
Epigenetic Regulation of DLK1-DIO3 Region in Thyroid Carcinoma
 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Methylation Array
2.3. MAPK Pathway Inhibition
2.4. TCGA Methylation Data Analysis
2.5. Bisulfite-qPCR
2.6. Overall Survival Analysis
2.7. Treatment with 5-Aza-dC and TSA
2.8. Quantitative Reverse Transcription PCR (RT-qPCR)
2.9. Chromatin Immunoprecipitation (ChIP) for Histone Modifications
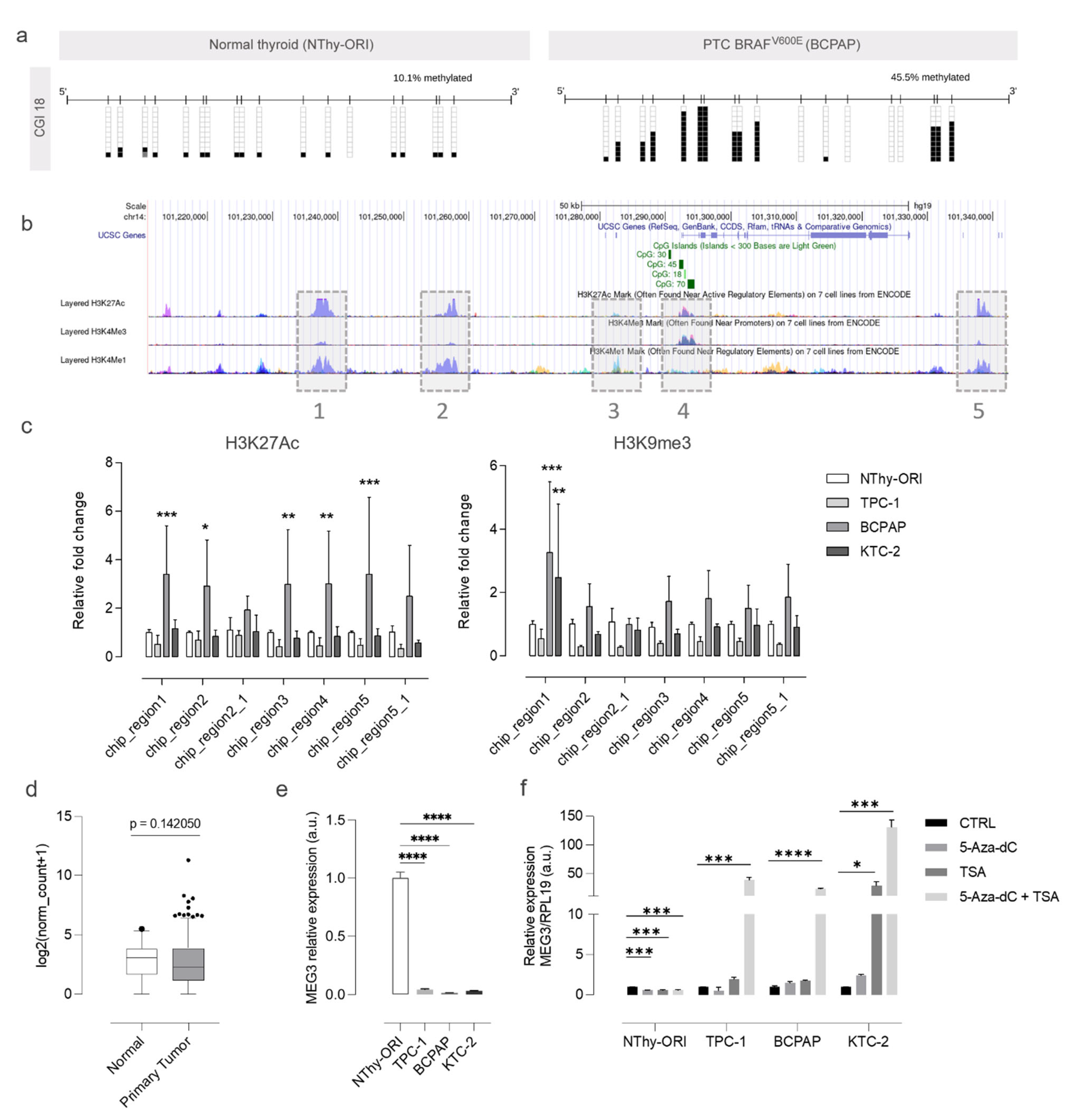
3. Results
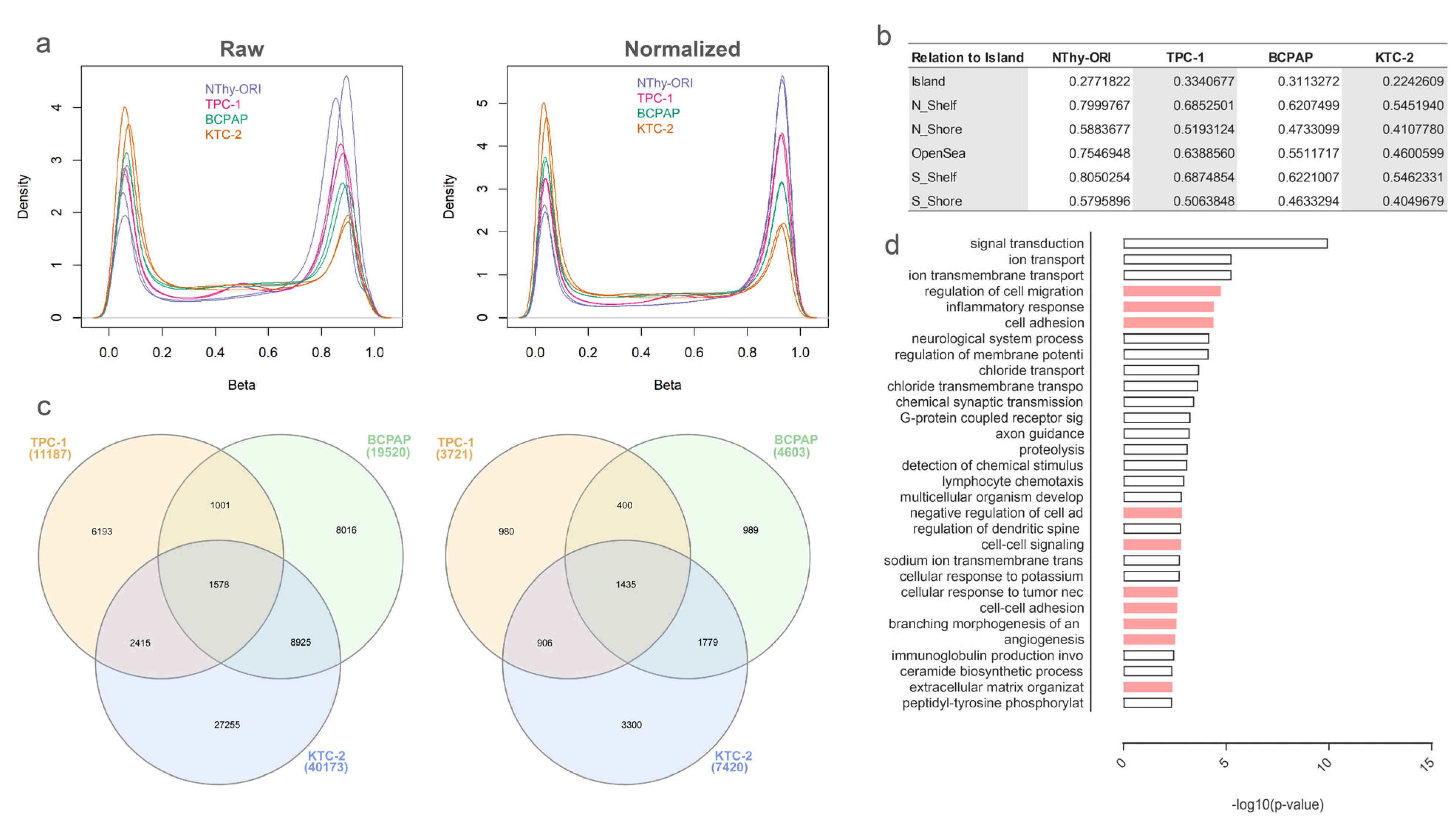
3.1. DNA Methylation of the DLK1-DIO3 Region Is Disrupted in Thyroid Cancer-Derived Cell Lines
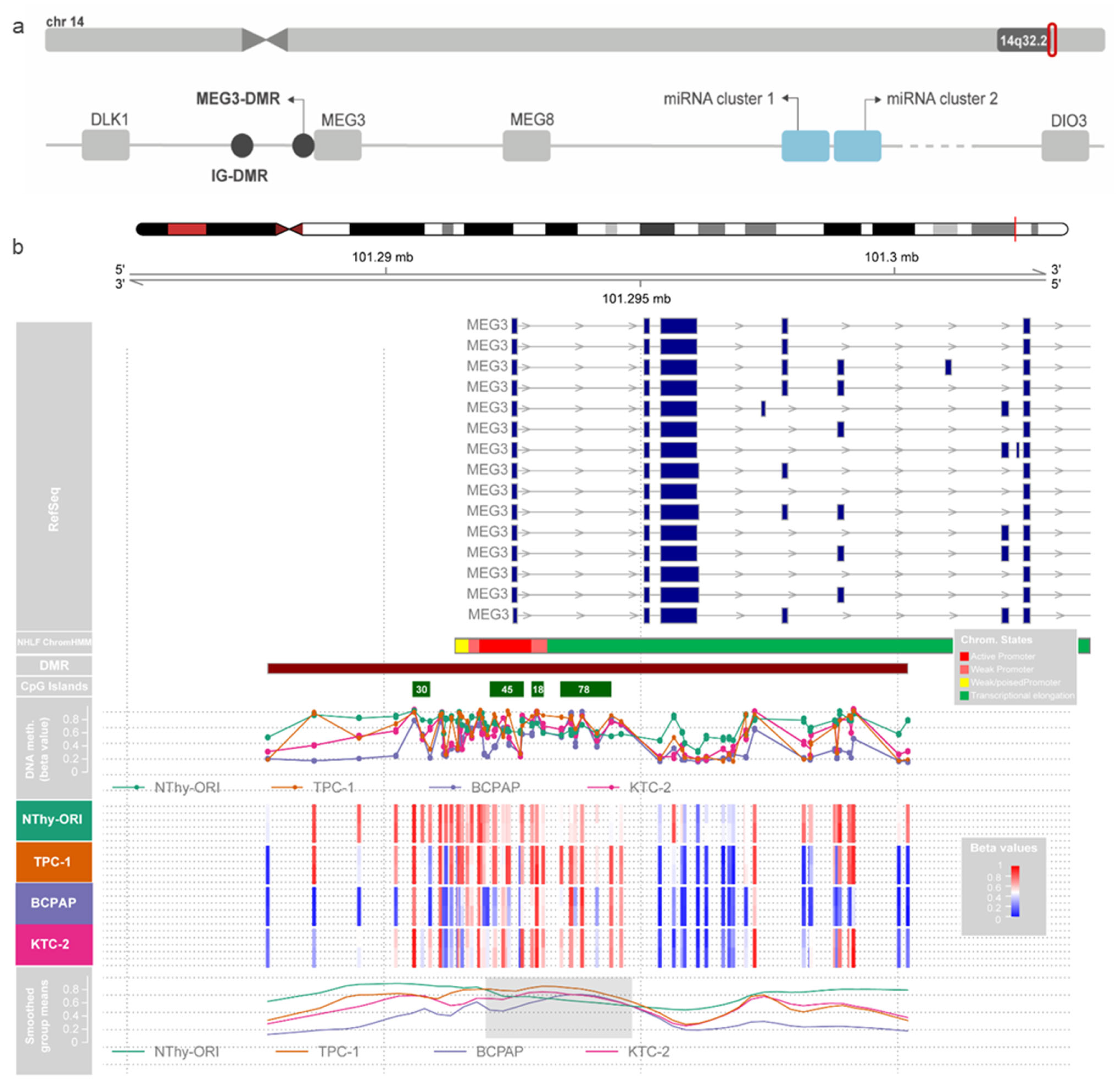
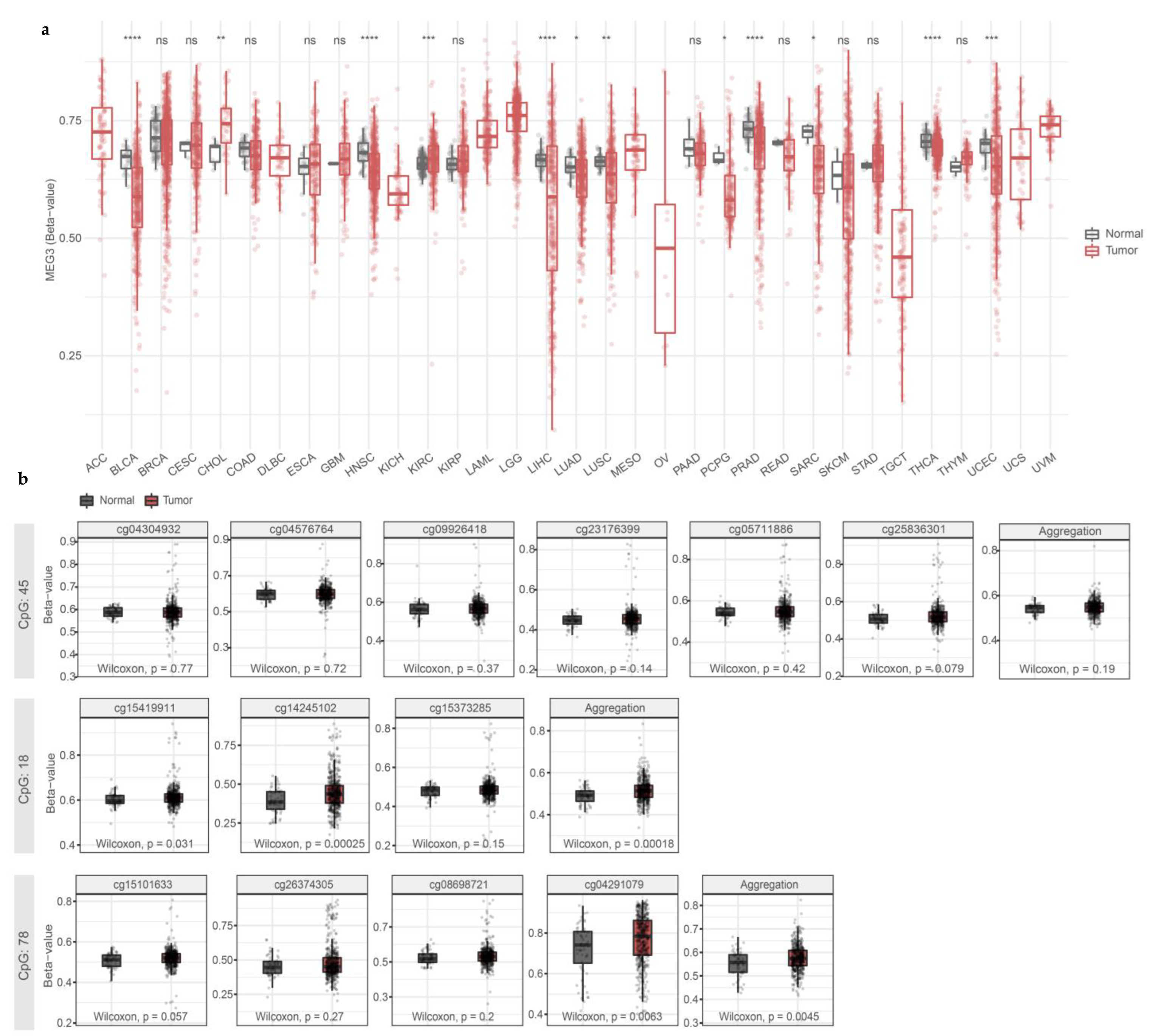
3.2. Hypermethylation of MEG3-DMR CGIs in Human Papillary Thyroid Carcinoma Samples from TCGA
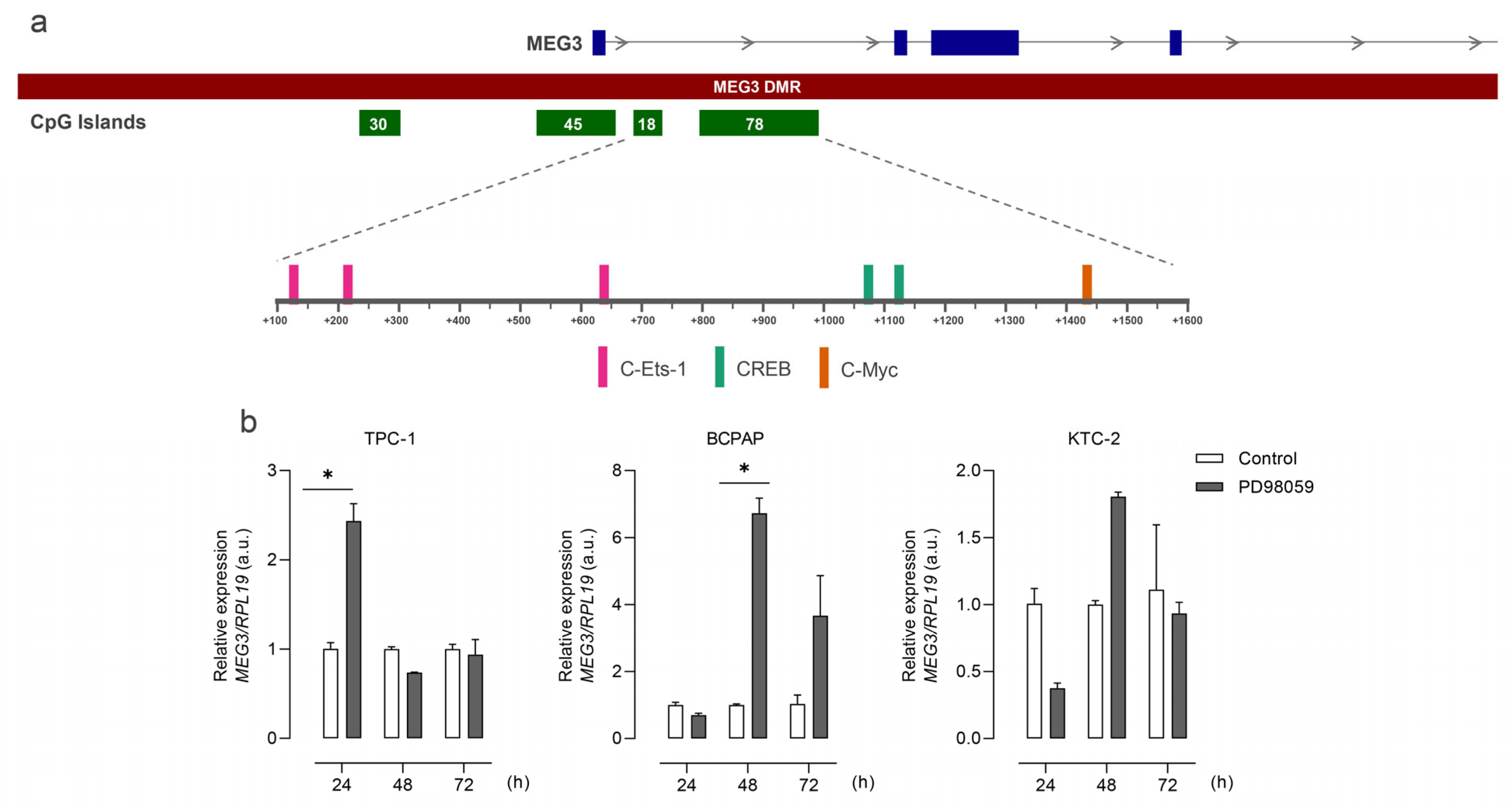
3.3. Silencing of MEG3 Is Mediated by DNA Methylation and Histone Acetylation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- American Cancer Society Cancer Facts & Figures; American Cancer Society: Atlanta, GA, USA, 2023.
- Fagin, J.A.; Wells, S.A. Biologic and Clinical Perspectives on Thyroid Cancer. N. Engl. J. Med. 2016, 375, 1054–1067. [Google Scholar] [CrossRef] [PubMed]
- Bhaumik, D.; Scott, G.K.; Schokrpur, S.; Patil, C.K.; Campisi, J.; Benz, C.C. Expression of MicroRNA-146 Suppresses NF-ΚB Activity with Reduction of Metastatic Potential in Breast Cancer Cells. Oncogene 2008, 27, 5643–5647. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.-K.; Yang, K.D.; Chou, F.-F.; Huang, C.-C.; Lan, Y.-W.; Lee, Y.-F.; Kang, H.-Y.; Liu, R.-T. Prognostic Implications of MiR-146b Expression and Its Functional Role in Papillary Thyroid Carcinoma. J. Clin. Endocrinol. Metab. 2013, 98, E196–E205. [Google Scholar] [CrossRef] [PubMed]
- Geraldo, M.V.; Yamashita, A.S.; Kimura, E.T. MicroRNA MiR-146b-5p Regulates Signal Transduction of TGF-Β by Repressing SMAD4 in Thyroid Cancer. Oncogene 2012, 31, 1910–1922. [Google Scholar] [CrossRef] [PubMed]
- Benetatos, L.; Hatzimichael, E.; Londin, E.; Vartholomatos, G.; Loher, P.; Rigoutsos, I.; Briasoulis, E. The MicroRNAs within the DLK1-DIO3 Genomic Region: Involvement in Disease Pathogenesis. Cell. Mol. Life Sci. 2013, 70, 795–814. [Google Scholar] [CrossRef]
- Geraldo, M.V.; Nakaya, H.I.; Kimura, E. Down-Regulation of 14q32-Encoded MiRNAs and Tumor Suppressor Role for MiR-654-3p in Papillary Thyroid Cancer. Oncotarget 2017, 8, 9597–9607. [Google Scholar] [CrossRef] [PubMed]
- Alves, L.F.; Geraldo, M.V. MiR-495-3p Regulates Cell Migration and Invasion in Papillary Thyroid Carcinoma. Front. Oncol. 2023, 13, 1039654. [Google Scholar] [CrossRef] [PubMed]
- Marson, L.A.; Alves, L.F.; Sielski, M.S.; Vicente, C.P.; Kimura, E.T.; Geraldo, M.V. DLK1-DIO3 Region as a Source of Candidate Tumor Suppressor MiRNAs in Papillary Thyroid Carcinoma. PREPRINT 2020, 53, 1689–1699. [Google Scholar] [CrossRef]
- Kojima, S.; Shiochi, N.; Sato, K.; Yamaura, M.; Ito, T.; Yamamura, N.; Goto, N.; Odamoto, M.; Kobayashi, S.; Kimura, T.; et al. Epigenome Editing Reveals Core DNA Methylation for Imprinting Control in the Dlk1-Dio3 Imprinted Domain. Nucleic Acids Res. 2022, 50, 5080–5094. [Google Scholar] [CrossRef]
- Luo, Z.; Lin, C.; Woodfin, A.R.; Bartom, E.T.; Gao, X.; Smith, E.R.; Shilatifard, A. Regulation of the Imprinted Dlk1-Dio3 Locus by Allele-Specific Enhancer Activity. Genes Dev. 2016, 30, 92. [Google Scholar] [CrossRef] [PubMed]
- Manodoro, F.; Marzec, J.; Chaplin, T.; Miraki-Moud, F.; Moravcsik, E.; Jovanovic, J.V.; Wang, J.; Iqbal, S.; Taussig, D.; Grimwade, D.; et al. Loss of Imprinting at the 14q32 Domain Is Associated with MicroRNA Overexpression in Acute Promyelocytic Leukemia. Blood 2014, 123, 2066–2074. [Google Scholar] [CrossRef] [PubMed]
- Pan, T.; Ding, H.; Jin, L.; Zhang, S.; Wu, D.; Pan, W.; Dong, M.; Ma, X.; Chen, Z. DNMT1-Mediated Demethylation of LncRNA MEG3 Promoter Suppressed Breast Cancer Progression by Repressing Notch1 Signaling Pathway. Cell Cycle 2022, 21, 2323–2337. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Hara, S.; Kato, T.; Tamano, M.; Muramatsu, A.; Asahara, H.; Takada, S. A Tandem Repeat Array in IG-DMR Is Essential for Imprinting of Paternal Allele at the Dlk1-Dio3 Domain during Embryonic Development. Hum. Mol. Genet. 2018, 27, 3283–3292. [Google Scholar] [CrossRef] [PubMed]
- Peters, T.J.; Buckley, M.J.; Statham, A.L.; Pidsley, R.; Samaras, K.; V Lord, R.; Clark, S.J.; Molloy, P.L. De Novo Identification of Differentially Methylated Regions in the Human Genome. Epigenetics Chromatin 2015, 8, 6. [Google Scholar] [CrossRef] [PubMed]
- Peters, T.J.; Buckley, M.J.; Chen, Y.; Smyth, G.K.; Goodnow, C.C.; Clark, S.J. Calling Differentially Methylated Regions from Whole Genome Bisulphite Sequencing with DMRcate. Nucleic Acids Res. 2021, 49, e109. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A Flexible and Comprehensive Bioconductor Package for the Analysis of Infinium DNA Methylation Microarrays. Bioinformatics 2014, 30, 1363–1369. [Google Scholar] [CrossRef]
- Li, Y.; Ge, D.; Lu, C. The SMART App: An Interactive Web Application for Comprehensive DNA Methylation Analysis and Visualization. Epigenetics Chromatin 2019, 12, 71. [Google Scholar] [CrossRef] [PubMed]
- Ernst, J.; Kheradpour, P.; Mikkelsen, T.S.; Shoresh, N.; Ward, L.D.; Epstein, C.B.; Zhang, X.; Wang, L.; Issner, R.; Coyne, M.; et al. Mapping and Analysis of Chromatin State Dynamics in Nine Human Cell Types. Nature 2011, 473, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Grabe, N. AliBaba2: Context Specific Identification of Transcription Factor Binding Sites. Silico Biol. 2002, 2, S1–S15. [Google Scholar]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor Package for Integrative Analysis of TCGA Data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef] [PubMed]
- Therneau, T.M. A Package for Survival Analysis in R. Available online: https://cran.r-project.org/package=survival (accessed on 25 April 2024).
- Kassambara, A.; Kosinski, M.; Biecek, P. Drawing Survival Curves Using “ggplot2” [R Package Survminer], Version 3.7-0. Available online: https://CRAN.R-project.org/package=survival (accessed on 25 April 2024).
- Modhukur, V.; Iljasenko, T.; Metsalu, T.; Lokk, K.; Laisk-Podar, T.; Vilo, J. MethSurv: A Web Tool to Perform Multivariable Survival Analysis Using DNA Methylation Data. Epigenomics 2018, 10, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.P.; Youngson, N.; Takada, S.; Seitz, H.; Reik, W.; Paulsen, M.; Cavaille, J.; Ferguson-Smith, A.C. Asymmetric Regulation of Imprinting on the Maternal and Paternal Chromosomes at the Dlk1-Gtl2 Imprinted Cluster on Mouse Chromosome 12. Nat. Genet. 2003, 35, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Takada, S.; Paulsen, M.; Tevendale, M.; Tsai, C.E.; Kelsey, G.; Cattanach, B.M.; Ferguson-Smith, A.C. Epigenetic Analysis of the Dlk1–Gtl2 Imprinted Domain on Mouse Chromosome 12: Implications for Imprinting Control from Comparison with Igf2–H19. Hum. Mol. Genet. 2002, 11, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Kagami, M.; Sekita, Y.; Nishimura, G.; Irie, M.; Kato, F.; Okada, M.; Yamamori, S.; Kishimoto, H.; Nakayama, M.; Tanaka, Y.; et al. Deletions and Epimutations Affecting the Human 14q32.2 Imprinted Region in Individuals with Paternal and Maternal Upd(14)-like Phenotypes. Nat. Genet. 2008, 40, 237–242. [Google Scholar] [CrossRef] [PubMed]
- da Rocha, S.T.; Edwards, C.A.; Ito, M.; Ogata, T.; Ferguson-Smith, A.C. Genomic Imprinting at the Mammalian Dlk1-Dio3 Domain. Trends Genet. 2008, 24, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Cheunsuchon, P.; Nakayama, Y.; Lawlor, M.W.; Zhong, Y.; Rice, K.A.; Zhang, L.; Zhang, X.; Gordon, F.E.; Lidov, H.G.W.; et al. Activation of Paternally Expressed Genes and Perinatal Death Caused by Deletion of the Gtl2 Gene. Development 2010, 137, 2643–2652. [Google Scholar] [CrossRef]
- Zhu, W.; Botticelli, E.M.; Kery, R.E.; Mao, Y.; Wang, X.; Yang, A.; Wang, X.; Zhou, J.; Zhang, X.; Soberman, R.J.; et al. Meg3-DMR, Not the Meg3 Gene, Regulates Imprinting of the Dlk1-Dio3 Locus. Dev. Biol. 2019, 455, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Hiura, H.; Hattori, H.; Kobayashi, N.; Okae, H.; Chiba, H.; Miyauchi, N.; Kitamura, A.; Kikuchi, H.; Yoshida, H.; Arima, T. Genome-Wide MicroRNA Expression Profiling in Placentae from Frozen-Thawed Blastocyst Transfer. Clin. Epigenetics 2017, 9, 79. [Google Scholar] [CrossRef] [PubMed]
- Zafon, C.; Gil, J.; Pérez-González, B.; Jordà, M. DNA Methylation in Thyroid Cancer. Endocr. Relat. Cancer 2019, 26, R415–R439. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M.; Silva, J.M.; Dominguez, G.; Bonilla, F.; Matias-Guiu, X.; Lerma, E.; Bussaglia, E.; Prat, J.; Harkes, I.C.; Repasky, E.A.; et al. Promoter Hypermethylation and BRCA1 Inactivation in Sporadic Breast and Ovarian Tumors. J. Natl. Cancer Inst. 2000, 92, 564–569. [Google Scholar] [CrossRef] [PubMed]
- Saiselet, M.; Floor, S.; Tarabichi, M.; Dom, G.; Hébrant, A.; van Staveren, W.C.G.; Maenhaut, C. Thyroid Cancer Cell Lines: An Overview. Front. Endocrinol. 2012, 3, 133. [Google Scholar] [CrossRef] [PubMed]
- Van Staveren, W.C.G.; Solís, D.W.; Delys, L.; Duprez, L.; Andry, G.; Franc, B.; Thomas, G.; Libert, F.; Dumont, J.E.; Detours, V.; et al. Human Thyroid Tumor Cell Lines Derived from Different Tumor Types Present a Common Dedifferentiated Phenotype. Cancer Res. 2007, 67, 8113–8120. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wang, X.; Zhu, C.; Wang, K. A Review of Current Evidence about LncRNA MEG3: A Tumor Suppressor in Multiple Cancers. Front. Cell Dev. Biol. 2022, 10, 997633. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yue, P.; Zhou, T.; Zhang, F.; Wang, H.; Chen, X. LncRNA MEG3 Enhances 131I Sensitivity in Thyroid Carcinoma via Sponging MiR-182. Biomed. Pharmacother. 2018, 105, 1232–1239. [Google Scholar] [CrossRef] [PubMed]
- Dadafarin, S.; Rodríguez, T.C.; Carnazza, M.A.; Tiwari, R.K.; Moscatello, A.; Geliebter, J. MEG3 Expression Indicates Lymph Node Metastasis and Presence of Cancer-Associated Fibroblasts in Papillary Thyroid Cancer. Cells 2022, 11, 3181. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alves, L.F.; da Silva, I.N.; de Mello, D.C.; Fuziwara, C.S.; Guil, S.; Esteller, M.; Geraldo, M.V. Epigenetic Regulation of DLK1-DIO3 Region in Thyroid Carcinoma. Cells 2024, 13, 1001. https://doi.org/10.3390/cells13121001
Alves LF, da Silva IN, de Mello DC, Fuziwara CS, Guil S, Esteller M, Geraldo MV. Epigenetic Regulation of DLK1-DIO3 Region in Thyroid Carcinoma. Cells. 2024; 13(12):1001. https://doi.org/10.3390/cells13121001
Chicago/Turabian StyleAlves, Letícia F., Isabelle N. da Silva, Diego C. de Mello, Cesar S. Fuziwara, Sonia Guil, Manel Esteller, and Murilo V. Geraldo. 2024. "Epigenetic Regulation of DLK1-DIO3 Region in Thyroid Carcinoma" Cells 13, no. 12: 1001. https://doi.org/10.3390/cells13121001