y.; Pang, Q. NOX4-Derived ROS Mediates TGF-β1-Induced Metabolic Reprogramming during Epithelial-Mesenchymal Transition through the PI3K/AKT/HIF-1α Pathway in Glioblastoma. Oxid. Med. Cell. Longev. 2021, 2021, 5549047. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Talasila, K.M.; Røsland, G.V.; Hagland, H.R.; Eskilsson, E.; Flønes, I.H.; Fritah, S.; Azuaje, F.; Atai, N.; Harter, P.N.; Mittelbronn, M.; et al. The angiogenic switch leads to a metabolic shift in human glioblastoma. Neuro-Oncology 2017, 19, 383–393. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yang, M.; Chen, X.; Zhang, J.; Xiong, E.; Wang, Q.; Fang, W.; Li, L.; Fei, F.; Gong, A. ME2 Promotes Proneural-Mesenchymal Transition and Lipogenesis in Glioblastoma. Front. Oncol. 2021, 11, 715593. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lau, J.; Ilkhanizadeh, S.; Wang, S.; Miroshnikova, Y.A.; Salvatierra, N.A.; Wong, R.A.; Schmidt, C.; Weaver, V.M.; Weiss, W.A.; Persson, A.I. STAT3 Blockade Inhibits Radiation-Induced Malignant Progression in Glioma. Cancer Res. 2015, 75, 4302–4311. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Minata, M.; Audia, A.; Shi, J.; Lu, S.; Bernstock, J.; Pavlyukov, M.S.; Das, A.; Kim, S.H.; Shin, Y.J.; Lee, Y.; et al. Phenotypic Plasticity of Invasive Edge Glioma Stem-like Cells in Response to Ionizing Radiation. Cell Rep. 2019, 26, 1893–1905.e7. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wood, M.D.; Reis, G.F.; Reuss, D.E.; Phillips, J.J. Protein Analysis of Glioblastoma Primary and Posttreatment Pairs Suggests a Mesenchymal Shift at Recurrence. J. Neuropathol. Exp. Neurol. 2016, 75, 925–935. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Timke, C.; Zieher, H.; Roth, A.; Hauser, K.; Lipson, K.E.; Weber, K.J.; Debus, J.; Abdollahi, A.; Huber, P.E. Combination of vascular endothelial growth factor receptor/platelet-derived growth factor receptor inhibition markedly improves radiation tumor therapy. Clin. Cancer Res. 2008, 14, 2210–2219. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.C.; Liu, J.Y.; Li, J.; Zhang, J.; Xu, Y.Q.; Zhang, H.W.; Qiu, L.B.; Ding, G.R.; Su, X.M.; Mei, S.; et al. Ionizing radiation promotes migration and invasion of cancer cells through transforming growth factor-beta-mediated epithelial-mesenchymal transition. Int. J. Radiat. Oncol. Biol. Phys. 2011, 81, 1530–1537. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, X.; Xu, R.; Ji, J.; Xu, Y.; Han, M.; Wei, Y.; Huang, B.; Chen, A.; Zhang, Q.; et al. YM155 decreases radiation-induced invasion and reverses epithelial-mesenchymal transition by targeting STAT3 in glioblastoma. J. Transl. Med. 2018, 16, 79. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Moreno, M.; Pedrosa, L.; Paré, L.; Pineda, E.; Bejarano, L.; Martínez, J.; Balasubramaniyan, V.; Ezhilarasan, R.; Kallarackal, N.; Kim, S.H.; et al. GPR56/ADGRG1 Inhibits Mesenchymal Differentiation and Radioresistance in Glioblastoma. Cell Rep. 2017, 21, 2183–2197. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Han, G.; Li, Y.; Yue, Z.; Wang, L.; Liu, J. FOXO1 associated with sensitivity to chemotherapy drugs and glial-mesenchymal transition in glioma. J. Cell. Biochem. 2019, 120, 882–893. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.E.; Mazor, T.; Hong, C.; Barnes, M.; Aihara, K.; McLean, C.Y.; Fouse, S.D.; Yamamoto, S.; Ueda, H.; Tatsuno, K.; et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science 2014, 343, 189–193. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Prins, R.M.; Soto, H.; Konkankit, V.; Odesa, S.K.; Eskin, A.; Yong, W.H.; Nelson, S.F.; Liau, L.M. Gene expression profile correlates with T-cell infiltration and relative survival in glioblastoma patients vaccinated with dendritic cell immunotherapy. Clin. Cancer Res. 2011, 17, 1603–1615. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tejero, R.; Huang, Y.; Katsyv, I.; Kluge, M.; Lin, J.Y.; Tome-Garcia, J.; Daviaud, N.; Wang, Y.; Zhang, B.; Tsankova, N.M.; et al. Gene signatures of quiescent glioblastoma cells reveal mesenchymal shift and interactions with niche microenvironment. eBioMedicine 2019, 42, 252–269. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, J.; Zhou, F.; Li, Y.; Li, Q.; Wu, Z.; Yu, L.; Yuan, F.; Liu, J.; Tian, Y.; Cao, Y.; et al. Cdc20 overexpression is involved in temozolomide-resistant glioma cells with epithelial-mesenchymal transition. Cell Cycle (Georget. Tex.) 2017, 16, 2355–2365. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Dumas, A.A.; Pomella, N.; Rosser, G.; Guglielmi, L.; Vinel, C.; Millner, T.O.; Rees, J.; Aley, N.; Sheer, D.; Wei, J.; et al. Microglia promote glioblastoma via mTOR-mediated immunosuppression of the tumour microenvironment. EMBO J. 2020, 39, e103790. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chinot, O.L.; Wick, W.; Mason, W.; Henriksson, R.; Saran, F.; Nishikawa, R.; Carpentier, A.F.; Hoang-Xuan, K.; Kavan, P.; Cernea, D.; et al. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 709–722. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wick, W.; Platten, M.; Wick, A.; Hertenstein, A.; Radbruch, A.; Bendszus, M.; Winkler, F. Current status and future directions of anti-angiogenic therapy for gliomas. Neuro-Oncology 2016, 18, 315–328. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Piao, Y.; Liang, J.; Holmes, L.; Zurita, A.J.; Henry, V.; Heymach, J.V.; de Groot, J.F. Glioblastoma resistance to anti-VEGF therapy is associated with myeloid cell infiltration, stem cell accumulation, and a mesenchymal phenotype. Neuro-Oncology 2012, 14, 1379–1392. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Puchalski, R.B.; Shah, N.; Miller, J.; Dalley, R.; Nomura, S.R.; Yoon, J.G.; Smith, K.A.; Lankerovich, M.; Bertagnolli, D.; Bickley, K.; et al. An anatomic transcriptional atlas of human glioblastoma. Science 2018, 360, 660–663. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Fink, J.R.; Muzi, M.; Peck, M.; Krohn, K.A. Multimodality Brain Tumor Imaging: MR Imaging, PET, and PET/MR Imaging. J. Nucl. Med. 2015, 56, 1554–1561. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Horská, A.; Barker, P.B. Imaging of brain tumors: MR spectroscopy and metabolic imaging. Neuroimaging Clin. N. Am. 2010, 20, 293–310. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jensen, T.R.; Schmainda, K.M. Computer-aided detection of brain tumor invasion using multiparametric MRI. J. Magn. Reson. Imaging 2009, 30, 481–489. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Civita, P.; Franceschi, S.; Aretini, P.; Ortenzi, V.; Menicagli, M.; Lessi, F.; Pasqualetti, F.; Naccarato, A.G.; Mazzanti, C.M. Laser Capture Microdissection and RNA-Seq Analysis: High Sensitivity Approaches to Explain Histopathological Heterogeneity in Human Glioblastoma FFPE Archived Tissues. Front. Oncol. 2019, 9, 482. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- De Palma, M.; Biziato, D.; Petrova, T.V. Microenvironmental regulation of tumour angiogenesis. Nat. Rev. Cancer 2017, 17, 457–474. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.L. Hypoxia--a key regulatory factor in tumour growth. Nat. Rev. Cancer 2002, 2, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, A.R.; Hill, R.; Pilkington, G.J.; Madureira, P.A. The Role of Hypoxia in Glioblastoma Invasion. Cells 2017, 6, 45. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Joseph, J.V.; Conroy, S.; Pavlov, K.; Sontakke, P.; Tomar, T.; Eggens-Meijer, E.; Balasubramaniyan, V.; Wagemakers, M.; den Dunnen, W.F.; Kruyt, F.A. Hypoxia enhances migration and invasion in glioblastoma by promoting a mesenchymal shift mediated by the HIF1α-ZEB1 axis. Cancer Lett. 2015, 359, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.; Song, S.; Chen, W.; Zhang, J.; Yang, H.; Chen, Y. Hypoxia-induced EPHB2 promotes invasive potential of glioblastoma. Int. J. Clin. Exp. Pathol. 2019, 12, 539–548. [Google Scholar] [PubMed] [PubMed Central]
- Jin, P.; Shin, S.H.; Chun, Y.S.; Shin, H.W.; Shin, Y.J.; Lee, Y.; Kim, D.; Nam, D.H.; Park, J.W. Astrocyte-derived CCL20 reinforces HIF-1-mediated hypoxic responses in glioblastoma by stimulating the CCR6-NF-κB signaling pathway. Oncogene 2018, 37, 3070–3087. [Google Scholar] [CrossRef] [PubMed]
- Niklasson, M.; Bergström, T.; Jarvius, M.; Sundström, A.; Nyberg, F.; Haglund, C.; Larsson, R.; Westermark, B.; Segerman, B.; Segerman, A. Mesenchymal transition and increased therapy resistance of glioblastoma cells is related to astrocyte reactivity. J. Pathol. 2019, 249, 295–307. [Google Scholar] [CrossRef] [PubMed]
- Doucette, T.; Rao, G.; Rao, A.; Shen, L.; Aldape, K.; Wei, J.; Dziurzynski, K.; Gilbert, M.; Heimberger, A.B. Immune heterogeneity of glioblastoma subtypes: Extrapolation from the cancer genome atlas. Cancer Immunol. Res. 2013, 1, 112–122. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rutledge, W.C.; Kong, J.; Gao, J.; Gutman, D.A.; Cooper, L.A.; Appin, C.; Park, Y.; Scarpace, L.; Mikkelsen, T.; Cohen, M.L.; et al. Tumor-infiltrating lymphocytes in glioblastoma are associated with specific genomic alterations and related to transcriptional class. Clin. Cancer Res. 2013, 19, 4951–4960. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Beier, C.P.; Kumar, P.; Meyer, K.; Leukel, P.; Bruttel, V.; Aschenbrenner, I.; Riemenschneider, M.J.; Fragoulis, A.; Rümmele, P.; Lamszus, K.; et al. The cancer stem cell subtype determines immune infiltration of glioblastoma. Stem Cells Dev. 2012, 21, 2753–2761. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Engler, J.R.; Robinson, A.E.; Smirnov, I.; Hodgson, J.G.; Berger, M.S.; Gupta, N.; James, C.D.; Molinaro, A.; Phillips, J.J. Increased microglia/macrophage gene expression in a subset of adult and pediatric astrocytomas. PLoS ONE 2012, 7, e43339. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sørensen, M.D.; Dahlrot, R.H.; Boldt, H.B.; Hansen, S.; Kristensen, B.W. Tumour-associated microglia/macrophages predict poor prognosis in high-grade gliomas and correlate with an aggressive tumour subtype. Neuropathol. Appl. Neurobiol. 2018, 44, 185–206. [Google Scholar] [CrossRef] [PubMed]
- Kaffes, I.; Szulzewsky, F.; Chen, Z.; Herting, C.J.; Gabanic, B.; Velázquez Vega, J.E.; Shelton, J.; Switchenko, J.M.; Ross, J.L.; McSwain, L.F.; et al. Human Mesenchymal glioblastomas are characterized by an increased immune cell presence compared to Proneural and Classical tumors. Oncoimmunology 2019, 8, e1655360. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zanotto-Filho, A.; Gonçalves, R.M.; Klafke, K.; de Souza, P.O.; Dillenburg, F.C.; Carro, L.; Gelain, D.P.; Moreira, J.C. Inflammatory landscape of human brain tumors reveals an NFκB dependent cytokine pathway associated with mesenchymal glioblastoma. Cancer Lett. 2017, 390, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Chanoch-Myers, R.; Mathewson, N.D.; Myskiw, C.; Atta, L.; Bussema, L.; Eichhorn, S.W.; Greenwald, A.C.; Kinker, G.S.; Rodman, C.; et al. Interactions between cancer cells and immune cells drive transitions to mesenchymal-like states in glioblastoma. Cancer Cell 2021, 39, 779–792.e11. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Arbab, A.S.; Rashid, M.H.; Angara, K.; Borin, T.F.; Lin, P.C.; Jain, M.; Achyut, B.R. Major Challenges and Potential Microenvironment-Targeted Therapies in Glioblastoma. Int. J. Mol. Sci. 2017, 18, 2732. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ye, Z.; Ai, X.; Zhao, L.; Fei, F.; Wang, P.; Zhou, S. Phenotypic plasticity of myeloid cells in glioblastoma development, progression, and therapeutics. Oncogene 2021, 40, 6059–6070. [Google Scholar] [CrossRef] [PubMed]
- Broekman, M.L.; Maas, S.L.N.; Abels, E.R.; Mempel, T.R.; Krichevsky, A.M.; Breakefield, X.O. Multidimensional communication in the microenvirons of glioblastoma. Nat. Rev. Neurol. 2018, 14, 482–495. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Brandenburg, S.; Müller, A.; Turkowski, K.; Radev, Y.T.; Rot, S.; Schmidt, C.; Bungert, A.D.; Acker, G.; Schorr, A.; Hippe, A.; et al. Resident microglia rather than peripheral macrophages promote vascularization in brain tumors and are source of alternative pro-angiogenic factors. Acta Neuropathol. 2016, 131, 365–378. [Google Scholar] [CrossRef] [PubMed]
- Poon, C.C.; Sarkar, S.; Yong, V.W.; Kelly, J.J.P. Glioblastoma-associated microglia and macrophages: Targets for therapies to improve prognosis. Brain 2017, 140, 1548–1560. [Google Scholar] [CrossRef] [PubMed]
- Cooper, L.A.; Gutman, D.A.; Chisolm, C.; Appin, C.; Kong, J.; Rong, Y.; Kurc, T.; Van Meir, E.G.; Saltz, J.H.; Moreno, C.S.; et al. The tumor microenvironment strongly impacts master transcriptional regulators and gene expression class of glioblastoma. Am. J. Pathol. 2012, 180, 2108–2119. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Caverzán, M.D.; Beaugé, L.; Oliveda, P.M.; Cesca González, B.; Bühler, E.M.; Ibarra, L.E. Exploring Monocytes-Macrophages in Immune Microenvironment of Glioblastoma for the Design of Novel Therapeutic Strategies. Brain Sci. 2023, 13, 542. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pombo Antunes, A.R.; Scheyltjens, I.; Duerinck, J.; Neyns, B.; Movahedi, K.; Van Ginderachter, J.A. Understanding the glioblastoma immune microenvironment as basis for the development of new immunotherapeutic strategies. eLife 2020, 9, e52176. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Li, C.W.; Xia, W.; Huo, L.; Lim, S.O.; Wu, Y.; Hsu, J.L.; Chao, C.H.; Yamaguchi, H.; Yang, N.K.; Ding, Q.; et al. Epithelial-mesenchymal transition induced by TNF-α requires NF-κB-mediated transcriptional upregulation of Twist1. Cancer Res. 2012, 72, 1290–1300. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Taniguchi, K.; Karin, M. NF-κB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef] [PubMed]
- De Craene, B.; Berx, G. Regulatory networks defining EMT during cancer initiation and progression. Nat. Rev. Cancer 2013, 13, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Min, C.; Eddy, S.F.; Sherr, D.H.; Sonenshein, G.E. NF-kappaB and epithelial to mesenchymal transition of cancer. J. Cell. Biochem. 2008, 104, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Mao, R.; Yang, J. NF-κB and STAT3 signaling pathways collaboratively link inflammation to cancer. Protein Cell 2013, 4, 176–185. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Carro, M.S.; Lim, W.K.; Alvarez, M.J.; Bollo, R.J.; Zhao, X.; Snyder, E.Y.; Sulman, E.P.; Anne, S.L.; Doetsch, F.; Colman, H.; et al. The transcriptional network for mesenchymal transformation of brain tumours. Nature 2010, 463, 318–325. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bhat, K.P.; Salazar, K.L.; Balasubramaniyan, V.; Wani, K.; Heathcock, L.; Hollingsworth, F.; James, J.D.; Gumin, J.; Diefes, K.L.; Kim, S.H.; et al. The transcriptional coactivator TAZ regulates mesenchymal differentiation in malignant glioma. Genes Dev. 2011, 25, 2594–2609. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yee, P.P.; Wei, Y.; Kim, S.Y.; Lu, T.; Chih, S.Y.; Lawson, C.; Tang, M.; Liu, Z.; Anderson, B.; Thamburaj, K.; et al. Neutrophil-induced ferroptosis promotes tumor necrosis in glioblastoma progression. Nat. Commun. 2020, 11, 5424. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Vigneswaran, K.; Boyd, N.H.; Oh, S.Y.; Lallani, S.; Boucher, A.; Neill, S.G.; Olson, J.J.; Read, R.D. YAP/TAZ Transcriptional Coactivators Create Therapeutic Vulnerability to Verteporfin in EGFR-mutant Glioblastoma. Clin. Cancer Res. 2021, 27, 1553–1569. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yamini, B. NF-κB, Mesenchymal Differentiation and Glioblastoma. Cells 2018, 7, 125. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, D.; Berglund, A.E.; Kenchappa, R.S.; MacAulay, R.J.; Mule, J.J.; Etame, A.B. BIRC3 is a biomarker of mesenchymal habitat of glioblastoma, and a mediator of survival adaptation in hypoxia-driven glioblastoma habitats. Sci. Rep. 2017, 7, 9350. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, D.; Berglund, A.; Kenchappa, R.S.; Forsyth, P.A.; Mule, J.J.; Etame, A.B. BIRC3 is a novel driver of therapeutic resistance in Glioblastoma. Sci. Rep. 2016, 6, 21710. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Oh, Y.T.; Kim, J.Y.; Kim, S.S.; Choi, E.; Kim, T.H.; Hong, J.H.; Chang, N.; Cho, H.J.; Sa, J.K.; et al. Transglutaminase 2 Inhibition Reverses Mesenchymal Transdifferentiation of Glioma Stem Cells by Regulating C/EBPβ Signaling. Cancer Res. 2017, 77, 4973–4984. [Google Scholar] [CrossRef] [PubMed]
- Chow, K.H.; Park, H.J.; George, J.; Yamamoto, K.; Gallup, A.D.; Graber, J.H.; Chen, Y.; Jiang, W.; Steindler, D.A.; Neilson, E.G.; et al. S100A4 Is a Biomarker and Regulator of Glioma Stem Cells That Is Critical for Mesenchymal Transition in Glioblastoma. Cancer Res. 2017, 77, 5360–5373. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jiang, Y.; Zhou, J.; Hou, D.; Luo, P.; Gao, H.; Ma, Y.; Chen, Y.S.; Li, L.; Zou, D.; Zhang, H.; et al. Prosaposin is a biomarker of mesenchymal glioblastoma and regulates mesenchymal transition through the TGF-β1/Smad signaling pathway. J. Pathol. 2019, 249, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Brodie, S.; Lee, H.K.; Jiang, W.; Cazacu, S.; Xiang, C.; Poisson, L.M.; Datta, I.; Kalkanis, S.; Ginsberg, D.; Brodie, C. The novel long non-coding RNA TALNEC2, regulates tumor cell growth and the stemness and radiation response of glioma stem cells. Oncotarget 2017, 8, 31785–31801. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Du, X.; Tu, Y.; Liu, S.; Zhao, P.; Bao, Z.; Li, C.; Li, J.; Pan, M.; Ji, J. LINC00511 contributes to glioblastoma tumorigenesis and epithelial-mesenchymal transition via LINC00511/miR-524-5p/YB1/ZEB1 positive feedback loop. J. Cell. Mol. Med. 2020, 24, 1474–1487. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Li, C.; Zheng, H.; Hou, W.; Bao, H.; Xiong, J.; Che, W.; Gu, Y.; Sun, H.; Liang, P. Long non-coding RNA linc00645 promotes TGF-β-induced epithelial-mesenchymal transition by regulating miR-205-3p-ZEB1 axis in glioma. Cell Death Dis. 2019, 10, 717. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Fu, Q.; Li, S.; Zhou, Q.; Yalikun, K.; Yisireyili, D.; Xia, M. Low LINC00599 expression is a poor prognostic factor in glioma. Biosci. Rep. 2019, 39, BSR20190232. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- He, X.; Liu, Z.; Peng, Y.; Yu, C. MicroRNA-181c inhibits glioblastoma cell invasion, migration and mesenchymal transition by targeting TGF-β pathway. Biochem. Biophys. Res. Commun. 2016, 469, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zeng, A.; Liu, S.; Li, R.; Wang, X.; Yan, W.; Li, H.; You, Y. Genome-wide identification of epithelial-mesenchymal transition-associated microRNAs reveals novel targets for glioblastoma therapy. Oncol. Lett. 2018, 15, 7625–7630. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Huang, B.S.; Luo, Q.Z.; Han, Y.; Huang, D.; Tang, Q.P.; Wu, L.X. MiR-223/PAX6 Axis Regulates Glioblastoma Stem Cell Proliferation and the Chemo Resistance to TMZ via Regulating PI3K/Akt Pathway. J. Cell. Biochem. 2017, 118, 3452–3461. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Yao, J.; Zhang, Z.; Zhang, Y.; Zhang, Z.; Liu, J.; Tan, W.; Sun, C.; Chen, L.; Yu, X. miR-96 inhibits EMT by targeting AEG-1 in glioblastoma cancer cells. Mol. Med. Rep. 2018, 17, 2964–2972. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Li, L.; Shao, M.Y.; Zou, S.C.; Xiao, Z.F.; Chen, Z.C. MiR-101-3p inhibits EMT to attenuate proliferation and metastasis in glioblastoma by targeting TRIM44. J. Neurooncol 2019, 141, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Urdiciain, A.; Erausquin, E.; Meléndez, B.; Rey, J.A.; Idoate, M.A.; Castresana, J.S. Tubastatin A, an inhibitor of HDAC6, enhances temozolomide-induced apoptosis and reverses the malignant phenotype of glioblastoma cells. Int. J. Oncol. 2019, 54, 1797–1808. [Google Scholar] [CrossRef] [PubMed]
- Vinchure, O.S.; Sharma, V.; Tabasum, S.; Ghosh, S.; Singh, R.P.; Sarkar, C.; Kulshreshtha, R. Polycomb complex mediated epigenetic reprogramming alters TGF-β signaling via a novel EZH2/miR-490/TGIF2 axis thereby inducing migration and EMT potential in glioblastomas. Int. J. Cancer 2019, 145, 1254–1269. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Berglund, A.E.; Etame, A.B. The Impact of Epigenetic Modifications on Adaptive Resistance Evolution in Glioblastoma. Int. J. Mol. Sci. 2021, 22, 8324. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yu, T.; Wang, Y.; Hu, Q.; Wu, W.; Wu, Y.; Wei, W.; Han, D.; You, Y.; Lin, N.; Liu, N. The EZH2 inhibitor GSK343 suppresses cancer stem-like phenotypes and reverses mesenchymal transition in glioma cells. Oncotarget 2017, 8, 98348–98359. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liu, Z.; Wang, Z.; Chen, D.; Liu, X.; Yu, G.; Zhang, Y.; Chen, C.; Xu, R.; Wang, Y.; Liu, R.E. Paeoniflorin Inhibits EMT and Angiogenesis in Human Glioblastoma via K63-Linked C-Met Polyubiquitination-Dependent Autophagic Degradation. Front. Oncol. 2022, 12, 785345. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Burghardt, I.; Tritschler, F.; Opitz, C.A.; Frank, B.; Weller, M.; Wick, W. Pirfenidone inhibits TGF-beta expression in malignant glioma cells. Biochem. Biophys. Res. Commun. 2007, 354, 542–547. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Jin, X.; Sohn, Y.W.; Jin, X.; Jeon, H.Y.; Kim, E.J.; Ham, S.W.; Jeon, H.M.; Chang, S.Y.; Oh, S.Y.; et al. Tumoral RANKL activates astrocytes that promote glioma cell invasion through cytokine signaling. Cancer Lett. 2014, 353, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Zhou, L.; Han, N.; Zhang, M.; Lyu, X. Wnt/β-catenin pathway involvement in ionizing radiation-induced invasion of U87 glioblastoma cells. Strahlenther. Onkol. 2015, 191, 672–680. [Google Scholar] [CrossRef] [PubMed]
- Yi, G.Z.; Liu, Y.W.; Xiang, W.; Wang, H.; Chen, Z.Y.; Xie, S.D.; Qi, S.T. Akt and β-catenin contribute to TMZ resistance and EMT of MGMT negative malignant glioma cell line. J. Neurol. Sci. 2016, 367, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Chen, Y.; Li, Y.; Lyu, X.; Cui, J.; Cheng, Y.; Zhao, L.; Zhao, G. Metformin inhibits TGF-β1-induced epithelial-to-mesenchymal transition-like process and stem-like properties in GBM via AKT/mTOR/ZEB1 pathway. Oncotarget 2018, 9, 7023–7035. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yuan, X.; Wei, W.; Bao, Q.; Chen, H.; Jin, P.; Jiang, W. Metformin inhibits glioma cells stemness and epithelial-mesenchymal transition via regulating YAP activity. Biomed. Pharmacother. 2018, 102, 263–270. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Tumor Region | Leading Edge and Infiltrating Tumor | Perinecrotic Zone | Pseudopalisading around Necrosis | Hyperplastic Blood Vessels in Cellular Tumor | Microvascular Proliferation |
|---|---|---|---|---|---|
| Top 10 Gene Expression Profile | VSNL1 | PI3 | IL8 | COL3A1 | ESM1 |
| CCK | IL8 | VEGFA | LOC100506027 | COL3A1 | |
| SNAP25 | CCL20 | HILPDA | LUM | IBSP | |
| GABRA1 | SLPI | NDRG1 | COL1A1 | CRIP1 | |
| CRYM | SAA1 | ADM | ACTG2 | LOC100506027 | |
| GNG3 | PTX3 | CA9 | ESM1 | HIGD1B | |
| SYT1 | SAA2 | CA12 | ACTA2 | RGS5 | |
| NEFL | TREM1 | ANGPTL4 | COL6A3 | ITGA1 | |
| SYNPR | CHI3L1 | HK2 | COL1A2 | OR51E1 | |
| GABRA2 | MMP7 | CHI3L1 | DCN | MMP9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Q.; Berglund, A.E.; Macaulay, R.J.; Etame, A.B. The Role of Mesenchymal Reprogramming in Malignant Clonal Evolution and Intra-Tumoral Heterogeneity in Glioblastoma. Cells 2024, 13, 942. https://doi.org/10.3390/cells13110942
Wu Q, Berglund AE, Macaulay RJ, Etame AB. The Role of Mesenchymal Reprogramming in Malignant Clonal Evolution and Intra-Tumoral Heterogeneity in Glioblastoma. Cells. 2024; 13(11):942. https://doi.org/10.3390/cells13110942
Chicago/Turabian StyleWu, Qiong, Anders E. Berglund, Robert J. Macaulay, and Arnold B. Etame. 2024. "The Role of Mesenchymal Reprogramming in Malignant Clonal Evolution and Intra-Tumoral Heterogeneity in Glioblastoma" Cells 13, no. 11: 942. https://doi.org/10.3390/cells13110942