

Early Inhibition of Phosphodiesterase 4B (PDE4B) Instills Cognitive Resilience in APPswe/PS1dE9 Mice
 , , , , , , , , , and
, , , , , , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Primary Microglia Isolation
2.3. Synaptosome Isolation and Labeling
2.4. Phagocytosis Assay
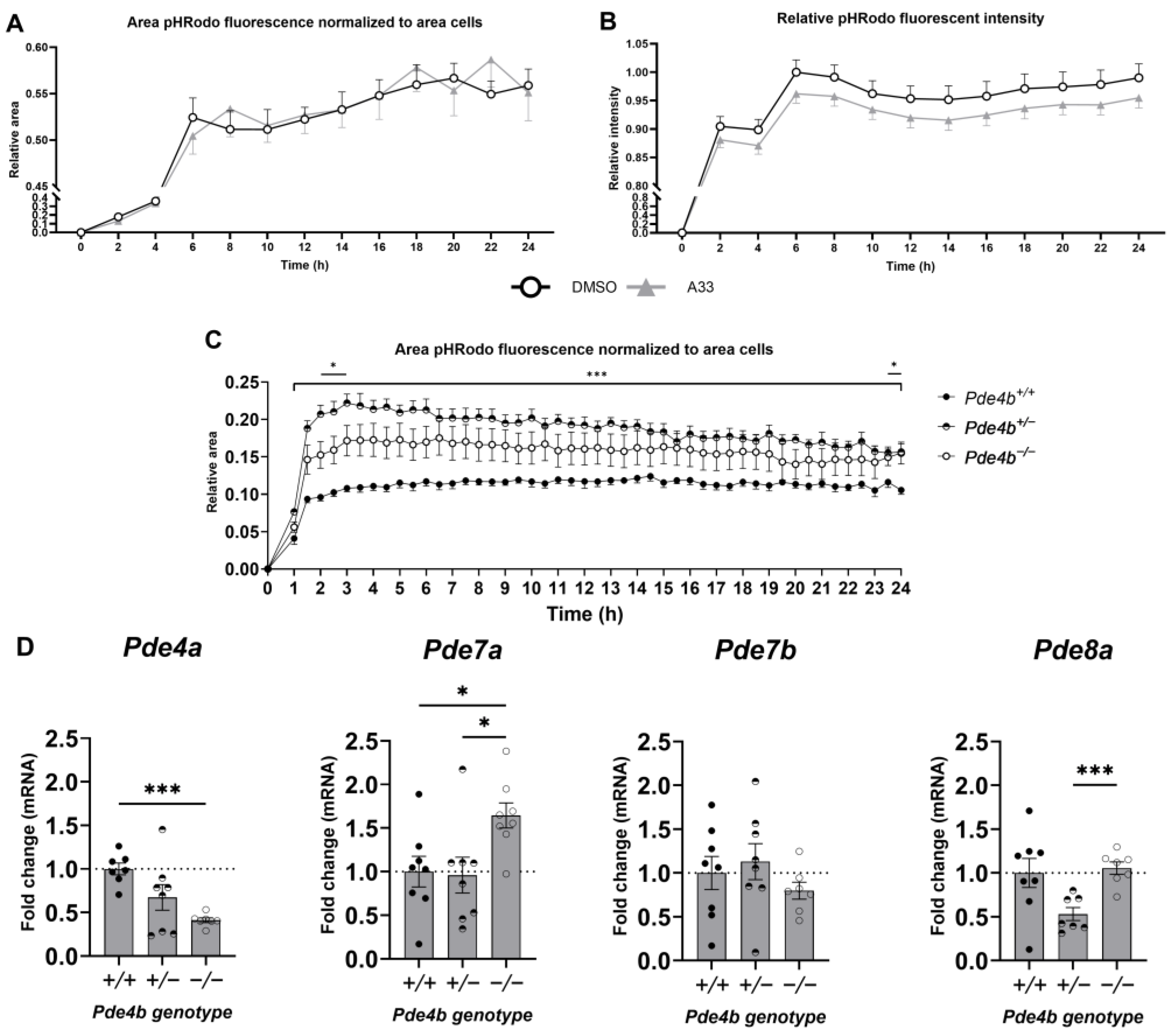
2.5. qPCR
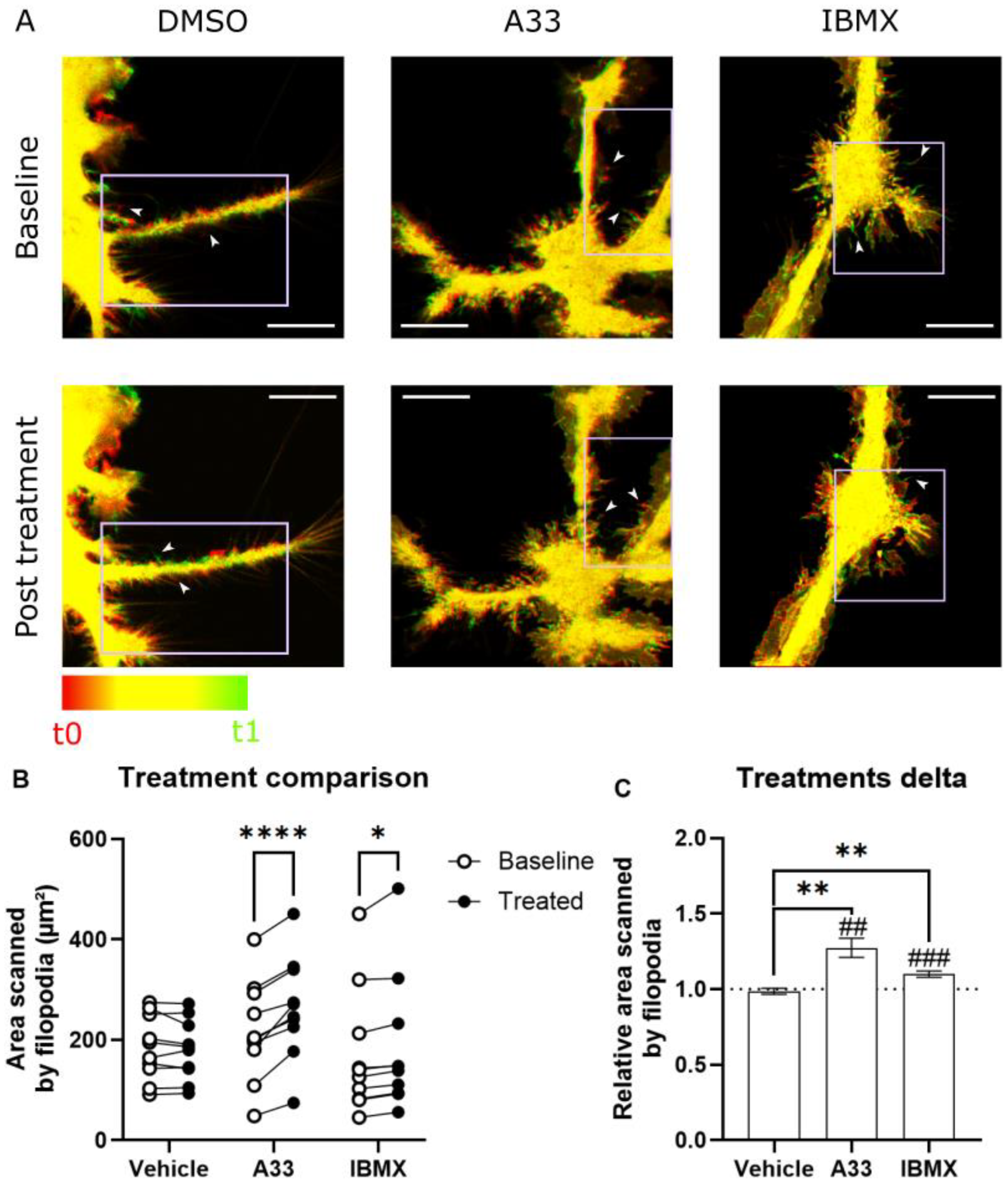
2.6. Surveillance
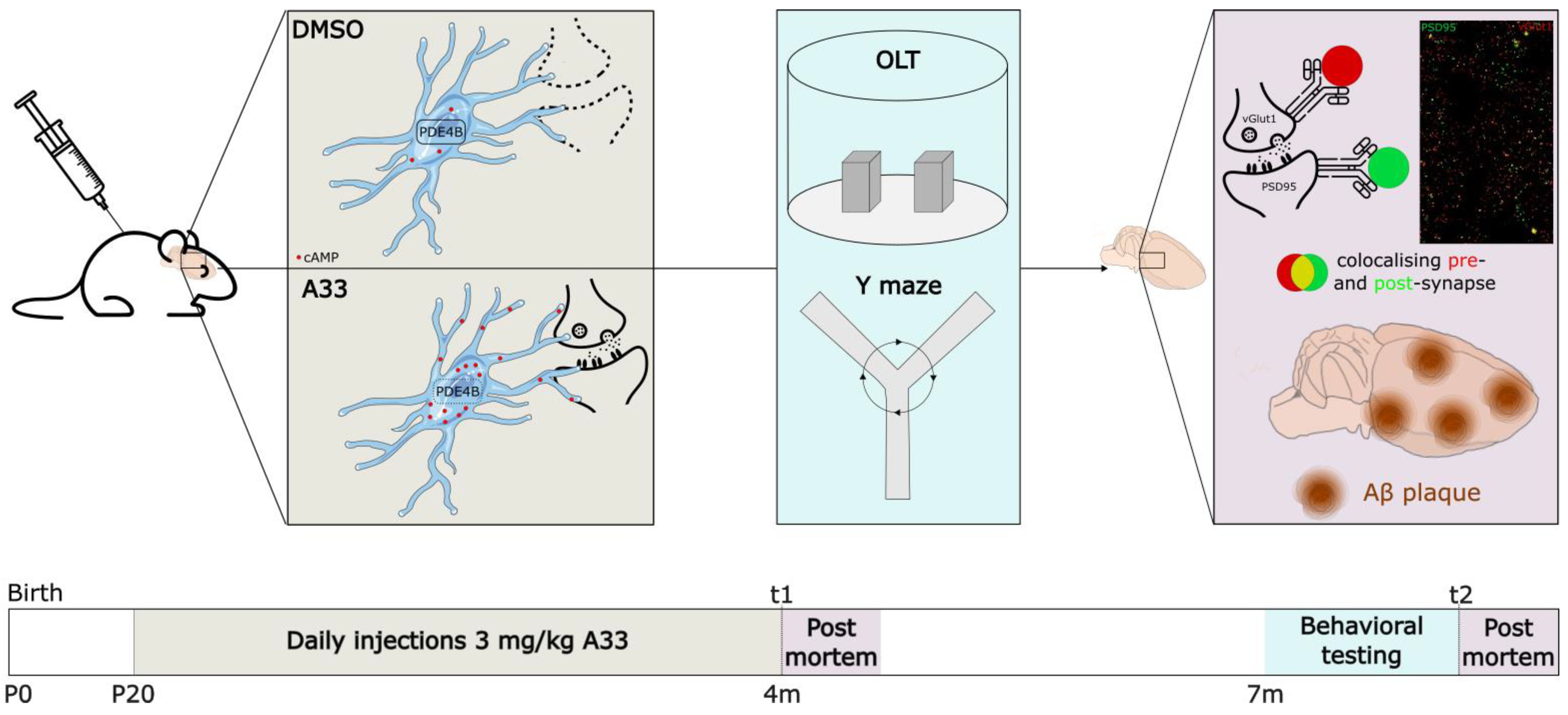
2.7. In Vivo Experimental Design
2.8. Behavioral Testing
2.9. Immunohistochemistry
2.10. ELISA of Distinct Pools of Aβ
2.11. Statistical Analysis
3. Results
3.1. Long-Term Inhibition of PDE4B Improves Cognition in 8-Month-Old APPswe/PS1dE9 Mice
3.2. Sustained Inhibition of PDE4B, Using A33, Does Not Alter Synapse Density of 4-Months-Old APPswe/PS1dE9 Mice but Normalizes Synapse Density in 8-Months-Old APPswe/PS1dE9 Mice
3.3. Inhibiting PDE4B Using A33 Reduces SDS-Soluble Aβ42 Levels but Does Not Affect Plaque Load
3.4. Inhibition of PDE4B Does Not Lower Microglial Phagocytic Capacity of Synaptosomes In Vitro
3.5. Microglia Increase Surveillance of Their Micro-Environment upon PDE4B Inhibition In Vitro
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- 2023 Alzheimer’s Disease Facts and Figures. Available online: https://alz-journals.onlinelibrary.wiley.com/doi/10.1002/alz.13016 (accessed on 23 April 2024).
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J.; Van Der Flier, W.M. Alzheimer’s disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef]
- Tao, Q.-Q.; Lin, R.-R.; Wu, Z.-Y. Early Diagnosis of Alzheimer’s Disease: Moving Toward a Blood-Based Biomarkers Era. Clin. Interv. Aging 2023, 18, 353–358. [Google Scholar] [CrossRef]
- Sperling, R.A.; Aisen, P.S.; Beckett, L.A.; Bennett, D.A.; Craft, S.; Fagan, A.M.; Iwatsubo, T.; Jack, C.R.; Kaye, J.; Montine, T.J.; et al. Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011, 7, 280–292. [Google Scholar] [CrossRef]
- Dubois, B.; Hampel, H.; Feldman, H.H.; Scheltens, P.; Aisen, P.; Andrieu, S.; Bakardjian, H.; Benali, H.; Bertram, L.; Blennow, K.; et al. Preclinical Alzheimer’s disease: Definition, natural history, and diagnostic criteria. Alzheimer’s Dement. 2016, 12, 292–323. [Google Scholar] [CrossRef]
- Gauthier, S.; Reisberg, B.; Zaudig, M.; Petersen, R.C.; Ritchie, K.; Broich, K.; Belleville, S.; Brodaty, H.; Bennett, D.; Chertkow, H.; et al. Mild cognitive impairment. Lancet 2006, 367, 1262–1270. [Google Scholar] [CrossRef]
- Terry, R.D.; Masliah, E.; Salmon, D.P.; Butters, N.; DeTeresa, R.; Hill, R.; Hansen, L.A.; Katzman, R. Physical basis of cognitive alterations in alzheimer’s disease: Synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 1991, 30, 572–580. [Google Scholar] [CrossRef]
- Mecca, A.P.; O’Dell, R.S.; Sharp, E.S.; Banks, E.R.; Bartlett, H.H.; Zhao, W.; Lipior, S.; Diepenbrock, N.G.; Chen, M.K.; Naganawa, M.; et al. Synaptic density and cognitive performance in Alzheimer’s disease: A PET imaging study with [11C]UCB-J. Alzheimer’s Dement. 2022, 18, 2527–2536. [Google Scholar] [CrossRef]
- DeKosky, S.T.; Scheff, S.W. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: Correlation with cognitive severity. Ann. Neurol. 1990, 27, 457–464. [Google Scholar] [CrossRef]
- Colom-Cadena, M.; Spires-Jones, T.; Zetterberg, H.; Blennow, K.; Caggiano, A.; DeKosky, S.T.; Fillit, H.; Harrison, J.E.; Schneider, L.S.; Scheltens, P.; et al. The clinical promise of biomarkers of synapse damage or loss in Alzheimer’s disease. Alzheimer’s Res. Ther. 2020, 12, 21. [Google Scholar] [CrossRef]
- Lee, E.; Chung, W.-S. Glial Control of Synapse Number in Healthy and Diseased Brain. Front. Cell. Neurosci. 2019, 13, 42. [Google Scholar] [CrossRef]
- Rajendran, L.; Paolicelli, R.C. Microglia-Mediated Synapse Loss in Alzheimer’s Disease. J. Neurosci. 2018, 38, 2911–2919. [Google Scholar] [CrossRef]
- De Schepper, S.; Ge, J.Z.; Crowley, G.; Ferreira, L.S.S.; Garceau, D.; Toomey, C.E.; Sokolova, D.; Rueda-Carrasco, J.; Shin, S.-H.; Kim, J.-S.; et al. Perivascular cells induce microglial phagocytic states and synaptic engulfment via SPP1 in mouse models of Alzheimer’s disease. Nat. Neurosci. 2023, 26, 406–415. [Google Scholar] [CrossRef]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 2018, 217, 459–472. [Google Scholar] [CrossRef]
- Wilson, D.M.; Cookson, M.R.; Van Den Bosch, L.; Zetterberg, H.; Holtzman, D.M.; Dewachter, I. Hallmarks of neurodegenerative diseases. Cell 2023, 186, 693–714. [Google Scholar] [CrossRef]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s Disease: The Amyloid Cascade Hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, H.; Li, R.; Sterling, K.; Song, W. Amyloid β-based therapy for Alzheimer’s disease: Challenges, successes and future. Signal Transduct. Target. Ther. 2023, 8, 248. [Google Scholar] [CrossRef]
- Cai, W.; Wu, T.; Chen, N. The Amyloid-Beta Clearance: From Molecular Targets to Glial and Neural Cells. Biomolecules 2023, 13, 313. [Google Scholar] [CrossRef]
- Bernier, L.P.; Bohlen, C.J.; York, E.M.; Choi, H.B.; Kamyabi, A.; Dissing-Olesen, L.; Hefendehl, J.K.; Collins, H.Y.; Stevens, B.; Barres, B.A.; et al. Nanoscale Surveillance of the Brain by Microglia via cAMP-Regulated Filopodia. Cell Rep. 2019, 27, 2895–2908.e4. [Google Scholar] [CrossRef]
- Rossi, A.G.; McCutcheon, J.C.; Roy, N.; Chilvers, E.R.; Haslett, C.; Dransfield, I. Regulation of macrophage phagocytosis of apoptotic cells by cAMP. J. Immunol. 1998, 160, 3562–3568. [Google Scholar] [CrossRef]
- Qin, L.; Bouchard, R.; Pugazhenthi, S. Regulation of cyclic AMP response element-binding protein during neuroglial interactions. J. Neurochem. 2016, 136, 918–930. [Google Scholar] [CrossRef] [PubMed]
- Tavares, L.P.; Negreiros-Lima, G.L.; Lima, K.M.; Silva, P.M.R.E.; Pinho, V.; Teixeira, M.M.; Sousa, L.P. Blame the signaling: Role of cAMP for the resolution of inflammation. Pharmacol. Res. 2020, 159, 105030. [Google Scholar] [CrossRef] [PubMed]
- Murthy, V.S.; Mangot, A.G. Psychiatric aspects of phosphodiesterases: An overview. Indian J. Pharmacol. 2015, 47, 594–599. [Google Scholar] [CrossRef]
- Prickaerts, J.; Heckman, P.R.A.; Blokland, A. Investigational phosphodiesterase inhibitors in phase I and phase II clinical trials for Alzheimer’s disease. Expert Opin. Investig. Drugs 2017, 26, 1033–1048. [Google Scholar] [CrossRef] [PubMed]
- Schepers, M.; Paes, D.; Tiane, A.; Rombaut, B.; Piccart, E.; van Veggel, L.; Gervois, P.; Wolfs, E.; Lambrichts, I.; Brullo, C.; et al. Selective PDE4 subtype inhibition provides new opportunities to intervene in neuroinflammatory versus myelin damaging hallmarks of multiple sclerosis. Brain Behav. Immun. 2023, 109, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Pearse, D.D.; Hughes, Z.A. PDE4B as a microglia target to reduce neuroinflammation. Glia 2016, 64, 1698–1709. [Google Scholar] [CrossRef] [PubMed]
- Fox, D.; Burgin, A.B.; Gurney, M.E. Structural basis for the design of selective phosphodiesterase 4B inhibitors. Cell. Signal. 2014, 26, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Wilson, N.M.; Gurney, M.E.; Dietrich, W.D.; Atkins, C.M. Therapeutic benefits of phosphodiesterase 4B inhibition after traumatic brain injury. PLoS ONE 2017, 12, e0178013. [Google Scholar] [CrossRef] [PubMed]
- Titus, D.J.; Wilson, N.M.; Freund, J.E.; Carballosa, M.M.; Sikah, K.E.; Furones, C.; Dietrich, W.D.; Gurney, M.E.; Atkins, C.M. Chronic Cognitive Dysfunction after Traumatic Brain Injury Is Improved with a Phosphodiesterase 4B Inhibitor. J. Neurosci. Off. J. Soc. Neurosci. 2016, 36, 7095–7108. [Google Scholar] [CrossRef]
- Gurney, M.E.; D’Amato, E.C.; Burgin, A.B. Phosphodiesterase-4 (PDE4) molecular pharmacology and Alzheimer’s disease. Neurother. J. Am. Soc. Exp. NeuroTher. 2015, 12, 49–56. [Google Scholar] [CrossRef]
- Santiago, A.; Soares, L.M.; Schepers, M.; Milani, H.; Vanmierlo, T.; Prickaerts, J.; de Oliveira, R.M.W. Roflumilast promotes memory recovery and attenuates white matter injury in aged rats subjected to chronic cerebral hypoperfusion. Neuropharmacology 2018, 138, 360–370. [Google Scholar] [CrossRef] [PubMed]
- Kelly, M.P. Cyclic nucleotide signaling changes associated with normal aging and age-related diseases of the brain. Cell. Signal. 2018, 42, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Myers, S.A.; Gobejishvili, L.; Saraswat Ohri, S.; Garrett Wilson, C.; Andres, K.R.; Riegler, A.S.; Donde, H.; Joshi-Barve, S.; Barve, S.; Whittemore, S.R. Following spinal cord injury, PDE4B drives an acute, local inflammatory response and a chronic, systemic response exacerbated by gut dysbiosis and endotoxemia. Neurobiol. Dis. 2019, 124, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.; Aliberti, J.; Graemmel, P.; Sunshine, M.J.; Kreutzberg, G.W.; Sher, A.; Littman, D.R. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol. Cell. Biol. 2000, 20, 4106–4114. [Google Scholar] [CrossRef] [PubMed]
- Bohlen, C.J.; Bennett, F.C.; Bennett, M.L. Isolation and Culture of Microglia. Curr. Protoc. Immunol. 2019, 125, e70. [Google Scholar] [CrossRef] [PubMed]
- Simms, D.; Cizdziel, P.E.; Chomczynski, P. TRIzol: A new reagent for optimal single-step isolation of RNA. Focus 1993, 15, 532–535. [Google Scholar]
- Ricciarelli, R.; Brullo, C.; Prickaerts, J.; Arancio, O.; Villa, C.; Rebosio, C.; Calcagno, E.; Balbi, M.; van Hagen, B.T.; Argyrousi, E.K.; et al. Memory-enhancing effects of GEBR-32a, a new PDE4D inhibitor holding promise for the treatment of Alzheimer’s disease. Sci. Rep. 2017, 7, 46320. [Google Scholar] [CrossRef] [PubMed]
- Soto, P.L.; Young, M.E.; Dimarco, G.M.; George, B.; Melnikova, T.; Savonenko, A.V.; Harris, B.N. Longitudinal assessment of cognitive function in the APPswe/PS1dE9 mouse model of Alzheimer’s-related beta-amyloidosis. Neurobiol. Aging 2023, 128, 85–99. [Google Scholar] [CrossRef] [PubMed]
- Vanmierlo, T.; Rutten, K.; Dederen, J.; Bloks, V.W.; van Vark-van der Zee, L.C.; Kuipers, F.; Kiliaan, A.; Blokland, A.; Sijbrands, E.J.; Steinbusch, H.; et al. Liver X receptor activation restores memory in aged AD mice without reducing amyloid. Neurobiol. Aging 2011, 32, 1262–1272. [Google Scholar] [CrossRef] [PubMed]
- Sierksma, A.S.R.; Rutten, K.; Sydlik, S.; Rostamian, S.; Steinbusch, H.W.M.; Van Den Hove, D.L.A.; Prickaerts, J. Chronic phosphodiesterase type 2 inhibition improves memory in the APPswe/PS1dE9 mouse model of Alzheimer’s disease. Neuropharmacology 2013, 64, 124–136. [Google Scholar] [CrossRef]
- McLeod, F.; Marzo, A.; Podpolny, M.; Galli, S.; Salinas, P. Evaluation of Synapse Density in Hippocampal Rodent Brain Slices. J. Vis. Exp. 2017, 128, e56153. [Google Scholar] [CrossRef] [PubMed]
- Khushi, M.; Napier, C.E.; Smyth, C.M.; Reddel, R.R.; Arthur, J.W. MatCol: A tool to measure fluorescence signal colocalisation in biological systems. Sci. Rep. 2017, 7, 8879. [Google Scholar] [CrossRef] [PubMed]
- Steinerman, J.R.; Irizarry, M.; Scarmeas, N.; Raju, S.; Brandt, J.; Albert, M.; Blacker, D.; Hyman, B.; Stern, Y. Distinct pools of beta-amyloid in Alzheimer disease-affected brain: A clinicopathologic study. Arch. Neurol. 2008, 65, 906–912. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef] [PubMed]
- Vigneault, É.; Poirel, O.; Riad, M.; Prud’Homme, J.; Dumas, S.; Turecki, G.; Fasano, C.; Mechawar, N.; El Mestikawy, S. Distribution of vesicular glutamate transporters in the human brain. Front. Neuroanat. 2015, 9, 23. [Google Scholar] [CrossRef] [PubMed]
- Levy, A.M.; Gomez-Puertas, P.; Tümer, Z. Neurodevelopmental Disorders Associated with PSD-95 and Its Interaction Partners. Int. J. Mol. Sci. 2022, 23, 4390. [Google Scholar] [CrossRef] [PubMed]
- Cameron, B.; Landreth, G.E. Inflammation, microglia, and alzheimer’s disease. Neurobiol. Dis. 2010, 37, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.B.; De Lannoy, I.A.M.; Pang, K.S. Measuring Amyloid-β Peptide Concentrations in Murine Brain with Improved ELISA Assay. Curr. Protoc. 2021, 1, e253. [Google Scholar] [CrossRef]
- Salanga, C.M.; Salanga, M.C. Genotype to Phenotype: CRISPR Gene Editing Reveals Genetic Compensation as a Mechanism for Phenotypic Disjunction of Morphants and Mutants. Int. J. Mol. Sci. 2021, 22, 3472. [Google Scholar] [CrossRef]
- Schepers, M.; Vanmierlo, T. Novel insights in phosphodiesterase 4 subtype inhibition to target neuroinflammation and stimulate remyelination. Neural Regen. Res. 2024, 19, 493–494. [Google Scholar] [CrossRef]
- Jackson, P.K. cAMP Signaling in Nanodomains. Cell 2020, 182, 1379–1381. [Google Scholar] [CrossRef] [PubMed]
- Musheshe, N.; Schmidt, M.; Zaccolo, M. cAMP: From Long-Range Second Messenger to Nanodomain Signalling. Trends Pharmacol. Sci. 2018, 39, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Blair, C.M.; Baillie, G.S. Reshaping cAMP nanodomains through targeted disruption of compartmentalised phosphodiesterase signalosomes. Biochem. Soc. Trans. 2019, 47, 1405–1414. [Google Scholar] [CrossRef] [PubMed]
- Gallop, J.L. Filopodia and their links with membrane traffic and cell adhesion. Semin. Cell Dev. Biol. 2020, 102, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Jiang, J.; Tan, Y.; Chen, S. Microglia in neurodegenerative diseases: Mechanism and potential therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 359. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Bestard-Lorigados, I.; Song, W. The synapse as a treatment avenue for Alzheimer’s Disease. Mol. Psychiatry 2022, 27, 2940–2949. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Yao, H.; Xu, Y.; Hao, R.; Zhang, W.; Liu, H.; Huang, Y.; Guo, W.; Lu, B. Therapeutic potential of a TrkB agonistic antibody for Alzheimer’s disease. Theranostics 2020, 10, 6854–6874. [Google Scholar] [CrossRef] [PubMed]
- Viana da Silva, S.; Zhang, P.; Haberl, M.G.; Labrousse, V.; Grosjean, N.; Blanchet, C.; Frick, A.; Mulle, C. Hippocampal Mossy Fibers Synapses in CA3 Pyramidal Cells Are Altered at an Early Stage in a Mouse Model of Alzheimer’s Disease. J. Neurosci. 2019, 39, 4193–4205. [Google Scholar] [CrossRef]
- Henstridge, C.M.; Tzioras, M.; Paolicelli, R.C. Glial Contribution to Excitatory and Inhibitory Synapse Loss in Neurodegeneration. Front. Cell. Neurosci. 2019, 13, 63. [Google Scholar] [CrossRef]
- Zhou, M.; Cornell, J.; Salinas, S.; Huang, H.-Y. Microglia regulation of synaptic plasticity and learning and memory. Neural Regen. Res. 2022, 17, 705. [Google Scholar] [CrossRef]
- Yasuda, M.; Nagappan-Chettiar, S.; Johnson-Venkatesh, E.M.; Umemori, H. An activity-dependent determinant of synapse elimination in the mammalian brain. Neuron 2021, 109, 1333–1349.e6. [Google Scholar] [CrossRef] [PubMed]
- Parkhurst, C.N.; Yang, G.; Ninan, I.; Savas, J.N.; Yates, J.R.; Lafaille, J.J.; Hempstead, B.L.; Littman, D.R.; Gan, W.B. Microglia Promote Learning-Dependent Synapse Formation through Brain-Derived Neurotrophic Factor. Cell 2013, 155, 1596–1609. [Google Scholar] [CrossRef] [PubMed]
- Bittner, T.; Burgold, S.; Dorostkar, M.M.; Fuhrmann, M.; Wegenast-Braun, B.M.; Schmidt, B.; Kretzschmar, H.; Herms, J. Amyloid plaque formation precedes dendritic spine loss. Acta Neuropathol. 2012, 124, 797–807. [Google Scholar] [CrossRef] [PubMed]
- Gong, B.; Vitolo, O.V.; Trinchese, F.; Liu, S.; Shelanski, M.; Arancio, O. Persistent improvement in synaptic and cognitive functions in an Alzheimer mouse model after rolipram treatment. J. Clin. Investig. 2004, 114, 1624–1634. [Google Scholar] [CrossRef] [PubMed]
- Sierksma, A.S.R.; Van Den Hove, D.L.A.; Pfau, F.; Philippens, M.; Bruno, O.; Fedele, E.; Ricciarelli, R.; Steinbusch, H.W.M.; Vanmierlo, T.; Prickaerts, J. Improvement of spatial memory function in APPswe/PS1dE9 mice after chronic inhibition of phosphodiesterase type 4D. Neuropharmacology 2014, 77, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Sebastiani, G.; Morissette, C.; Lagacé, C.; Boulé, M.; Ouellette, M.-J.; McLaughlin, R.W.; Lacombe, D.; Gervais, F.; Tremblay, P. The cAMP-specific phosphodiesterase 4B mediates Aβ-induced microglial activation. Neurobiol. Aging 2006, 27, 691–701. [Google Scholar] [CrossRef] [PubMed]
- Stevens, B.; Allen, N.J.; Vazquez, L.E.; Howell, G.R.; Christopherson, K.S.; Nouri, N.; Micheva, K.D.; Mehalow, A.K.; Huberman, A.D.; Stafford, B.; et al. The classical complement cascade mediates CNS synapse elimination. Cell 2007, 131, 1164–1178. [Google Scholar] [CrossRef] [PubMed]
- Gunner, G.; Cheadle, L.; Johnson, K.M.; Ayata, P.; Badimon, A.; Mondo, E.; Nagy, M.A.; Liu, L.; Bemiller, S.M.; Kim, K.W.; et al. Sensory lesioning induces microglial synapse elimination via ADAM10 and fractalkine signaling. Nat. Neurosci. 2019, 22, 1075–1088. [Google Scholar] [CrossRef] [PubMed]
- Serezani, C.H.; Ballinger, M.N.; Aronoff, D.M.; Peters-Golden, M. Cyclic AMP: Master regulator of innate immune cell function. Am. J. Respir. Cell Mol. Biol. 2008, 39, 127–132. [Google Scholar] [CrossRef]
- Aronoff, D.M.; Canetti, C.; Serezani, C.H.; Luo, M.; Peters-Golden, M. Cutting Edge: Macrophage Inhibition by Cyclic AMP (cAMP): Differential Roles of Protein Kinase A and Exchange Protein Directly Activated by cAMP-1. J. Immunol. 2005, 174, 595–599. [Google Scholar] [CrossRef]
- Makranz, C.; Cohen, G.; Reichert, F.; Kodama, T.; Rotshenker, S. cAMP cascade (PKA, Epac, adenylyl cyclase, Gi, and phosphodiesterases) regulates myelin phagocytosis mediated by complement receptor-3 and scavenger receptor-AI/II in microglia and macrophages. Glia 2006, 53, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Epstein, P.M. Different phosphodiesterases (PDEs) regulate distinct phosphoproteomes during cAMP signaling. Proc. Natl. Acad. Sci. USA 2017, 114, 7741–7743. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.L.; Bennett, F.C.; Liddelow, S.A.; Ajami, B.; Zamanian, J.L.; Fernhoff, N.B.; Mulinyawe, S.B.; Bohlen, C.J.; Adil, A.; Tucker, A.; et al. New tools for studying microglia in the mouse and human CNS. Proc. Natl. Acad. Sci. USA 2016, 113, E1738–E1746. [Google Scholar] [CrossRef] [PubMed]
- Benitez-Fernandez, R.; Gil, C.; Guaza, C.; Mestre, L.; Martinez, A. The Dual PDE7-GSK3beta Inhibitor, VP3.15, as Neuroprotective Disease-Modifying Treatment in a Model of Primary Progressive Multiple Sclerosis. Int. J. Mol. Sci. 2022, 23, 14378. [Google Scholar] [CrossRef] [PubMed]
- Paterniti, I.; Mazzon, E.; Gil, C.; Impellizzeri, D.; Palomo, V.; Redondo, M.; Perez, D.I.; Esposito, E.; Martinez, A.; Cuzzocrea, S. PDE 7 inhibitors: New potential drugs for the therapy of spinal cord injury. PLoS ONE 2011, 6, e15937. [Google Scholar] [CrossRef] [PubMed]
- Davalos, D.; Grutzendler, J.; Yang, G.; Kim, J.V.; Zuo, Y.; Jung, S.; Littman, D.R.; Dustin, M.L.; Gan, W.-B. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 2005, 8, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Badimon, A.; Strasburger, H.J.; Ayata, P.; Chen, X.; Nair, A.; Ikegami, A.; Hwang, P.; Chan, A.T.; Graves, S.M.; Uweru, J.O.; et al. Negative feedback control of neuronal activity by microglia. Nature 2020, 586, 417–423. [Google Scholar] [CrossRef]
- Umpierre, A.D.; Bystrom, L.L.; Ying, Y.; Liu, Y.U.; Worrell, G.; Wu, L.-J. Microglial calcium signaling is attuned to neuronal activity in awake mice. eLife 2020, 9, e56502. [Google Scholar] [CrossRef] [PubMed]
- Cangalaya, C.; Wegmann, S.; Sun, W.; Diez, L.; Gottfried, A.; Richter, K.; Stoyanov, S.; Pakan, J.; Fischer, K.-D.; Dityatev, A. Real-time mechanisms of exacerbated synaptic remodeling by microglia in acute models of systemic inflammation and tauopathy. Brain Behav. Immun. 2023, 110, 245–259. [Google Scholar] [CrossRef]
- Hashemiaghdam, A.; Mroczek, M. Microglia heterogeneity and neurodegeneration: The emerging paradigm of the role of immunity in Alzheimer’s disease. J. Neuroimmunol. 2020, 341, 577185. [Google Scholar] [CrossRef]
- Tibbo, A.J.; Baillie, G.S. Phosphodiesterase 4B: Master Regulator of Brain Signaling. Cells 2020, 9, 1254. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-Sequencing Transcriptome and Splicing Database of Glia, Neurons, and Vascular Cells of the Cerebral Cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef] [PubMed]
- Lyon, K.A.; Allen, N.J. From Synapses to Circuits, Astrocytes Regulate Behavior. Front. Neural Circuits 2022, 15, 786293. [Google Scholar] [CrossRef] [PubMed]
- Campbell, S.L.; van Groen, T.; Kadish, I.; Smoot, L.H.M.; Bolger, G.B. Altered phosphorylation, electrophysiology, and behavior on attenuation of PDE4B action in hippocampus. BMC Neurosci. 2017, 18, 77. [Google Scholar] [CrossRef] [PubMed]
- McGirr, A.; Lipina, T.V.; Mun, H.S.; Georgiou, J.; Al-Amri, A.H.; Ng, E.; Zhai, D.; Elliott, C.; Cameron, R.T.; Mullins, J.G.; et al. Specific Inhibition of Phosphodiesterase-4B Results in Anxiolysis and Facilitates Memory Acquisition. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2016, 41, 1080–1092. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, P.; Güngör, H.; Anongjanya, P.; Tweedy, C.; Parkin, E.; Johnston, J.; Carr, I.M.; Dawson, N.; Clapcote, S.J. Protective effect of PDE4B subtype-specific inhibition in an App knock-in mouse model for Alzheimer’s disease. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2024, 114, 1624–1634. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Zhong, J.; Niu, B.; Zhong, Q.; Xiao, J.; Xie, J.; Lin, M.; Zhou, Z.; Xu, J.; Wang, H. Inhibition of Phosphodiesterase 4 by FCPR03 Alleviates Chronic Unpredictable Mild Stress-Induced Depressive-Like Behaviors and Prevents Dendritic Spine Loss in Mice Hippocampi. Int. J. Neuropsychopharmacol. 2018, 22, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Kater, M.S.J.; Huffels, C.F.M.; Oshima, T.; Renckens, N.S.; Middeldorp, J.; Boddeke, E.W.G.M.; Smit, A.B.; Eggen, B.J.L.; Hol, E.M.; Verheijen, M.H.G. Prevention of microgliosis halts early memory loss in a mouse model of Alzheimer’s disease. Brain Behav. Immun. 2023, 107, 225–241. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Xiao, M.; He, W.; Zhao, Y. Minocycline upregulates cyclic AMP response element binding protein and brain-derived neurotrophic factor in the hippocampus of cerebral ischemia rats and improves behavioral deficits. Neuropsychiatr. Dis. Treat. 2015, 11, 507–516. [Google Scholar] [CrossRef]
- Baillie, G.S.; Tejeda, G.S.; Kelly, M.P. Therapeutic targeting of 3′,5′-cyclic nucleotide phosphodiesterases: Inhibition and beyond. Nat. Rev. Drug Discov. 2019, 18, 770–796. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| Pde4a | GCCTTGCACTGAGGAAACTC | GGCTGTCTCCTGCTTCAAAC |
| Pde7a | TGGAGGCTCAGATAGGTGCT | CCAGTTCCGACATGGGTTAC |
| Pde7b | ATCGCTTGACAAATGGGAAC | GGGTTGTGACCGTGGTAATC |
| Pde8a | TGGCTGTGCTCTACAACGAC | CCGGTAGTCATTCCTCTCCA |
| Cypa | GCCTCTCCTTCGAGCTGTT | AAGTCACCACCCTGGCA |
| Ywhaz | AGCCGAGCTGTCTAACCGAG | GCCAACTAGCGGTAGTAGTCA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rombaut, B.; Schepers, M.; Tiane, A.; Mussen, F.; Koole, L.; Kessels, S.; Trippaers, C.; Jacobs, R.; Wouters, K.; Willems, E.; et al. Early Inhibition of Phosphodiesterase 4B (PDE4B) Instills Cognitive Resilience in APPswe/PS1dE9 Mice. Cells 2024, 13, 1000. https://doi.org/10.3390/cells13121000
Rombaut B, Schepers M, Tiane A, Mussen F, Koole L, Kessels S, Trippaers C, Jacobs R, Wouters K, Willems E, et al. Early Inhibition of Phosphodiesterase 4B (PDE4B) Instills Cognitive Resilience in APPswe/PS1dE9 Mice. Cells. 2024; 13(12):1000. https://doi.org/10.3390/cells13121000
Chicago/Turabian StyleRombaut, Ben, Melissa Schepers, Assia Tiane, Femke Mussen, Lisa Koole, Sofie Kessels, Chloë Trippaers, Ruben Jacobs, Kristiaan Wouters, Emily Willems, and et al. 2024. "Early Inhibition of Phosphodiesterase 4B (PDE4B) Instills Cognitive Resilience in APPswe/PS1dE9 Mice" Cells 13, no. 12: 1000. https://doi.org/10.3390/cells13121000