
Tigilanol Tiglate-Induced Changes in Secretome Profiles Alter C-Met Phosphorylation and Cell Surface Protein Expression in H357 Head and Neck Cancer Cells
 , , , , and
, , , , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials
3. Methods
3.1. Tissue Culture
3.2. Western Blotting and Quantification of Proteins in Cell Lysates
3.3. Flow Cytometry
3.4. ELISA for Quantifying MET-ECD and Syndecan-1 ECD
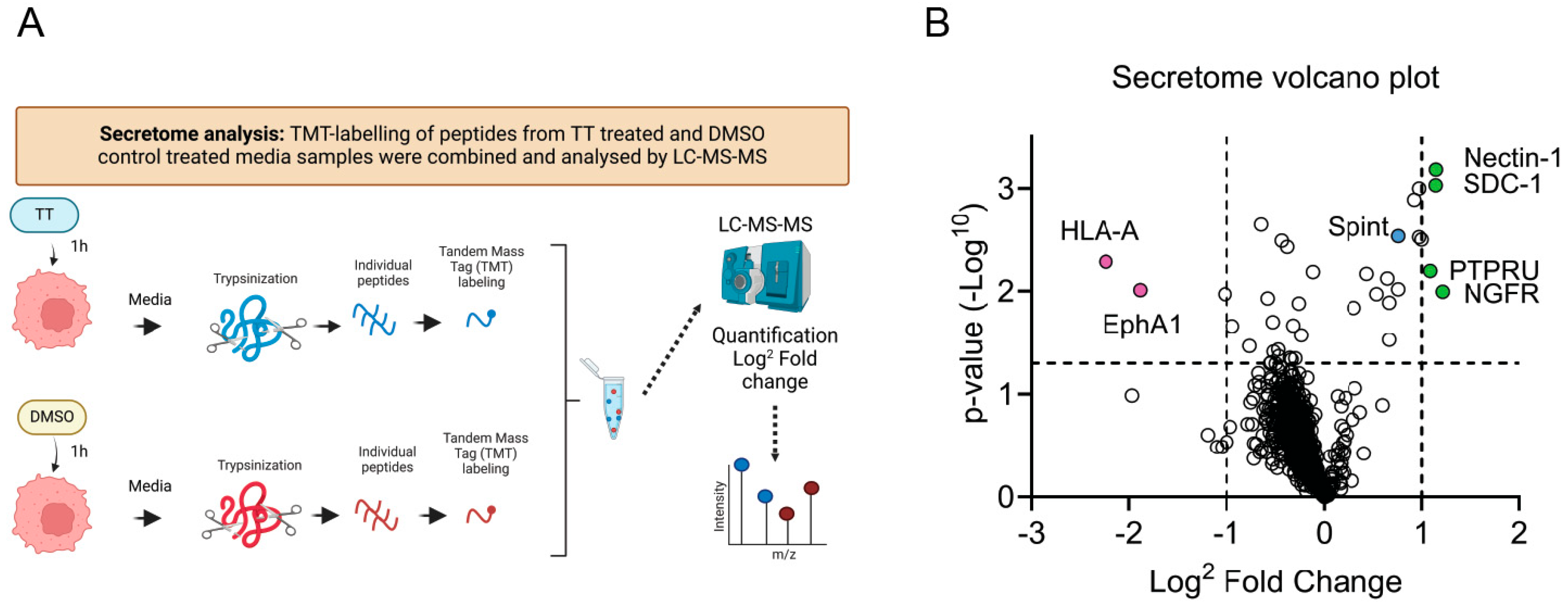
3.5. Secretome Proteomic Analysis
TMT Labelling and High pH Reversed-Phase Chromatography
3.6. Nano-LC Mass Spectrometry
3.7. Proteomic Data Analysis
3.8. Protein Annotation
3.9. Statistical Analysis
3.10. Volcano Plots
4. Results
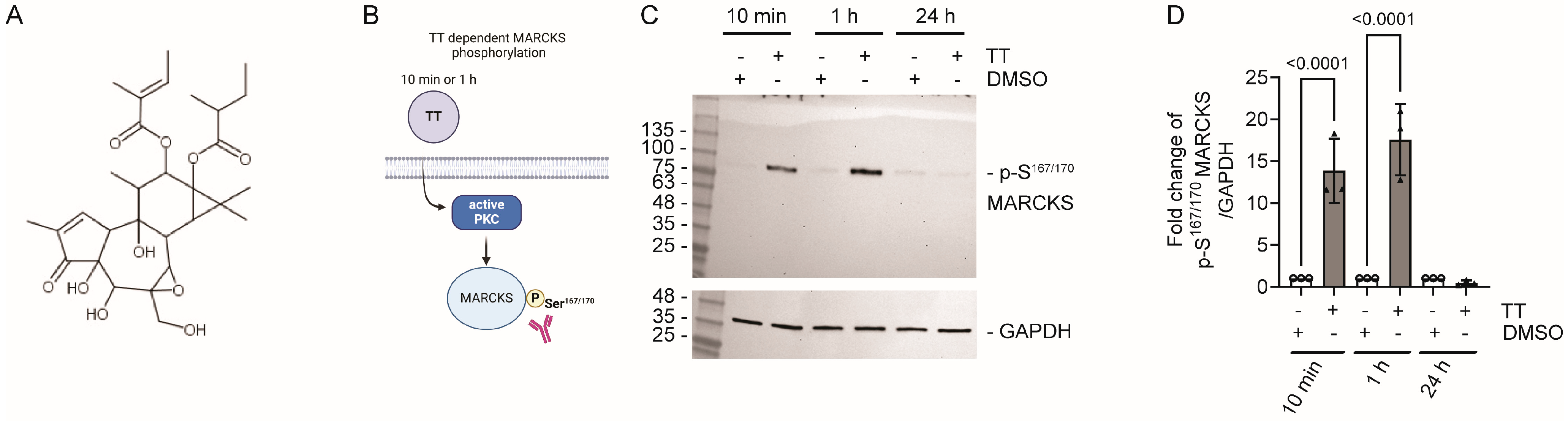
4.1. TT Treatment Leads to MARCKS Phosphorylation on S167/170
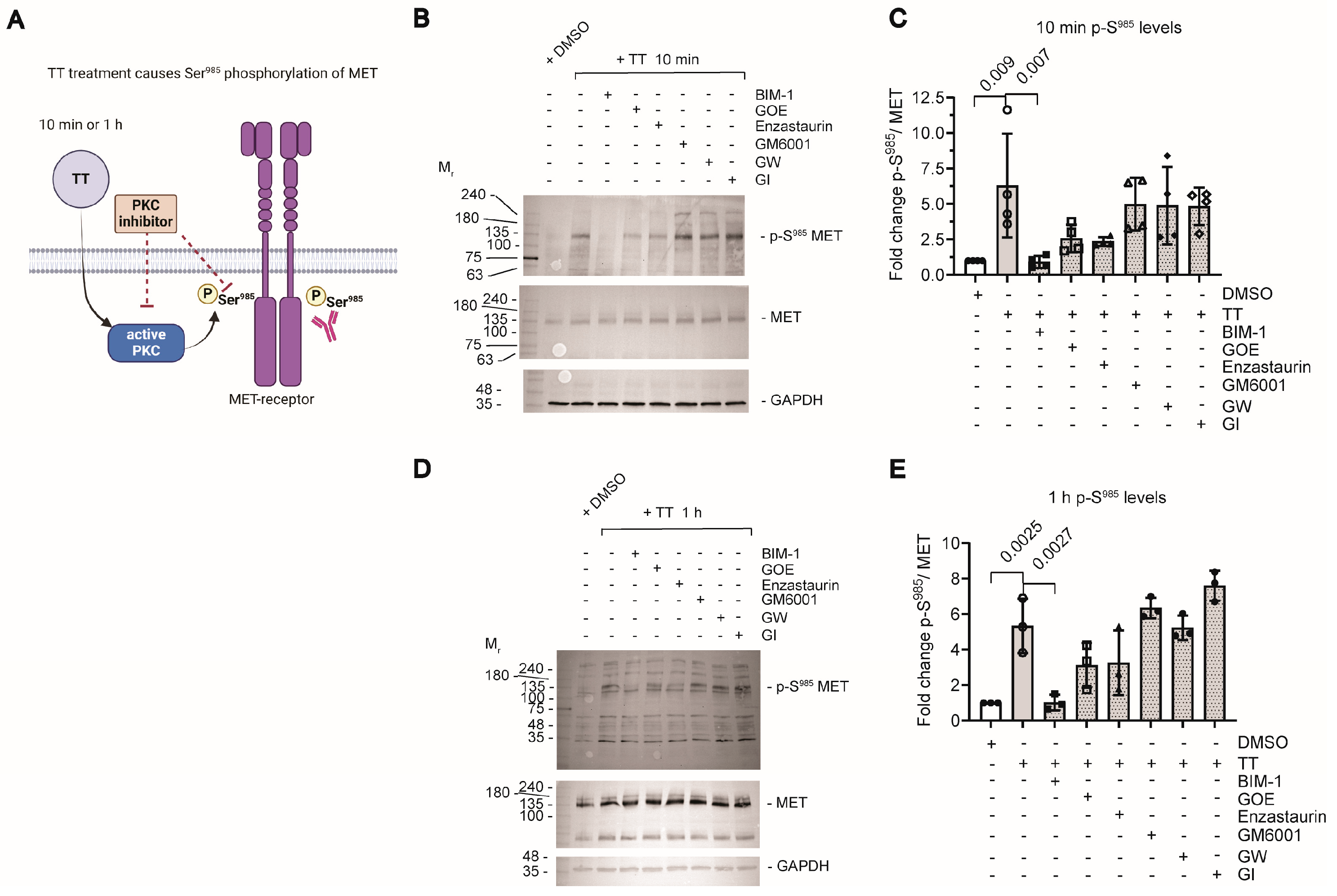
4.2. TT Treatment Induces Ser985 Phosphorylation of the MET Receptor
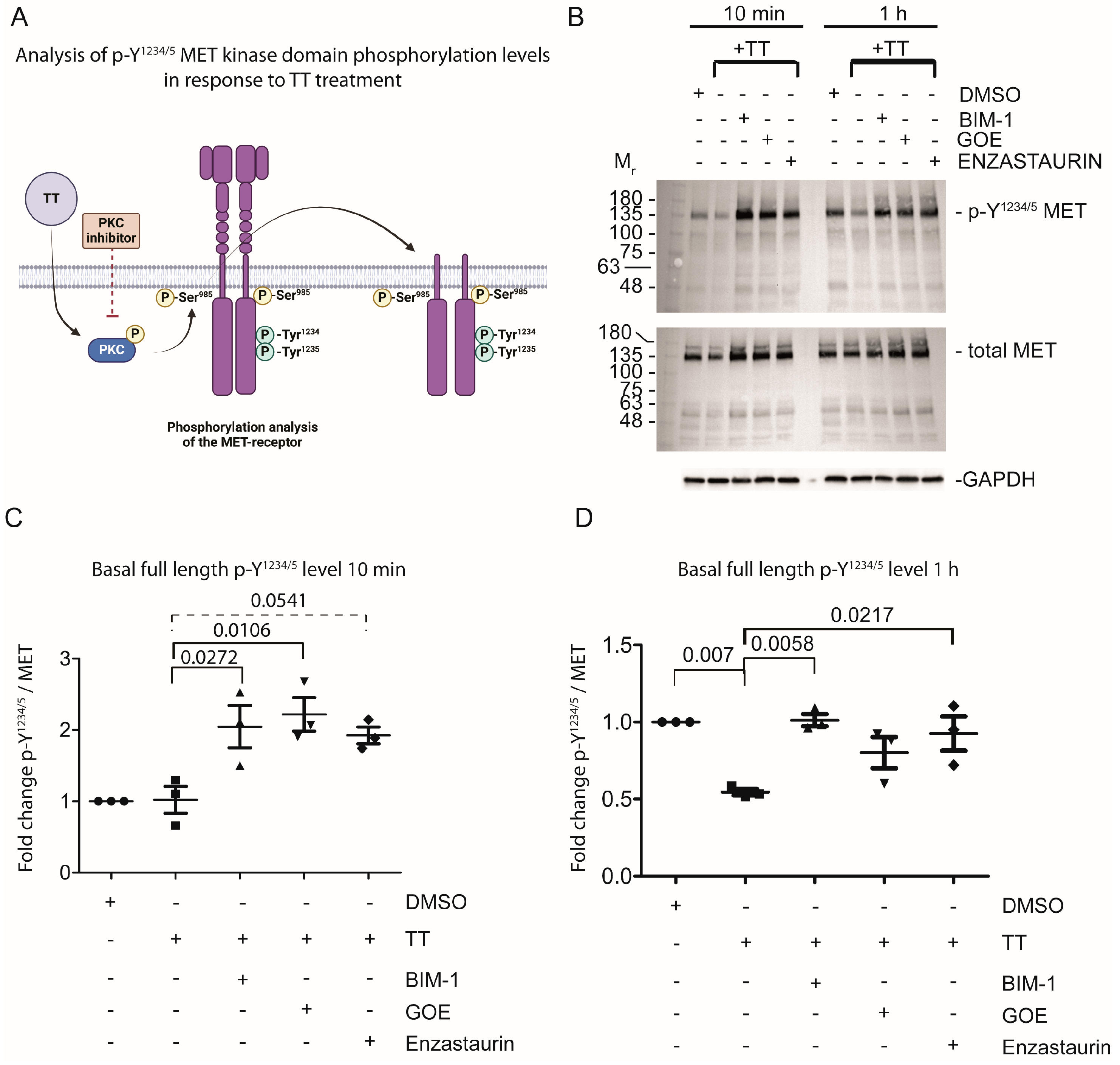
4.3. Prolonged TT Treatment Reduces Basal Y1234/5 Phosphorylation in the Kinase Domain of the MET Receptor in H357 Cells
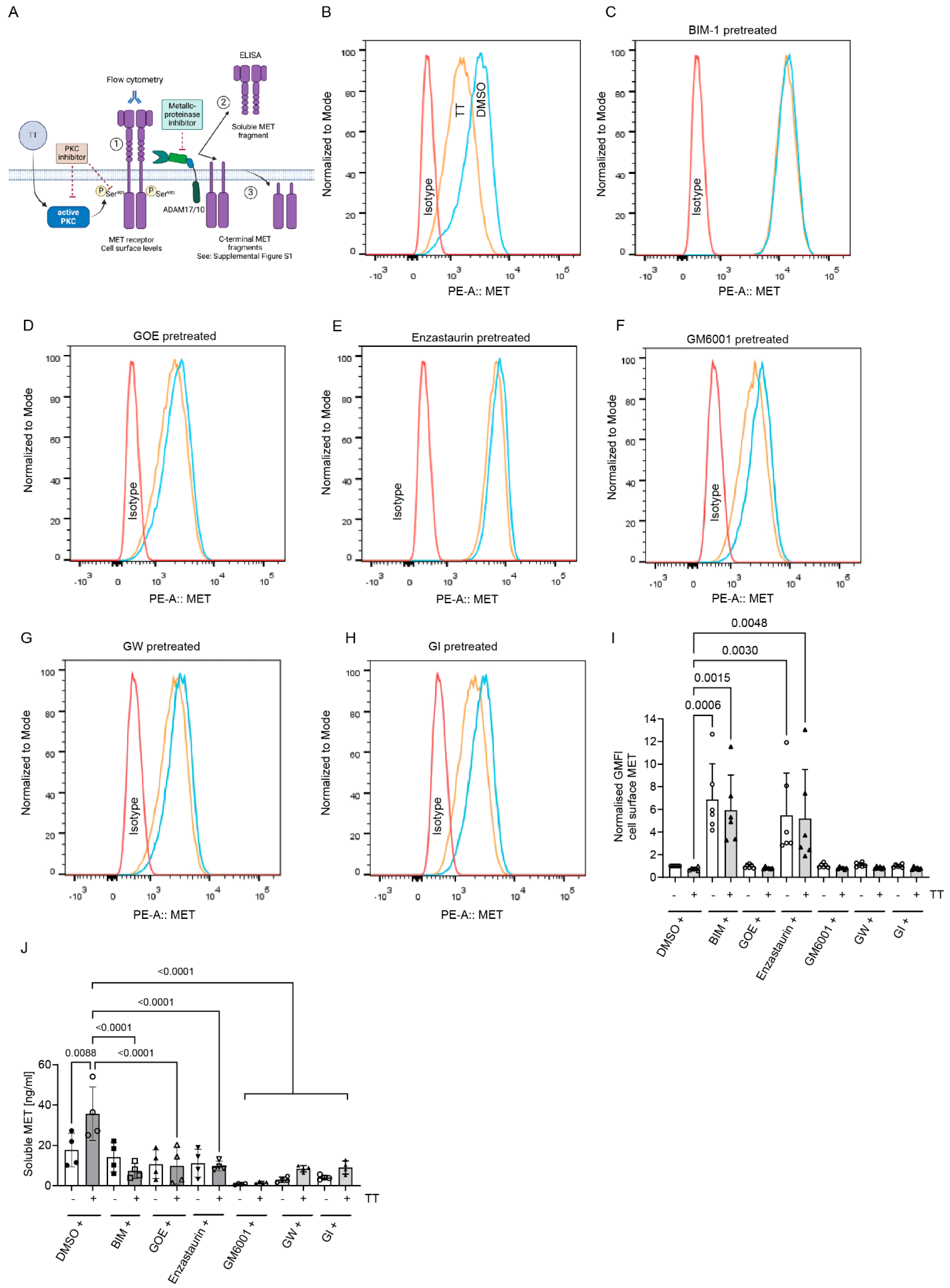
4.4. Does TT Regulate MET Cell Surface Levels and Cleavage?
4.5. Secretome Analysis of H357 Cell Treated with TT
5. Discussion
5.1. Early Secretome Targets of TT Treatment Identified by TMT Proteomics
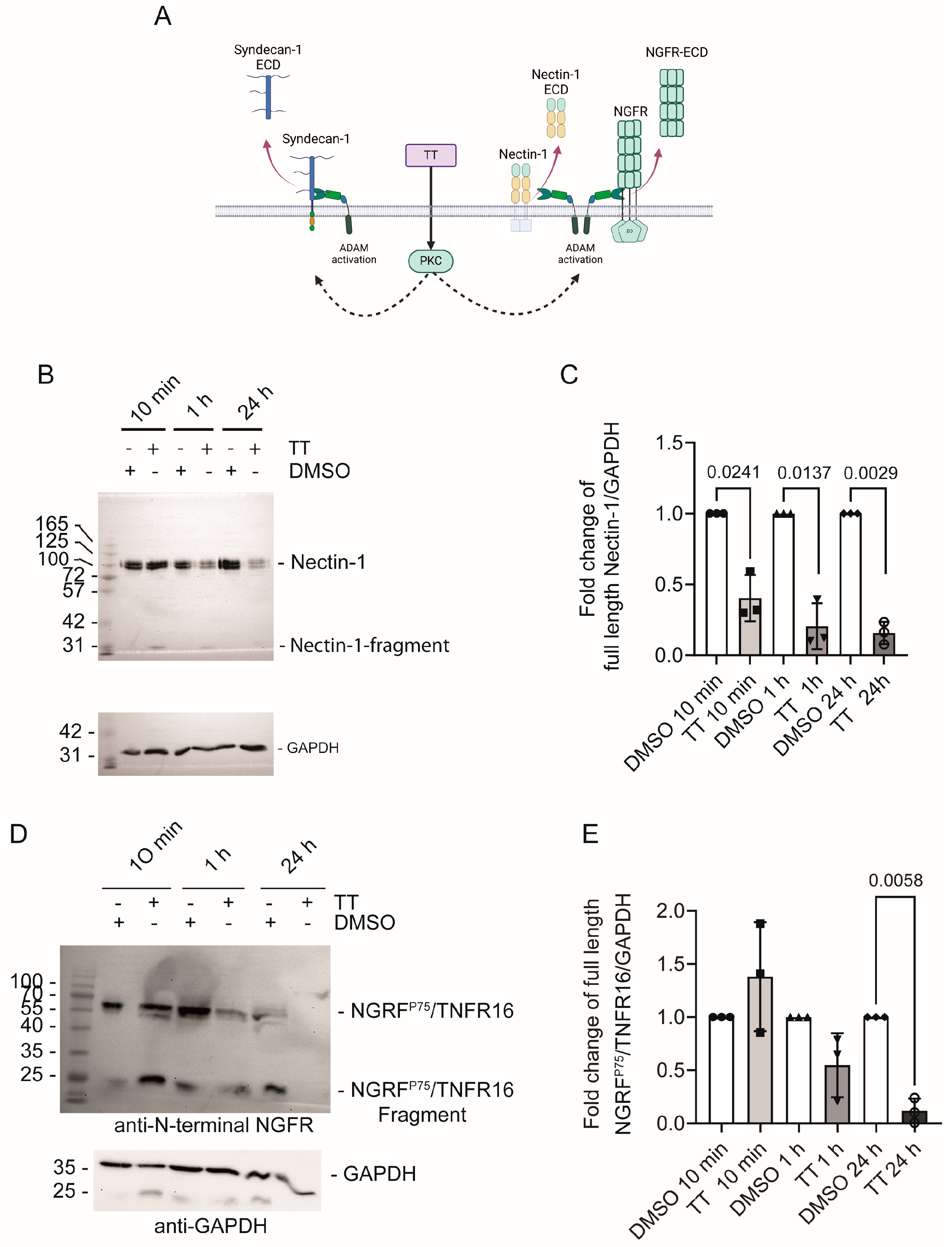
5.1.1. Nectin-1
5.1.2. NGFRp75/TNFR16
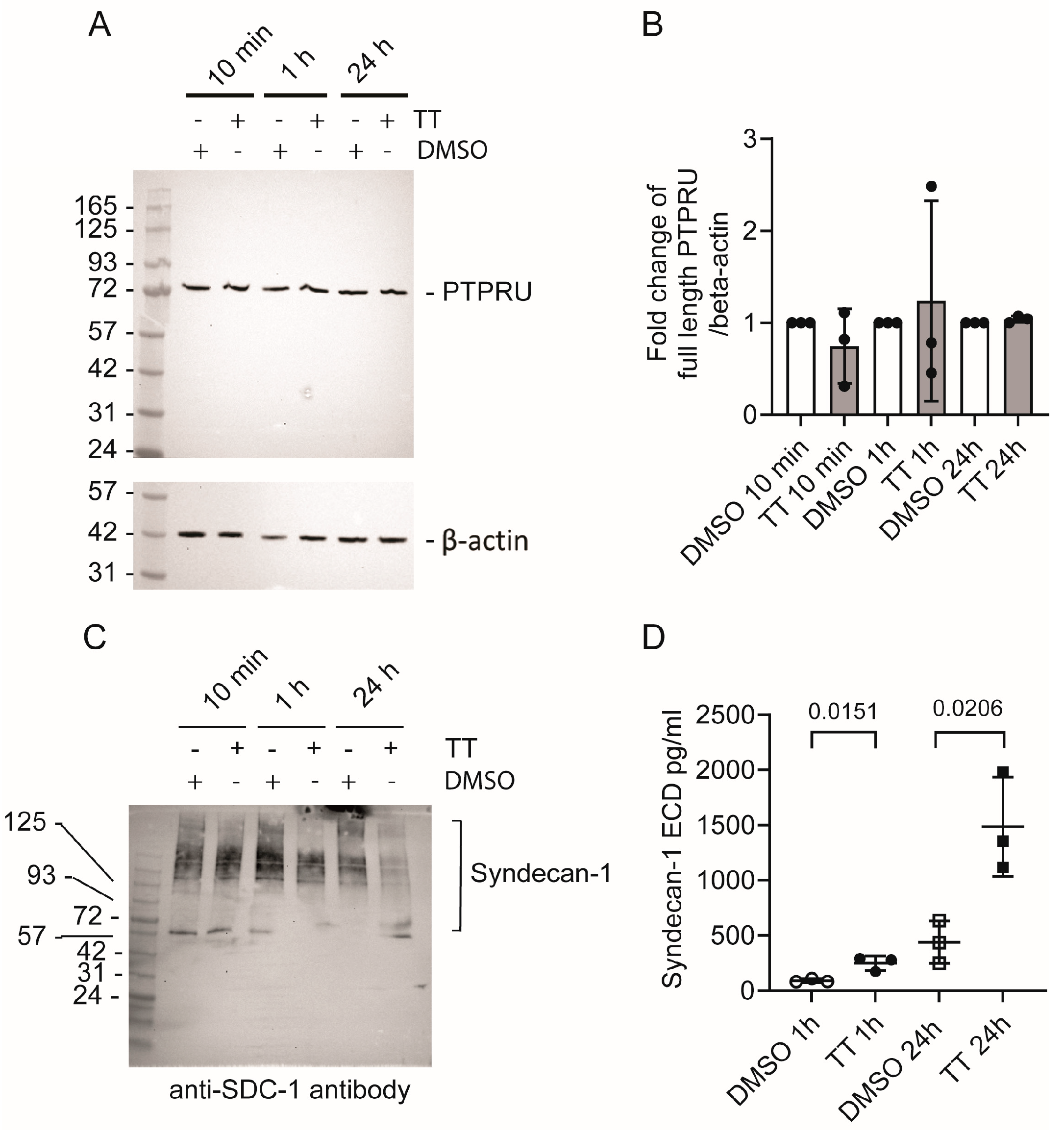
5.1.3. PTPRU
5.1.4. Syndecan-1
5.2. Potential Long-Term Implications on Head and Neck Cancer Treatment Strategies
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Boyle, G.M.; D’Souza, M.M.; Pierce, C.J.; Adams, R.A.; Cantor, A.S.; Johns, J.P.; Maslovskaya, L.; Gordon, V.A.; Reddell, P.W.; Parsons, P.G. Intra-lesional injection of the novel PKC activator EBC-46 rapidly ablates tumors in mouse models. PLoS ONE 2014, 9, e108887. [Google Scholar] [CrossRef] [PubMed]
- Cullen, J.K.; Boyle, G.M.; Yap, P.Y.; Elmlinger, S.; Simmons, J.L.; Broit, N.; Johns, J.; Ferguson, B.; Maslovskaya, L.A.; Savchenko, A.I.; et al. Activation of PKC supports the anticancer activity of tigilanol tiglate and related epoxytiglianes. Sci. Rep. 2021, 11, 207. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.A.; Oudin, M.J.; Sullivan, R.J.; Wang, S.J.; Meyer, A.S.; Im, H.; Frederick, D.T.; Tadros, J.; Griffith, L.G.; Lee, H.; et al. Reduced Proteolytic Shedding of Receptor Tyrosine Kinases Is a Post-Translational Mechanism of Kinase Inhibitor Resistance. Cancer Discov. 2016, 6, 382–399. [Google Scholar] [CrossRef] [PubMed]
- De Ridder, T.R.; Campbell, J.E.; Burke-Schwarz, C.; Clegg, D.; Elliot, E.L.; Geller, S.; Kozak, W.; Pittenger, S.T.; Pruitt, J.B.; Riehl, J.; et al. Randomized controlled clinical study evaluating the efficacy and safety of intratumoral treatment of canine mast cell tumors with tigilanol tiglate (EBC-46). J. Vet. Intern. Med. 2021, 35, 415–429. [Google Scholar] [CrossRef] [PubMed]
- Reddell, P.; De Ridder, T.R.; Morton, J.M.; Jones, P.D.; Campbell, J.E.; Brown, G.; Johannes, C.M.; Schmidt, P.F.; Gordon, V. Wound formation, wound size, and progression of wound healing after intratumoral treatment of mast cell tumors in dogs with tigilanol tiglate. J. Vet. Intern. Med. 2021, 35, 430–441. [Google Scholar] [CrossRef] [PubMed]
- Cullen, J.K.; Yap, P.Y.; Ferguson, B.; Bruce, Z.C.; Koyama, M.; Handoko, H.; Hendrawan, K.; Simmons, J.L.; Brooks, K.M.; Johns, J.; et al. Tigilanol tiglate is an oncolytic small molecule that induces immunogenic cell death and enhances the response of both target and non-injected tumors to immune checkpoint blockade. J. Immunother. Cancer 2024, 12, e006602. [Google Scholar] [CrossRef] [PubMed]
- Moses, R.L.; Boyle, G.M.; Howard-Jones, R.A.; Errington, R.J.; Johns, J.P.; Gordon, V.; Reddell, P.; Steadman, R.; Moseley, R. Novel epoxy-tiglianes stimulate skin keratinocyte wound healing responses and re-epithelialization via protein kinase C activation. Biochem. Pharmacol. 2020, 178, 114048. [Google Scholar] [CrossRef]
- Jones, P.D.; Campbell, J.E.; Brown, G.; Johannes, C.M.; Reddell, P. Recurrence-free interval 12 months after local treatment of mast cell tumors in dogs using intratumoral injection of tigilanol tiglate. J. Vet. Intern. Med. 2021, 35, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Panizza, B.J.; de Souza, P.; Cooper, A.; Roohullah, A.; Karapetis, C.S.; Lickliter, J.D. Phase I dose-escalation study to determine the safety, tolerability, preliminary efficacy and pharmacokinetics of an intratumoral injection of tigilanol tiglate (EBC-46). EBioMedicine 2019, 50, 433–441. [Google Scholar] [CrossRef]
- Newton, A.C. Protein kinase C as a tumor suppressor. Semin. Cancer Biol. 2018, 48, 18–26. [Google Scholar] [CrossRef]
- Wanger, T.M.; Dewitt, S.; Collins, A.; Maitland, N.J.; Poghosyan, Z.; Knäuper, V. Differential regulation of TROP2 release by PKC isoforms through vesicles and ADAM17. Cell Signal. 2015, 27, 1325–1335. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Irie, K.; Hirota, T.; Sakisaka, T.; Nakanishi, H.; Takai, Y. Ectodomain shedding of nectin-1alpha by SF/HGF and TPA in MDCK cells. Biochem. Biophys. Res. Commun. 2002, 299, 472–478. [Google Scholar] [CrossRef] [PubMed]
- Schelter, F.; Kobuch, J.; Moss, M.L.; Becherer, J.D.; Comoglio, P.M.; Boccaccio, C.; Kruger, A. A disintegrin and metalloproteinase-10 (ADAM-10) mediates DN30 antibody-induced shedding of the met surface receptor. J. Biol. Chem. 2010, 285, 26335–26340. [Google Scholar] [CrossRef] [PubMed]
- Vicario, A.; Kisiswa, L.; Tann, J.Y.; Kelly, C.E.; Ibanez, C.F. Neuron-type-specific signaling by the p75NTR death receptor is regulated by differential proteolytic cleavage. J. Cell Sci. 2015, 128, 1507–1517. [Google Scholar] [CrossRef] [PubMed]
- Herrlich, P.; Herrlich, A. ADAM Metalloprotease-Released Cancer Biomarkers. Trends Cancer 2017, 3, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Zunke, F.; Rose-John, S. The shedding protease ADAM17: Physiology and pathophysiology. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 2059–2070. [Google Scholar] [CrossRef] [PubMed]
- Gandino, L.; Longati, P.; Medico, E.; Prat, M.; Comoglio, P.M. Phosphorylation of serine 985 negatively regulates the hepatocyte growth factor receptor kinase. J. Biol. Chem. 1994, 269, 1815–1820. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Luo, J.; Chang, J.; Rekhtman, N.; Arcila, M.; Drilon, A. MET-dependent solid tumours-molecular diagnosis and targeted therapy. Nat. Rev. Clin. Oncol. 2020, 17, 569–587. [Google Scholar] [CrossRef] [PubMed]
- Shattuck, D.L.; Miller, J.K.; Carraway, K.L., 3rd; Sweeney, C. Met receptor contributes to trastuzumab resistance of Her2-overexpressing breast cancer cells. Cancer Res. 2008, 68, 1471–1477. [Google Scholar] [CrossRef]
- Ancot, F.; Leroy, C.; Muharram, G.; Lefebvre, J.; Vicogne, J.; Lemiere, A.; Kherrouche, Z.; Foveau, B.; Pourtier, A.; Melnyk, O.; et al. Shedding-generated Met receptor fragments can be routed to either the proteasomal or the lysosomal degradation pathway. Traffic 2012, 13, 1261–1272. [Google Scholar] [CrossRef]
- Pascovici, D.; Handler, D.C.; Wu, J.X.; Haynes, P.A. Multiple testing corrections in quantitative proteomics: A useful but blunt tool. Proteomics 2016, 16, 2448–2453. [Google Scholar] [CrossRef] [PubMed]
- Allen, L.A.; Aderem, A. Protein kinase C regulates MARCKS cycling between the plasma membrane and lysosomes in fibroblasts. EMBO J. 1995, 14, 1109–1121. [Google Scholar] [CrossRef] [PubMed]
- Parra, L.M.; Hartmann, M.; Schubach, S.; Li, Y.; Herrlich, P.; Herrlich, A. Distinct Intracellular Domain Substrate Modifications Selectively Regulate Ectodomain Cleavage of NRG1 or CD44. Mol. Cell Biol. 2015, 35, 3381–3395. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xia, M.; Jin, K.; Wang, S.; Wei, H.; Fan, C.; Wu, Y.; Li, X.; Li, X.; Li, G.; et al. Function of the c-Met receptor tyrosine kinase in carcinogenesis and associated therapeutic opportunities. Mol. Cancer 2018, 17, 45. [Google Scholar] [CrossRef] [PubMed]
- Tulasne, D.; Deheuninck, J.; Lourenco, F.C.; Lamballe, F.; Ji, Z.; Leroy, C.; Puchois, E.; Moumen, A.; Maina, F.; Mehlen, P.; et al. Proapoptotic function of the MET tyrosine kinase receptor through caspase cleavage. Mol. Cell Biol. 2004, 24, 10328–10339. [Google Scholar] [CrossRef] [PubMed]
- Nath, D.; Williamson, N.J.; Jarvis, R.; Murphy, G. Shedding of c-Met is regulated by crosstalk between a G-protein coupled receptor and the EGF receptor and is mediated by a TIMP-3 sensitive metalloproteinase. J. Cell Sci. 2001, 114, 1213–1220. [Google Scholar] [CrossRef]
- Fernandes, M.; Duplaquet, L.; Tulasne, D. Proteolytic cleavages of MET: The divide-and-conquer strategy of a receptor tyrosine kinase. BMB Rep. 2019, 52, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Garton, K.J.; Gough, P.J.; Blobel, C.P.; Murphy, G.; Greaves, D.R.; Dempsey, P.J.; Raines, E.W. Tumor necrosis factor-alpha-converting enzyme (ADAM17) mediates the cleavage and shedding of fractalkine (CX3CL1). J. Biol. Chem. 2001, 276, 37993–38001. [Google Scholar] [CrossRef]
- Garton, K.J.; Gough, P.J.; Philalay, J.; Wille, P.T.; Blobel, C.P.; Whitehead, R.H.; Dempsey, P.J.; Raines, E.W. Stimulated shedding of vascular cell adhesion molecule 1 (VCAM-1) is mediated by tumor necrosis factor-alpha-converting enzyme (ADAM 17). J. Biol. Chem. 2003, 278, 37459–37464. [Google Scholar] [CrossRef]
- Zheng, Y.; Saftig, P.; Hartmann, D.; Blobel, C. Evaluation of the contribution of different ADAMs to tumor necrosis factor alpha (TNFalpha) shedding and of the function of the TNFalpha ectodomain in ensuring selective stimulated shedding by the TNFalpha convertase (TACE/ADAM17). J. Biol. Chem. 2004, 279, 42898–42906. [Google Scholar] [CrossRef]
- Dang, M.; Dubbin, K.; D’Aiello, A.; Hartmann, M.; Lodish, H.; Herrlich, A. Epidermal growth factor (EGF) ligand release by substrate-specific a disintegrin and metalloproteases (ADAMs) involves different protein kinase C (PKC) isoenzymes depending on the stimulus. J. Biol. Chem. 2011, 286, 17704–17713. [Google Scholar] [CrossRef]
- Kim, J.; Lilliehook, C.; Dudak, A.; Prox, J.; Saftig, P.; Federoff, H.J.; Lim, S.T. Activity-dependent alpha-cleavage of nectin-1 is mediated by a disintegrin and metalloprotease 10 (ADAM10). J. Biol. Chem. 2010, 285, 22919–22926. [Google Scholar] [CrossRef]
- Pasqualon, T.; Pruessmeyer, J.; Jankowski, V.; Babendreyer, A.; Groth, E.; Schumacher, J.; Koenen, A.; Weidenfeld, S.; Schwarz, N.; Denecke, B.; et al. A cytoplasmic C-terminal fragment of Syndecan-1 is generated by sequential proteolysis and antagonizes Syndecan-1 dependent lung tumor cell migration. Oncotarget 2015, 6, 31295–31312. [Google Scholar] [CrossRef] [PubMed]
- Derksen, P.W.; Keehnen, R.M.; Evers, L.M.; van Oers, M.H.; Spaargaren, M.; Pals, S.T. Cell surface proteoglycan syndecan-1 mediates hepatocyte growth factor binding and promotes Met signaling in multiple myeloma. Blood 2002, 99, 1405–1410. [Google Scholar] [CrossRef]
- Hay, I.M.; Fearnley, G.W.; Rios, P.; Kohn, M.; Sharpe, H.J.; Deane, J.E. The receptor PTPRU is a redox sensitive pseudophosphatase. Nat. Commun. 2020, 11, 3219. [Google Scholar] [CrossRef]
- Parkhurst, C.N.; Zampieri, N.; Chao, M.V. Nuclear localization of the p75 neurotrophin receptor intracellular domain. J. Biol. Chem. 2010, 285, 5361–5368. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, M.L.; Wang, Z.; Park, P.W.; Murphy, G.; Bernfield, M. Shedding of syndecan-1 and -4 ectodomains is regulated by multiple signaling pathways and mediated by a TIMP-3-sensitive metalloproteinase. J. Cell Biol. 2000, 148, 811–824. [Google Scholar] [CrossRef] [PubMed]
- Pruessmeyer, J.; Martin, C.; Hess, F.M.; Schwarz, N.; Schmidt, S.; Kogel, T.; Hoettecke, N.; Schmidt, B.; Sechi, A.; Uhlig, S.; et al. A disintegrin and metalloproteinase 17 (ADAM17) mediates inflammation-induced shedding of syndecan-1 and -4 by lung epithelial cells. J. Biol. Chem. 2010, 285, 555–564. [Google Scholar] [CrossRef]
- Cheng, J.; Wu, K.; Armanini, M.; O’Rourke, N.; Dowbenko, D.; Lasky, L.A. A novel protein-tyrosine phosphatase related to the homotypically adhering kappa and mu receptors. J. Biol. Chem. 1997, 272, 7264–7277. [Google Scholar] [CrossRef]
- Hashigasako, A.; Machide, M.; Nakamura, T.; Matsumoto, K.; Nakamura, T. Bi-directional regulation of Ser-985 phosphorylation of c-met via protein kinase C and protein phosphatase 2A involves c-Met activation and cellular responsiveness to hepatocyte growth factor. J. Biol. Chem. 2004, 279, 26445–26452. [Google Scholar] [CrossRef]
- Peschard, P.; Fournier, T.M.; Lamorte, L.; Naujokas, M.A.; Band, H.; Langdon, W.Y.; Park, M. Mutation of the c-Cbl TKB domain binding site on the Met receptor tyrosine kinase converts it into a transforming protein. Mol. Cell 2001, 8, 995–1004. [Google Scholar] [CrossRef]
- Duplaquet, L.; Leroy, C.; Vinchent, A.; Paget, S.; Lefebvre, J.; Vanden Abeele, F.; Lancel, S.; Giffard, F.; Paumelle, R.; Bidaux, G.; et al. Control of cell death/survival balance by the MET dependence receptor. Elife 2020, 9, e50041. [Google Scholar] [CrossRef] [PubMed]
- Antal, C.E.; Hudson, A.M.; Kang, E.; Zanca, C.; Wirth, C.; Stephenson, N.L.; Trotter, E.W.; Gallegos, L.L.; Miller, C.J.; Furnari, F.B.; et al. Cancer-associated protein kinase C mutations reveal kinase’s role as tumor suppressor. Cell 2015, 160, 489–502. [Google Scholar] [CrossRef] [PubMed]
- Dowling, C.M.; Phelan, J.; Callender, J.A.; Cathcart, M.C.; Mehigan, B.; McCormick, P.; Dalton, T.; Coffey, J.C.; Newton, A.C.; O’Sullivan, J.; et al. Protein kinase C beta II suppresses colorectal cancer by regulating IGF-1 mediated cell survival. Oncotarget 2016, 7, 20919–20933. [Google Scholar] [CrossRef] [PubMed]
- Bourhill, T.; Narendran, A.; Johnston, R.N. Enzastaurin: A lesson in drug development. Crit. Rev. Oncol. Hematol. 2017, 112, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Hagege, A.; Saada-Bouzid, E.; Ambrosetti, D.; Rastoin, O.; Boyer, J.; He, X.; Rousset, J.; Montemagno, C.; Doyen, J.; Pedeutour, F.; et al. Targeting of c-MET and AXL by cabozantinib is a potential therapeutic strategy for patients with head and neck cell carcinoma. Cell Rep. Med. 2022, 3, 100659. [Google Scholar] [CrossRef] [PubMed]
- Craig, S.E.; Brady-Kalnay, S.M. Regulation of development and cancer by the R2B subfamily of RPTPs and the implications of proteolysis. Semin. Cell Dev. Biol. 2015, 37, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Takai, Y.; Miyoshi, J.; Ikeda, W.; Ogita, H. Nectins and nectin-like molecules: Roles in contact inhibition of cell movement and proliferation. Nat. Rev. Mol. Cell Biol. 2008, 9, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Hermans, D.; Rodriguez-Mogeda, C.; Kemps, H.; Bronckaers, A.; de Vries, H.E.; Broux, B. Nectins and Nectin-like molecules drive vascular development and barrier function. Angiogenesis 2023, 26, 349–362. [Google Scholar] [CrossRef]
- Honda, T.; Shimizu, K.; Kawakatsu, T.; Yasumi, M.; Shingai, T.; Fukuhara, A.; Ozaki-Kuroda, K.; Irie, K.; Nakanishi, H.; Takai, Y. Antagonistic and agonistic effects of an extracellular fragment of nectin on formation of E-cadherin-based cell-cell adhesion. Genes Cells 2003, 8, 51–63. [Google Scholar] [CrossRef]
- Truzzi, F.; Marconi, A.; Lotti, R.; Dallaglio, K.; French, L.E.; Hempstead, B.L.; Pincelli, C. Neurotrophins and their receptors stimulate melanoma cell proliferation and migration. J. Investig. Dermatol. 2008, 128, 2031–2040. [Google Scholar] [CrossRef] [PubMed]
- Chakravarthy, R.; Mnich, K.; Gorman, A.M. Nerve growth factor (NGF)-mediated regulation of p75(NTR) expression contributes to chemotherapeutic resistance in triple negative breast cancer cells. Biochem. Biophys. Res. Commun. 2016, 478, 1541–1547. [Google Scholar] [CrossRef] [PubMed]
- Saltari, A.; Dzung, A.; Quadri, M.; Tiso, N.; Facchinello, N.; Hernandez-Barranco, A.; Garcia-Silva, S.; Nogues, L.; Stoffel, C.I.; Cheng, P.F.; et al. Specific Activation of the CD271 Intracellular Domain in Combination with Chemotherapy or Targeted Therapy Inhibits Melanoma Progression. Cancer Res. 2021, 81, 6044–6057. [Google Scholar] [CrossRef] [PubMed]
- Szatmari, T.; Dobra, K. The role of syndecan-1 in cellular signaling and its effects on heparan sulfate biosynthesis in mesenchymal tumors. Front. Oncol. 2013, 3, 310. [Google Scholar] [CrossRef] [PubMed]
- Manon-Jensen, T.; Multhaupt, H.A.; Couchman, J.R. Mapping of matrix metalloproteinase cleavage sites on syndecan-1 and syndecan-4 ectodomains. FEBS J. 2013, 280, 2320–2331. [Google Scholar] [CrossRef]
- Seidel, C.; Borset, M.; Hjertner, O.; Cao, D.; Abildgaard, N.; Hjorth-Hansen, H.; Sanderson, R.D.; Waage, A.; Sundan, A. High levels of soluble syndecan-1 in myeloma-derived bone marrow: Modulation of hepatocyte growth factor activity. Blood 2000, 96, 3139–3146. [Google Scholar] [CrossRef]
- Guo, S.; Wu, X.; Lei, T.; Zhong, R.; Wang, Y.; Zhang, L.; Zhao, Q.; Huang, Y.; Shi, Y.; Wu, L. The Role and Therapeutic Value of Syndecan-1 in Cancer Metastasis and Drug Resistance. Front. Cell Dev. Biol. 2021, 9, 784983. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Antwi, F.D.; Awad, T.; Larin, M.; Heesom, K.; Lewis, P.; Reddell, P.; Poghosyan, Z.; Dewitt, S.; Moseley, R.; Knäuper, V. Tigilanol Tiglate-Induced Changes in Secretome Profiles Alter C-Met Phosphorylation and Cell Surface Protein Expression in H357 Head and Neck Cancer Cells. Cells 2024, 13, 982. https://doi.org/10.3390/cells13110982
Antwi FD, Awad T, Larin M, Heesom K, Lewis P, Reddell P, Poghosyan Z, Dewitt S, Moseley R, Knäuper V. Tigilanol Tiglate-Induced Changes in Secretome Profiles Alter C-Met Phosphorylation and Cell Surface Protein Expression in H357 Head and Neck Cancer Cells. Cells. 2024; 13(11):982. https://doi.org/10.3390/cells13110982
Chicago/Turabian StyleAntwi, Frank Dickson, Tufaha Awad, Meghan Larin, Kate Heesom, Phil Lewis, Paul Reddell, Zaruhi Poghosyan, Sharon Dewitt, Ryan Moseley, and Vera Knäuper. 2024. "Tigilanol Tiglate-Induced Changes in Secretome Profiles Alter C-Met Phosphorylation and Cell Surface Protein Expression in H357 Head and Neck Cancer Cells" Cells 13, no. 11: 982. https://doi.org/10.3390/cells13110982