
Tumour Microenvironment: The General Principles of Pathogenesis and Implications in Diffuse Large B Cell Lymphoma

Abstract
:1. Introduction
2. Components of Diffuse Large B-Cell Lymphoma
3. Tumour-Associated Macrophages (TAMs)
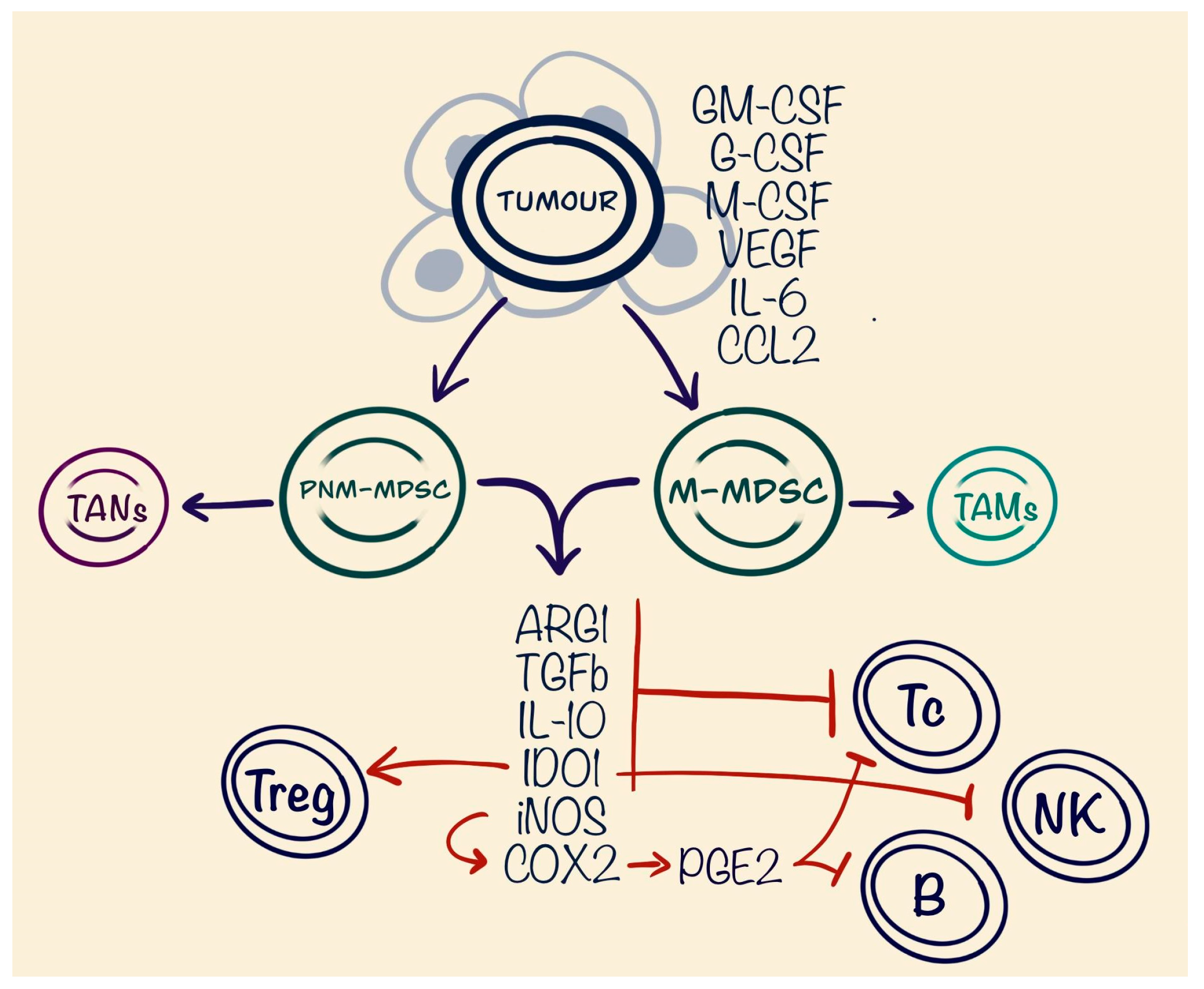
4. Myeloid-Derived Suppressor Cells (MDSCs)
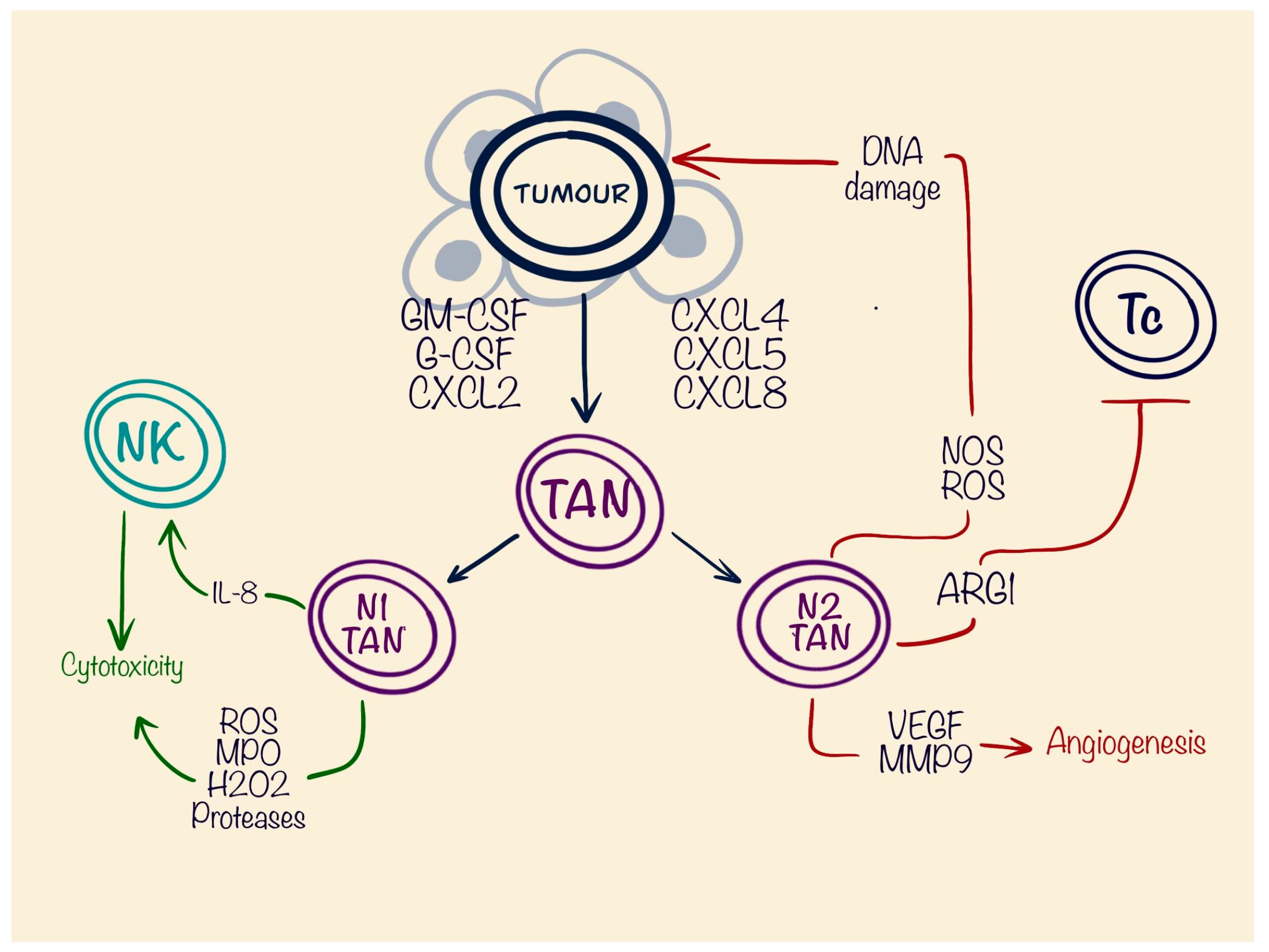
5. Tumour-Associated Neutrophils (TANs)
6. Cancer-Associated Fibroblasts (CAFs)
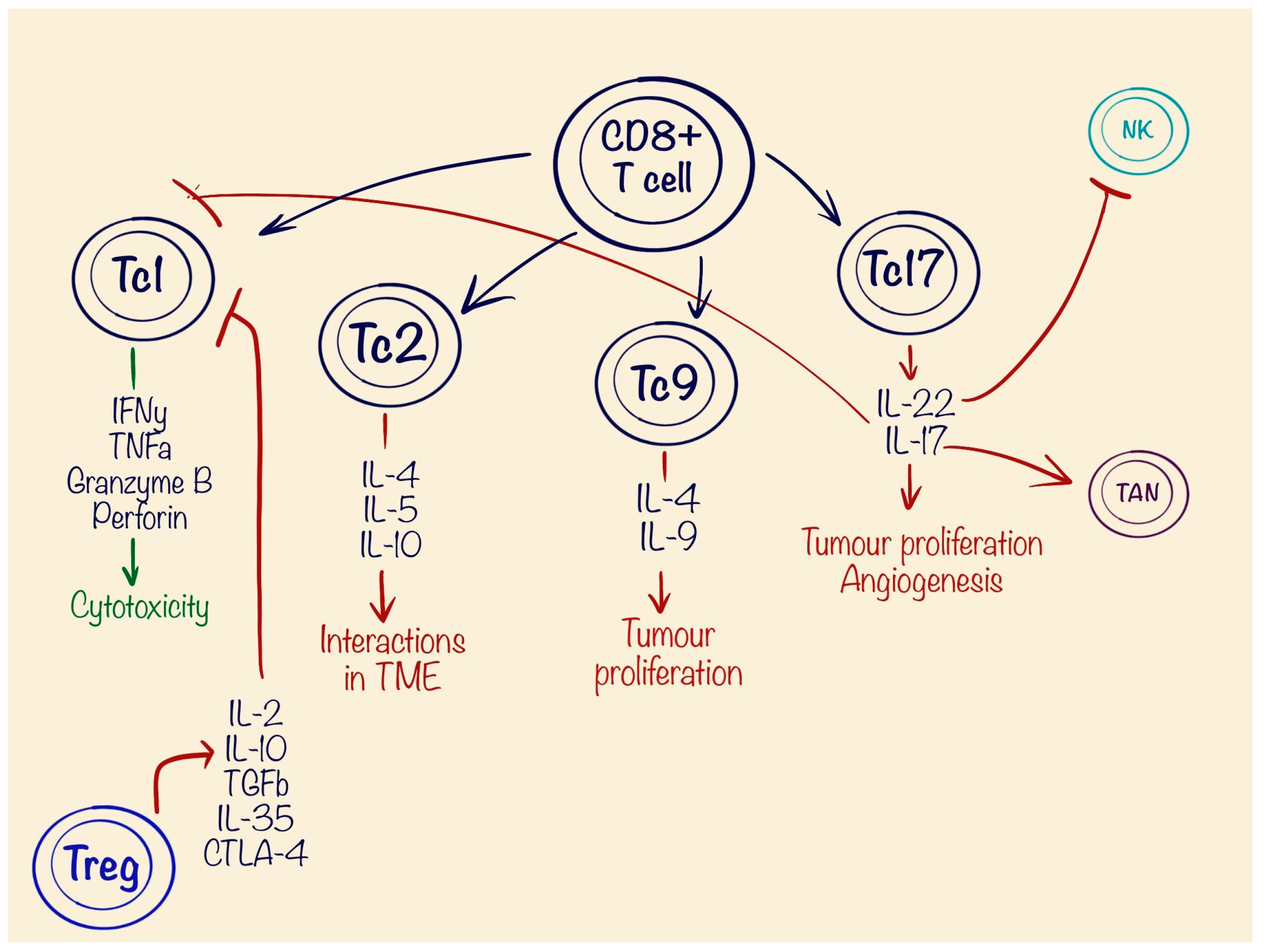
7. Role of T Lymphocytes in Tumour Pathogenesis
8. Targeting Tumour Microenvironment Elements in Diffuse Large B Cell Lymphoma
9. Role of the Tumour Microenvironment in Rare B Cell Lymphomas
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Munich Cancer Registry. Survival Statistics for Diffuse Large B-Cell Lymphoma (ICD-10 C83.3). Available online: https://www.tumorregister-muenchen.de/en/facts/surv/sC833_E-ICD-10-C83.3-Diff.-large-B-cell-lymphoma-survival.pdf (accessed on 24 April 2024).
- Wang, S.S. Epidemiology and etiology of diffuse large B-cell lymphoma. Semin. Hematol. 2023, 60, 255–266. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute. Diffuse Large B-Cell Lymphoma—Cancer Stat Facts. SEER. Available online: https://seer.cancer.gov/statfacts/html/dlbcl.html (accessed on 24 April 2024).
- Coiffier, B.; Lepage, E.; Brière, J.; Herbrecht, R.; Tilly, H.; Bouabdallah, R.; Morel, P.; Van Den Neste, E.; Salles, G.; Gaulard, P.; et al. CHOP Chemotherapy plus Rituximab Compared with CHOP Alone in Elderly Patients with Diffuse Large-B-Cell Lymphoma. N. Engl. J. Med. 2002, 346, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Raut, L.S.; Chakrabarti, P.P. Management of relapsed-refractory diffuse large B cell lymphoma. South Asian J. Cancer 2014, 3, 066–070. [Google Scholar] [CrossRef] [PubMed]
- Kesireddy, M.; Lunning, M. Relapsed or Refractory Diffuse Large B-Cell Lymphoma: “Dazed and Confused”. Oncology 2022, 36, 366–375. [Google Scholar] [CrossRef] [PubMed]
- Crump, M.; Neelapu, S.S.; Farooq, U.; Van Den Neste, E.; Kuruvilla, J.; Westin, J.; Link, B.K.; Hay, A.; Cerhan, J.R.; Zhu, L.; et al. Outcomes in refractory diffuse large B-cell lymphoma: Results from the international SCHOLAR-1 study. Blood 2017, 130, 1800–1808. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Chen, L. Tumor microenviroment and hepatocellular carcinoma metastasis. J. Gastroenterol. Hepatol. 2013, 28, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.W. Cell-of-Origin in Diffuse Large B-Cell Lymphoma: Are the Assays Ready for the Clinic? Am. Soc. Clin. Oncol. Educ. Book 2015, 35, e458–e466. [Google Scholar] [CrossRef] [PubMed]
- Camicia, R.; Winkler, H.C.; Hassa, P.O. Novel drug targets for personalized precision medicine in relapsed/refractory diffuse large B-cell lymphoma: A comprehensive review. Mol. Cancer 2015, 14, 1–62. [Google Scholar] [CrossRef]
- Nowakowski, G.S.; Czuczman, M.S. ABC, GCB, and Double-Hit Diffuse Large B-Cell Lymphoma: Does Subtype Make a Difference in Therapy Selection? Am. Soc. Clin. Oncol. Educ. Book 2015, 35, e449–e457. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, X.; Wang, X. Targeting the tumor microenvironment in B-cell lymphoma: Challenges and opportunities. J. Hematol. Oncol. 2021, 14, 1–17. [Google Scholar] [CrossRef]
- Cioroianu, A.I.; Stinga, P.I.; Sticlaru, L.; Cioplea, M.D.; Nichita, L.; Popp, C.; Staniceanu, F. Tumor Microenvironment in Diffuse Large B-Cell Lymphoma: Role and Prognosis. Anal. Cell. Pathol. 2019, 2019, 8586354. [Google Scholar] [CrossRef]
- Wu, K.; Lin, K.; Li, X.; Yuan, X.; Xu, P.; Ni, P.; Xu, D. Redefining Tumor-Associated Macrophage Subpopulations and Functions in the Tumor Microenvironment. Front. Immunol. 2020, 11, 1731. [Google Scholar] [CrossRef]
- Marini, O.; Spina, C.; Mimiola, E.; Cassaro, A.; Malerba, G.; Todeschini, G.; Perbellini, O.; Scupoli, M.; Carli, G.; Facchinelli, D.; et al. Identification of granulocytic myeloid-derived suppressor cells (G-MDSCs) in the peripheral blood of Hodgkin and non-Hodgkin lymphoma patients. Oncotarget 2016, 7, 27676–27688. [Google Scholar] [CrossRef]
- Manfroi, B.; Moreaux, J.; Righini, C.; Ghiringhelli, F.; Sturm, N.; Huard, B. Tumor-associated neutrophils correlate with poor prognosis in diffuse large B-cell lymphoma patients. Blood Cancer J. 2018, 8, 66. [Google Scholar] [CrossRef]
- Haro, M.; Orsulic, S. A Paradoxical Correlation of Cancer-Associated Fibroblasts with Survival Outcomes in B-Cell Lymphomas and Carcinomas. Front. Cell Dev. Biol. 2018, 6, 98. [Google Scholar] [CrossRef]
- Munir, M.T.; Kay, M.K.; Kang, M.H.; Rahman, M.; Al-Harrasi, A.; Choudhury, M.; Moustaid-Moussa, N.; Hussain, F.; Rahman, S.M. Tumor-Associated Macrophages as Multifaceted Regulators of Breast Tumor Growth. Int. J. Mol. Sci. 2021, 22, 6526. [Google Scholar] [CrossRef]
- Wang, H.; Tian, T.; Zhang, J. Tumor-Associated Macrophages (TAMs) in Colorectal Cancer (CRC): From Mechanism to Therapy and Prognosis. Int. J. Mol. Sci. 2021, 22, 8470. [Google Scholar] [CrossRef]
- Xu, M.; Wang, Y.; Xia, R.; Wei, Y.; Wei, X. Role of the CCL2-CCR2 signalling axis in cancer: Mechanisms and therapeutic targeting. Cell Prolif. 2021, 54, e13115. [Google Scholar] [CrossRef]
- Boutilier, A.J.; Elsawa, S.F. Macrophage Polarization States in the Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 6995. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, L.; Liu, J.; Dang, P.; Hu, S.; Yuan, W.; Sun, Z.; Liu, Y.; Wang, C. Roles of tumor-associated macrophages in anti-PD-1/PD-L1 immunotherapy for solid cancers. Mol. Cancer 2023, 22, 58. [Google Scholar] [CrossRef] [PubMed]
- Albini, A.; Bruno, A.; Noonan, D.M.; Mortara, L. Contribution to Tumor Angiogenesis from Innate Immune Cells Within the Tumor Microenvironment: Implications for Immunotherapy. Front. Immunol. 2018, 9, 527. [Google Scholar] [CrossRef]
- Salmaninejad, A.; Valilou, S.F.; Soltani, A.; Ahmadi, S.; Abarghan, Y.J.; Rosengren, R.J.; Sahebkar, A. Tumor-associated macrophages: Role in cancer development and therapeutic implications. Cell. Oncol. 2019, 42, 591–608. [Google Scholar] [CrossRef]
- Xie, Y.; Yang, H.; Yang, C.; He, L.; Zhang, X.; Peng, L.; Zhu, H.; Gao, L. Role and Mechanisms of Tumor-Associated Macrophages in Hematological Malignancies. Front. Oncol. 2022, 12, 933666. [Google Scholar] [CrossRef]
- Waqas, S.F.H.; Ampem, G.; Röszer, T. Analysis of IL-4/STAT6 Signaling in Macrophages. Methods Mol. Biol. 2019, 1966, 211–224. [Google Scholar] [CrossRef]
- Huber, R.; Meier, B.; Otsuka, A.; Fenini, G.; Satoh, T.; Gehrke, S.; Widmer, D.; Levesque, M.P.; Mangana, J.; Kerl, K.; et al. Tumour hypoxia promotes melanoma growth and metastasis via High Mobility Group Box-1 and M2-like macrophages. Sci. Rep. 2016, 6, 29914. [Google Scholar] [CrossRef]
- Carlini, V.; Noonan, D.M.; Abdalalem, E.; Goletti, D.; Sansone, C.; Calabrone, L.; Albini, A. The multifaceted nature of IL-10: Regulation, role in immunological homeostasis and its relevance to cancer, COVID-19 and post-COVID conditions. Front. Immunol. 2023, 14, 1161067. [Google Scholar] [CrossRef]
- Basak, U.; Sarkar, T.; Mukherjee, S.; Chakraborty, S.; Dutta, A.; Dutta, S.; Nayak, D.; Kaushik, S.; Das, T.; Sa, G. Tumor-associated macrophages: An effective player of the tumor microenvironment. Front. Immunol. 2023, 14, 1295257. [Google Scholar] [CrossRef]
- Lin, Y.; Xu, J.; Lan, H. Tumor-associated macrophages in tumor metastasis: Biological roles and clinical therapeutic applications. J. Hematol. Oncol. 2019, 12, 76. [Google Scholar] [CrossRef]
- Ries, C.H.; Cannarile, M.A.; Hoves, S.; Benz, J.; Wartha, K.; Runza, V.; Rey-Giraud, F.; Pradel, L.P.; Feuerhake, F.; Klaman, I.; et al. Targeting Tumor-Associated Macrophages with Anti-CSF-1R Antibody Reveals a Strategy for Cancer Therapy. Cancer Cell 2014, 25, 846–859. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Roca, C.A.; Cassier, P.A.; Italiano, A.; Cannarile, M.; Ries, C.; Brillouet, A.; Mueller, C.; Jegg, A.M.; Meneses-Lorente, G.; Baehner, M.; et al. Phase I study of RG7155, a novel anti-CSF1R antibody, in patients with advanced/metastatic solid tumors. J. Clin. Oncol. 2015, 33, 15. [Google Scholar] [CrossRef]
- Linehan, D.; Noel, M.S.; Hezel, A.F.; Wang-Gillam, A.; Eskens, F.; Sleijfer, S.; Desar, I.M.E.; Erdkamp, F.; Wilmink, J.; Diehl, J.; et al. Overall survival in a trial of orally administered CCR2 inhibitor CCX872 in locally advanced/metastatic pancreatic cancer: Correlation with blood monocyte counts. J. Clin. Oncol. 2018, 36, 92. [Google Scholar] [CrossRef]
- Wada, N.; Zaki, M.A.A.; Hori, Y.; Hashimoto, K.; Tsukaguchi, M.; Tatsumi, Y.; Ishikawa, J.; Tominaga, N.; Sakoda, H.; Take, H.; et al. Tumour-associated macrophages in diffuse large B-cell lymphoma: A study of the Osaka Lymphoma Study Group. Histopathology 2011, 60, 313–319. [Google Scholar] [CrossRef]
- Riihijärvi, S.; Fiskvik, I.; Taskinen, M.; Vajavaara, H.; Tikkala, M.; Yri, O.; Karjalainen-Lindsberg, M.-L.; Delabie, J.; Smeland, E.; Holte, H.; et al. Prognostic influence of macrophages in patients with diffuse large B-cell lymphoma: A correlative study from a Nordic phase II trial. Haematologica 2014, 100, 238–245. [Google Scholar] [CrossRef]
- Hasselblom, S.; Hansson, U.; Sigurdardottir, M.; Nilsson-Ehle, H.; Ridell, B.; Andersson, P. Expression of CD68+ tumor-associated macrophages in patients with diffuse large B-cell lymphoma and its relation to prognosis. Pathol. Int. 2008, 58, 529–532. [Google Scholar] [CrossRef]
- Chang, J.E.; Seo, S.; Kim, K.M.; Werndli, J.E.; Bottner, W.A.; Rodrigues, G.A.; Sanchez, F.A.; Saphner, T.J.; Longo, W.L.; Kahl, B.S. Rituximab and CHOP Chemotherapy Plus GM-CSF for Previously Untreated Diffuse Large B-Cell Lymphoma in the Elderly: A Wisconsin Oncology Network Study. Clin. Lymphoma Myeloma Leuk. 2010, 10, 379–384. [Google Scholar] [CrossRef]
- Yonemitsu, K.; Pan, C.; Fujiwara, Y.; Miyasato, Y.; Shiota, T.; Yano, H.; Hosaka, S.; Tamada, K.; Yamamoto, Y.; Komohara, Y. GM-CSF derived from the inflammatory microenvironment potentially enhanced PD-L1 expression on tumor-associated macrophages in human breast cancer. Sci. Rep. 2022, 12, 12007. [Google Scholar] [CrossRef]
- Wu, Y.; Yi, M.; Niu, M.; Mei, Q.; Wu, K. Myeloid-derived suppressor cells: An emerging target for anticancer immunotherapy. Mol. Cancer 2022, 21, 184. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Bronte, V.; Chen, S.-H.; Colombo, M.P.; Ochoa, A.; Ostrand-Rosenberg, S.; Schreiber, H. The Terminology Issue for Myeloid-Derived Suppressor Cells. Cancer Res. 2007, 67, 425. [Google Scholar] [CrossRef]
- Porembka, M.R.; Mitchem, J.B.; Belt, B.A.; Hsieh, C.-S.; Lee, H.-M.; Herndon, J.; Gillanders, W.E.; Linehan, D.C.; Goedegebuure, P. Pancreatic adenocarcinoma induces bone marrow mobilization of myeloid-derived suppressor cells which promote primary tumor growth. Cancer Immunol. Immunother. 2012, 61, 1373–1385. [Google Scholar] [CrossRef] [PubMed]
- Capietto, A.-H.; Kim, S.; Sanford, D.E.; Linehan, D.C.; Hikida, M.; Kumosaki, T.; Novack, D.V.; Faccio, R. Down-regulation of PLCγ2–β-catenin pathway promotes activation and expansion of myeloid-derived suppressor cells in cancer. J. Exp. Med. 2013, 210, 2257–2271. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016, 37, 208–220. [Google Scholar] [CrossRef]
- Veglia, F.; Perego, M.; Gabrilovich, D. Myeloid-derived suppressor cells coming of age. Nat. Immunol. 2018, 19, 108–119. [Google Scholar] [CrossRef]
- Jin, S.; Yang, Z.; Hao, X.; Tang, W.; Ma, W.; Zong, H. Roles of HMGB1 in regulating myeloid-derived suppressor cells in the tumor microenvironment. Biomark. Res. 2020, 8, 21. [Google Scholar] [CrossRef]
- Su, Y.-L.; Banerjee, S.; White, S.V.; Kortylewski, M. STAT3 in Tumor-Associated Myeloid Cells: Multitasking to Disrupt Immunity. Int. J. Mol. Sci. 2018, 19, 1803. [Google Scholar] [CrossRef]
- Hix, L.M.; Karavitis, J.; Khan, M.W.; Shi, Y.H.; Khazaie, K.; Zhang, M. Tumor STAT1 Transcription Factor Activity Enhances Breast Tumor Growth and Immune Suppression Mediated by Myeloid-derived Suppressor Cells. J. Biol. Chem. 2013, 288, 11676–11688. [Google Scholar] [CrossRef]
- Condamine, T.; Mastio, J.; I Gabrilovich, D. Transcriptional regulation of myeloid-derived suppressor cells. J. Leukoc. Biol. 2015, 98, 913–922. [Google Scholar] [CrossRef]
- Gabrilovich, D.I. Myeloid-Derived Suppressor Cells. Cancer Immunol. Res. 2017, 5, 3–8. [Google Scholar] [CrossRef]
- Bronte, V.; Brandau, S.; Chen, S.-H.; Colombo, M.P.; Frey, A.B.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S.; et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016, 7, 12150. [Google Scholar] [CrossRef]
- Ming, Z.; Zou, Z.; Cai, K.; Xu, Y.; Chen, X.; Yi, W.; Luo, J.; Luo, Z. ARG1 functions as a tumor suppressor in breast cancer. Acta Biochim. Biophys. Sin. 2020, 52, 1257–1264. [Google Scholar] [CrossRef] [PubMed]
- Menjivar, R.E.; Nwosu, Z.C.; Du, W.; Donahue, K.L.; Hong, H.S.; Espinoza, C.; Brown, K.; Velez-Delgado, A.; Yan, W.; Lima, F.; et al. Arginase 1 is a key driver of immune suppression in pancreatic cancer. eLife 2023, 12, e80721. [Google Scholar] [CrossRef] [PubMed]
- Sreeramkumar, V.; Fresno, M.; Cuesta, N. Prostaglandin E2 and T cells: Friends or foes? Immunol. Cell Biol. 2011, 90, 579–586. [Google Scholar] [CrossRef]
- Dahmani, A.; Delisle, J.-S. TGF-β in T Cell Biology: Implications for Cancer Immunotherapy. Cancers 2018, 10, 194. [Google Scholar] [CrossRef]
- Hornyák, L.; Dobos, N.; Koncz, G.; Karányi, Z.; Páll, D.; Szabó, Z.; Halmos, G.; Székvölgyi, L. The Role of Indoleamine-2,3-Dioxygenase in Cancer Development, Diagnostics, and Therapy. Front. Immunol. 2018, 9, 151. [Google Scholar] [CrossRef]
- Condamine, T.; Kumar, V.; Ramachandran, I.R.; Youn, J.-I.; Celis, E.; Finnberg, N.; El-Deiry, W.S.; Winograd, R.; Vonderheide, R.H.; English, N.R.; et al. ER stress regulates myeloid-derived suppressor cell fate through TRAIL-R–mediated apoptosis. J. Clin. Investig. 2014, 124, 2626–2639. [Google Scholar] [CrossRef]
- Palumbo, G.A.; Parrinello, N.L.; Giallongo, C.; D’amico, E.; Zanghì, A.; Puglisi, F.; Conticello, C.; Chiarenza, A.; Tibullo, D.; Di Raimondo, F.; et al. Monocytic Myeloid Derived Suppressor Cells in Hematological Malignancies. Int. J. Mol. Sci. 2019, 20, 5459. [Google Scholar] [CrossRef]
- Zhou, X.; Fang, D.; Liu, H.; Ou, X.; Zhang, C.; Zhao, Z.; Zhao, S.; Peng, J.; Cai, S.; He, Y.; et al. PMN-MDSCs accumulation induced by CXCL1 promotes CD8+ T cells exhaustion in gastric cancer. Cancer Lett. 2022, 532, 215598. [Google Scholar] [CrossRef]
- Sieminska, I.; Baran, J. Myeloid-Derived Suppressor Cells in Colorectal Cancer. Front. Immunol. 2020, 11, 1526. [Google Scholar] [CrossRef]
- Jen, E.Y.; Ko, C.-W.; Lee, J.E.; Del Valle, P.L.; Aydanian, A.; Jewell, C.; Norsworthy, K.J.; Przepiorka, D.; Nie, L.; Liu, J.; et al. FDA Approval: Gemtuzumab Ozogamicin for the Treatment of Adults with Newly Diagnosed CD33-Positive Acute Myeloid Leukemia. Clin. Cancer Res. 2018, 24, 3242–3246. [Google Scholar] [CrossRef]
- Meyer, C.; Sevko, A.; Ramacher, M.; Bazhin, A.V.; Falk, C.S.; Osen, W.; Borrello, I.; Kato, M.; Schadendorf, D.; Baniyash, M.; et al. Chronic inflammation promotes myeloid-derived suppressor cell activation blocking antitumor immunity in transgenic mouse melanoma model. Proc. Natl. Acad. Sci. USA 2011, 108, 17111–17116. [Google Scholar] [CrossRef]
- Zou, S.; Tong, Q.; Liu, B.; Huang, W.; Tian, Y.; Fu, X. Targeting STAT3 in Cancer Immunotherapy. Mol. Cancer 2020, 19, 1–19. [Google Scholar] [CrossRef]
- Azzaoui, I.; Uhel, F.; Rossille, D.; Pangault, C.; Dulong, J.; Le Priol, J.; Lamy, T.; Houot, R.; Le Gouill, S.; Cartron, G.; et al. T-cell defect in diffuse large B-cell lymphomas involves expansion of myeloid-derived suppressor cells. Blood 2016, 128, 1081–1092. [Google Scholar] [CrossRef]
- Xu, T.; Chai, J.; Wang, K.; Jia, Q.; Liu, Y.; Wang, Y.; Xu, J.; Yu, K.; Zhao, D.; Ma, J.; et al. Tumor Immune Microenvironment Components and Checkpoint Molecules in Anaplastic Variant of Diffuse Large B-Cell Lymphoma. Front. Oncol. 2021, 11, 638154. [Google Scholar] [CrossRef]
- Que, H.; Fu, Q.; Lan, T.; Tian, X.; Wei, X. Tumor-associated neutrophils and neutrophil-targeted cancer therapies. Biochim. Biophys. Acta BBA—Rev. Cancer 2022, 1877, 188762. [Google Scholar] [CrossRef]
- Jensen, T.O.; Schmidt, H.; Møller, H.J.; Donskov, F.; Høyer, M.; Sjoegren, P.; Christensen, J.; Steiniche, T. Intratumoral neutrophils and plasmacy-toid dendritic cells indicate poor prognosis and are associated with pSTAT3 expression in AJCC stage I/II melanoma. Cancer 2012, 118, 2476–2485. [Google Scholar] [CrossRef]
- Wislez, M.; Rabbe, N.; Marchal, J.; Milleron, B.; Crestani, B.; Mayaud, C.; Antoine, M.; Soler, P.; Cadranel, J. Hepatocyte growth factor production by neutrophils infiltrating bronchioloalveolar subtype pulmonary adenocarcinoma: Role in tumor progression and death. Cancer Res. 2003, 63, 1405–1412. [Google Scholar]
- Trellakis, S.; Bruderek, K.; Dumitru, C.A.; Gholaman, H.; Gu, X.; Bankfalvi, A.; Scherag, A.; Hütte, J.; Dominas, N.; Lehnerdt, G.F.; et al. Polymorphonuclear granulocytes in human head and neck cancer: Enhanced inflammatory activity, modulation by cancer cells and expansion in advanced disease. Int. J. Cancer 2010, 129, 2183–2193. [Google Scholar] [CrossRef]
- Hedrick, C.C.; Malanchi, I. Neutrophils in cancer: Heterogeneous and multifaceted. Nat. Rev. Immunol. 2021, 22, 173–187. [Google Scholar] [CrossRef]
- Ocana, A.; Nieto-Jiménez, C.; Pandiella, A.; Templeton, A.J. Neutrophils in cancer: Prognostic role and therapeutic strategies. Mol. Cancer 2017, 16, 137. [Google Scholar] [CrossRef]
- Dumitru, C.A.; Moses, K.; Trellakis, S.; Lang, S.; Brandau, S. Neutrophils and granulocytic myeloid-derived suppressor cells: Immunophenotyping, cell biology and clinical relevance in human oncology. Cancer Immunol. Immunother. 2012, 61, 1155–1167. [Google Scholar] [CrossRef]
- Jaillon, S.; Ponzetta, A.; Di Mitri, D.; Santoni, A.; Bonecchi, R.; Mantovani, A. Neutrophil diversity and plasticity in tumour progression and therapy. Nat. Rev. Cancer 2020, 20, 485–503. [Google Scholar] [CrossRef]
- Masucci, M.T.; Minopoli, M.; Carriero, M.V. Tumor Associated Neutrophils. Their Role in Tumorigenesis, Metastasis, Prognosis and Therapy. Front. Oncol. 2019, 9, 1146. [Google Scholar] [CrossRef]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef]
- Riise, R.E.; Bernson, E.; Aurelius, J.; Martner, A.; Pesce, S.; Della Chiesa, M.; Marcenaro, E.; Bylund, J.; Hellstrand, K.; Moretta, L.; et al. TLR-Stimulated Neutrophils Instruct NK Cells To Trigger Dendritic Cell Maturation and Promote Adaptive T Cell Responses. J. Immunol. 2015, 195, 1121–1128. [Google Scholar] [CrossRef]
- Yang, R.; Wang, X.; Ma, C.; Zhang, Z.; Liu, N.; Ma, X.; Zhang, Y.; Wang, X.; Liu, Y. Tumor-infiltrating neutrophils and peripheral neutrophil-to-lymphocyte ratio conversely predicted the prognosis of patients with non-small cell lung cancer. Cell. Immunol. 2022, 379, 104588. [Google Scholar] [CrossRef]
- Quaas, A.; Pamuk, A.; Klein, S.; Quantius, J.; Rehkaemper, J.; Barutcu, A.G.; Rueschoff, J.; Zander, T.; Gebauer, F.; Hillmer, A.; et al. Sex-specific prognostic effect of CD66b-positive tumor-infiltrating neutrophils (TANs) in gastric and esophageal adenocarcinoma. Gastric Cancer 2021, 24, 1213–1226. [Google Scholar] [CrossRef]
- Sandhu, J.K.; Privora, H.F.; Wenckebach, G.; Birnboim, H.C. Neutrophils, Nitric Oxide Synthase, and Mutations in the Mutatect Murine Tumor Model. Am. J. Pathol. 2000, 156, 509–518. [Google Scholar] [CrossRef]
- Haqqani, A.S.; Sandhu, J.K.; Birnboim, H.C. Expression of Interleukin-8 Promotes Neutrophil Infiltration and Genetic Instability in Mutatect Tumors. Neoplasia 2000, 2, 561–568. [Google Scholar] [CrossRef]
- Butin-Israeli, V.; Bui, T.M.; Wiesolek, H.L.; Mascarenhas, L.; Lee, J.J.; Mehl, L.C.; Knutson, K.R.; Adam, S.A.; Goldman, R.D.; Beyder, A.; et al. Neutrophil-induced genomic instability impedes resolution of inflammation and wound healing. J. Clin. Investig. 2019, 129, 712–726. [Google Scholar] [CrossRef]
- Moloney, J.N.; Cotter, T.G. ROS signalling in the biology of cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef]
- Antuamwine, B.B.; Bosnjakovic, R.; Hofmann-Vega, F.; Wang, X.; Theodosiou, T.; Iliopoulos, I.; Brandau, S. N1 versus N2 and PMN-MDSC: A critical appraisal of current concepts on tumor-associated neutrophils and new directions for human oncology. Immunol. Rev. 2022, 314, 250–279. [Google Scholar] [CrossRef]
- Deryugina, E.I.; Zajac, E.; Juncker-Jensen, A.; Kupriyanova, T.A.; Welter, L.; Quigley, J.P. Tissue-Infiltrating Neutrophils Constitute the Major In Vivo Source of Angiogenesis-Inducing MMP-9 in the Tumor Microenvironment. Neoplasia 2014, 16, 771–788. [Google Scholar] [CrossRef] [PubMed]
- Kotlov, N.; Bagaev, A.; Revuelta, M.V.; Phillip, J.M.; Cacciapuoti, M.T.; Antysheva, Z.; Svekolkin, V.; Tikhonova, E.; Miheecheva, N.; Kuzkina, N.; et al. Clinical and Biological Subtypes of B-cell Lymphoma Revealed by Microenvironmental Signatures. Cancer Discov. 2021, 11, 1468–1489. [Google Scholar] [CrossRef] [PubMed]
- Biffi, G.; Tuveson, D.A. Diversity and Biology of Cancer-Associated Fibroblasts. Physiol. Rev. 2020, 101, 147–176. [Google Scholar] [CrossRef] [PubMed]
- Ohlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef] [PubMed]
- Tran, L.L.; Dang, T.; Thomas, R.; Rowley, D.R. ELF3 mediates IL-1α induced differentiation of mesenchymal stem cells to inflammatory iCAFs. Stem Cells 2021, 39, 1766–1777. [Google Scholar] [CrossRef] [PubMed]
- Cords, L.; Tietscher, S.; Anzeneder, T.; Langwieder, C.; Rees, M.; de Souza, N.; Bodenmiller, B. Cancer-associated fibroblast classification in single-cell and spatial proteomics data. Nat. Commun. 2023, 14, 1–13. [Google Scholar] [CrossRef]
- Yang, X.; Lin, Y.; Shi, Y.; Li, B.; Liu, W.; Yin, W.; Dang, Y.; Chu, Y.; Fan, J.; He, R. FAP Promotes Immunosuppression by Cancer-Associated Fibroblasts in the Tumor Microenvironment via STAT3–CCL2 Signaling. Cancer Res. 2016, 76, 4124–4135. [Google Scholar] [CrossRef] [PubMed]
- Nurmik, M.; Ullmann, P.; Rodriguez, F.; Haan, S.; Letellier, E. In search of definitions: Cancer-associated fibroblasts and their markers. Int. J. Cancer 2019, 146, 895–905. [Google Scholar] [CrossRef]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef]
- Liu, X.; Yao, L.; Qu, J.; Liu, L.; Lu, N.; Wang, J.; Zhang, J. Cancer-associated fibroblast infiltration in gastric cancer: The discrepancy in subtypes pathways and immunosuppression. J. Transl. Med. 2021, 19, 1–16. [Google Scholar] [CrossRef]
- E Hegab, A.; Ozaki, M.; Kameyama, N.; Gao, J.; Kagawa, S.; Yasuda, H.; Soejima, K.; Yin, Y.; Guzy, R.D.; Nakamura, Y.; et al. Effect of FGF/FGFR pathway blocking on lung adenocarcinoma and its cancer-associated fibroblasts. J. Pathol. 2019, 249, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Kasashima, H.; Fukui, Y.; Tsujio, G.; Yashiro, M.; Maeda, K. The heterogeneity of cancer-associated fibroblast subpopulations: Their origins, biomarkers, and roles in the tumor microenvironment. Cancer Sci. 2022, 114, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Chakravarthy, A.; Khan, L.; Bensler, N.P.; Bose, P.; De Carvalho, D.D. TGF-beta-associated extracellular matrix genes link cancer-associated fibroblasts to immune evasion and immunotherapy failure. Nat. Commun. 2018, 9, 4692. [Google Scholar] [CrossRef] [PubMed]
- Desbois, M.; Wang, Y. Cancer-associated fibroblasts: Key players in shaping the tumor immune microenvironment. Immunol. Rev. 2021, 302, 241–258. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.J.; Datta, A.; Littlefield, C.; Kalra, A.; Chapron, C.; Wawersik, S.; Dagbay, K.B.; Brueckner, C.T.; Nikiforov, A.; Danehy, F.T.; et al. Selective inhibition of TGFβ1 activation overcomes primary resistance to checkpoint blockade therapy by altering tumor immune landscape. Sci. Transl. Med. 2020, 12, eaay8456. [Google Scholar] [CrossRef] [PubMed]
- Binabaj, M.M.; Asgharzadeh, F.; Rahmani, F.; Al-Asady, A.M.; Hashemzehi, M.; Soleimani, A.; Avan, A.; Mehraban, S.; Ghorbani, E.; Ryzhikov, M.; et al. Vactosertib potently improves anti-tumor properties of 5-FU for colon cancer. DARU J. Pharm. Sci. 2023, 31, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Myers, J.; Tomchuck, S.; Bonner, M.; Eid, S.; Kingsley, D.; VanHeyst, K.; Kim, S.-J.; Kim, B.-G.; Huang, A.Y. Oral TGF-βR1 inhibitor Vactosertib promotes osteosarcoma regression by targeting tumor proliferation and enhancing anti-tumor immunity. Res. Sq. 2023. preprint. [Google Scholar] [CrossRef]
- Lei, X.; Lei, Y.; Li, J.-K.; Du, W.-X.; Li, R.-G.; Yang, J.; Li, J.; Li, F.; Tan, H.-B. Immune cells within the tumor microenvironment: Biological functions and roles in cancer immunotherapy. Cancer Lett. 2019, 470, 126–133. [Google Scholar] [CrossRef]
- Kwok, G.; Yau, T.C.C.; Chiu, J.W.; Tse, E.; Kwong, Y.-L. Pembrolizumab (Keytruda). Hum. Vaccines Immunother. 2016, 12, 2777–2789. [Google Scholar] [CrossRef]
- Zhang, X.; Zhu, L.; Zhang, H.; Chen, S.; Xiao, Y. CAR-T Cell Therapy in Hematological Malignancies: Current Opportunities and Challenges. Front. Immunol. 2022, 13, 927153. [Google Scholar] [CrossRef]
- de Assis, L.H.; El Fassi, D.; Hutchings, M. Bispecific antibody therapies. Hematology 2023, 2023, 216–222. [Google Scholar] [CrossRef]
- Su, J.; Liu, X.; Guo, S.; Zhang, J.; Wei, X.; Li, X. Nanobodies: A new potential for prostate cancer treatment. J. Cancer Res. Clin. Oncol. 2023, 149, 6703–6710. [Google Scholar] [CrossRef] [PubMed]
- Mittrücker, H.-W.; Visekruna, A.; Huber, M. Heterogeneity in the Differentiation and Function of CD8+ T Cells. Arch. Immunol. Ther. Exp. 2014, 62, 449–458. [Google Scholar] [CrossRef]
- Colombo, M.P.; Trinchieri, G. Interleukin-12 in anti-tumor immunity and immunotherapy. Cytokine Growth Factor Rev. 2002, 13, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Germann, T.; Rüde, E. lnterleukin-12. Int. Arch. Allergy Immunol. 1995, 108, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Kemp, R.A.; Ronchese, F. Tumor-Specific Tc1, But Not Tc2, Cells Deliver Protective Antitumor Immunity. J. Immunol. 2001, 167, 6497–6502. [Google Scholar] [CrossRef]
- Visekruna, A.; Ritter, J.; Scholz, T.; Campos, L.; Guralnik, A.; Poncette, L.; Raifer, H.; Hagner, S.; Garn, H.; Staudt, V.; et al. Tc9 cells, a new subset of CD8+ T cells, support Th2-mediated airway inflammation. Eur. J. Immunol. 2012, 43, 606–618. [Google Scholar] [CrossRef]
- Wan, J.; Wu, Y.; Ji, X.; Huang, L.; Cai, W.; Su, Z.; Wang, S.; Xu, H. IL-9 and IL-9-producing cells in tumor immunity. Cell Commun. Signal. 2020, 18, 1–11. [Google Scholar] [CrossRef]
- He, Y.; Dong, L.; Cao, Y.; Bi, Y.; Liu, G. IL-9 and Th9 Cells in Tumor Immunity. Adv. Exp. Med. Biol. 2020, 1240, 35–46. [Google Scholar] [CrossRef]
- Lv, X.; Feng, L.; Ge, X.; Lu, K.; Wang, X. Interleukin-9 promotes cell survival and drug resistance in diffuse large B-cell lymphoma. J. Exp. Clin. Cancer Res. 2016, 35, 1–9. [Google Scholar] [CrossRef]
- Akbay, E.A.; Koyama, S.; Liu, Y.; Dries, R.; Bufe, L.E.; Silkes, M.; Alam, M.; Magee, D.M.; Jones, R.; Jinushi, M.; et al. Interleukin-17A Promotes Lung Tumor Progression through Neutrophil Attraction to Tumor Sites and Mediating Resistance to PD-1 Blockade. J. Thorac. Oncol. 2017, 12, 1268–1279. [Google Scholar] [CrossRef] [PubMed]
- Razi, S.; Noveiry, B.B.; Keshavarz-Fathi, M.; Rezaei, N. IL-17 and colorectal cancer: From carcinogenesis to treatment. Cytokine 2019, 116, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Briukhovetska, D.; Suarez-Gosalvez, J.; Voigt, C.; Markota, A.; Giannou, A.D.; Schübel, M.; Jobst, J.; Zhang, T.; Dörr, J.; Märkl, F.; et al. T cell-derived interleukin-22 drives the expression of CD155 by cancer cells to suppress NK cell function and promote metastasis. Immunity 2023, 56, 143–161. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Mikami, N.; Wing, J.B.; Tanaka, A.; Ichiyama, K.; Ohkura, N. Regulatory T Cells and Human Disease. Annu. Rev. Immunol. 2020, 38, 541–566. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Sakaguchi, S. Regulatory T cells in cancer immunotherapy. Cell Res. 2016, 27, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Shang, B.; Liu, Y.; Jiang, S.-J.; Liu, Y. Prognostic value of tumor-infiltrating FoxP3+ regulatory T cells in cancers: A systematic review and meta-analysis. Sci. Rep. 2015, 5, 15179. [Google Scholar] [CrossRef] [PubMed]
- Ohue, Y.; Nishikawa, H. Regulatory T (Treg) cells in cancer: Can Treg cells be a new therapeutic target? Cancer Sci. 2019, 110, 2080–2089. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.-J.; Zhao, J.-W.; Zhang, D.-H.; Zheng, A.-H.; Wu, G.-Q. Immunotherapy of Cancer by Targeting Regulatory T cells. Int. Immunopharmacol. 2022, 104, 108469. [Google Scholar] [CrossRef]
- Hatzioannou, A.; Boumpas, A.; Papadopoulou, M.; Papafragkos, I.; Varveri, A.; Alissafi, T.; Verginis, P. Regulatory T Cells in Autoimmunity and Cancer: A Duplicitous Lifestyle. Front. Immunol. 2021, 12, 731947. [Google Scholar] [CrossRef]
- Marchesi, F.; Cirillo, M.; Bianchi, A.; Gately, M.; Olimpieri, O.M.; Cerchiara, E.; Renzi, D.; Micera, A.; Balzamino, B.O.; Bonini, S.; et al. High density of CD68+/CD163+ tumour-associated macrophages (M2-TAM) at diagnosis is significantly correlated to unfavorable prognostic factors and to poor clinical outcomes in patients with diffuse large B-cell lymphoma. Hematol. Oncol. 2014, 33, 110–112. [Google Scholar] [CrossRef]
- Khalifa, K.A.; Badawy, H.M.; Radwan, W.M.; Shehata, M.A.; Bassuoni, M.A. CD14+HLA-DR low/− monocytes as indicator of disease aggressiveness in B-cell non-Hodgkin lymphoma. Int. J. Lab. Hematol. 2014, 36, 650–655. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.-Q.; Wen, Z.-F.; Huang, Q.-T.; Li, Q.; Zhai, Z.-M.; Li, Y.-L. CC chemokine receptor 2 (CCR2) expression promotes diffuse large B-Cell lymphoma survival and invasion. Mod. Pathol. 2022, 102, 1377–1388. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Bethesda (MD): National Library of Medicine (US). Identifier NCT00356460, Safety and Efficacy Study of GC1008 to Treat Renal Cell Carcinoma or Malignant Melanoma. 19 March 2014. Available online: https://clinicaltrials.gov/study/NCT00356460 (accessed on 30 May 2024).
- ClinicalTrials.gov. Bethesda (MD): National Library of Medicine (US). Identifier NCT01401062, Fresolimumab and Radiotherapy in Metastatic Breast Cancer; 5 March 2019. Available online: https://clinicaltrials.gov/study/NCT01401062 (accessed on 30 May 2024).
- ClinicalTrials.gov. Bethesda (MD): National Library of Medicine (US). Identifier NCT01112293, Anti-TGF Monoclonal Antibody (GC1008) in Relapsed Malignant Pleural Mesothelioma. 10 April 2020. Available online: https://clinicaltrials.gov/study/NCT01112293 (accessed on 30 May 2024).
- Paz-Ares, L.; Kim, T.M.; Vicente, D.; Felip, E.; Lee, D.H.; Lee, K.H.; Lin, C.-C.; Flor, M.J.; Di Nicola, M.; Alvarez, R.M.; et al. Bintrafusp Alfa, a Bifunctional Fusion Protein Targeting TGF-β and PD-L1, in Second-Line Treatment of Patients With NSCLC: Results from an Expansion Cohort of a Phase 1 Trial. J. Thorac. Oncol. 2020, 15, 1210–1222. [Google Scholar] [CrossRef]
- Cho, B.C.; Daste, A.; Ravaud, A.; Salas, S.; Isambert, N.; McClay, E.; Awada, A.; Borel, C.; Ojalvo, L.S.; Helwig, C.; et al. Bintrafusp alfa, a bifunctional fusion protein targeting TGF-beta and PD-L1, in advanced squamous cell carcinoma of the head and neck: Results from a phase I cohort. J. Immunother. Cancer 2019, 8, e000664. [Google Scholar] [CrossRef] [PubMed]
- Strauss, J.; E Gatti-Mays, M.; Cho, B.C.; Hill, A.; Salas, S.; McClay, E.; Redman, J.M.; A Sater, H.; Donahue, R.N.; Jochems, C.; et al. Bintrafusp alfa, a bifunctional fusion protein targeting TGF-β and PD-L1, in patients with human papillomavirus-associated malignancies. J. Immunother. Cancer 2020, 8, e001395. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Bethesda (MD): National Library of Medicine (US). Identifier NCT00368082, Autologous/Allogeneic TGFbeta-Resistant LMP-specific CTL, Lymphoma (TGF-beta) (TGF-beta). 29 November 2023. Available online: https://clinicaltrials.gov/study/NCT00368082 (accessed on 30 May 2024).
- Pilanc, P.; Wojnicki, K.; Roura, A.-J.; Cyranowski, S.; Ellert-Miklaszewska, A.; Ochocka, N.; Gielniewski, B.; Grzybowski, M.M.; Błaszczyk, R.; Stańczak, P.S.; et al. A Novel Oral Arginase 1/2 Inhibitor Enhances the Antitumor Effect of PD-1 Inhibition in Murine Experimental Gliomas by Altering the Immunosuppressive Environment. Front. Oncol. 2021, 11, 703465. [Google Scholar] [CrossRef] [PubMed]
- Lorentzen, C.L.; Martinenaite, E.; Kjeldsen, J.W.; Holmstroem, R.B.; Mørk, S.K.; Pedersen, A.W.; Ehrnrooth, E.; Andersen, M.H.; Svane, I.M. Arginase-1 targeting peptide vaccine in patients with metastatic solid tumors—A phase I trial. Front. Immunol. 2022, 13, 1023023. [Google Scholar] [CrossRef] [PubMed]
- Molecure, S.A. [Internet]. Molecure Announces Filing of Clinical Trial Application with Polish Regulator for OATD-02, a Novel Dual Arginase Inhibitor Being Developed for the Treatment of Cancer. Available online: https://molecure.com/molecure-announces-filing-of-clinical-trial-application-with-polish-regulator-for-oatd-02-a-novel-dual-arginase-inhibitor-being-developed-for-the-treatment-of-cancer/ (accessed on 30 May 2024).
- Sullivan, K.M.; Jiang, X.; Guha, P.; Lausted, C.; Carter, J.A.; Hsu, C.; Labadie, K.P.; Kohli, K.; Kenerson, H.L.; Daniel, S.K.; et al. Blockade of interleukin 10 potentiates antitumour immune function in human colorectal cancer liver metastases. Gut 2023, 72, 325–337. [Google Scholar] [CrossRef]
- Ogasawara, F.; Higuchi, T.; Nishimori, T.; Hashida, Y.; Kojima, K.; Daibata, M. Targeting VEGF with bevacizumab inhibits malignant effusion formation of primary human herpesvirus 8-unrelated effusion large B-cell lymphoma in vivo. J. Cell. Mol. Med. 2022, 26, 5580–5589. [Google Scholar] [CrossRef]
- Smyth, L.; Blunt, D.N.; Gatov, E.; Nagamuthu, C.; Croxford, R.; Mozessohn, L.; Cheung, M.C. Statin and cyclooxygenase-2 inhibitors improve survival in newly diagnosed diffuse large B-cell lymphoma: A large population-based study of 4913 subjects. Br. J. Haematol. 2020, 191, 396–404. [Google Scholar] [CrossRef]
- Qi, L.; Pan, X.; Chen, X.; Liu, P.; Chen, M.; Zhang, Q.; Hang, X.; Tang, M.; Wen, D.; Dai, L.; et al. COX-2/PGE2 upregulation contributes to the chromosome 17p-deleted lymphoma. Oncogenesis 2023, 12, 5. [Google Scholar] [CrossRef] [PubMed]
- Gallouet, A.-S.; Travert, M.; Bresson-Bepoldin, L.; Guilloton, F.; Pangault, C.; Caulet-Maugendre, S.; Lamy, T.; Tarte, K.; Guillaudeux, T. COX-2–Independent Effects of Celecoxib Sensitize Lymphoma B Cells to TRAIL-Mediated Apoptosis. Clin. Cancer Res. 2014, 20, 2663–2673. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Wang, L.; Ni, M.; Neuber, B.; Wang, S.; Gong, W.; Sauer, T.; Schubert, M.-L.; Hückelhoven-Krauss, A.; Xia, R.; et al. Dual Effects of Cyclooxygenase Inhibitors in Combination with CD19.CAR-T Cell Immunotherapy. Front. Immunol. 2021, 12, 670088. [Google Scholar] [CrossRef] [PubMed]
- Rubenstein, J.; Ferreri, A.J.M.; Pittaluga, S. Primary lymphoma of the central nervous system: Epidemiology, pathology and current approaches to diagnosis, prognosis and treatment. Leuk. Lymphoma 2008, 49, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Sun, X.; Wu, Y.; Cui, Q.; Sun, S.; Ji, N.; Liu, Y. Targeting the tumor microenvironment in primary central nervous system lymphoma: Implications for prognosis. J. Clin. Neurosci. 2024, 124, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.-X.; Fraser, E.; Merlini, M.; Geng, H.; Chen, L.; Lu, M.; Moshfegh, Y.; Cleveland, J.; Ryu, J.; Sharma, J.; et al. Regulation of CNS Lymphoma Progression By the Myeloid Microenvironment. Blood 2021, 138, 448. [Google Scholar] [CrossRef]
- Marcelis, L.; Antoranz, A.; Delsupehe, A.-M.; Biesemans, P.; Ferreiro, J.F.; Debackere, K.; Vandenberghe, P.; Verhoef, G.; Gheysens, O.; Cattoretti, G.; et al. In-depth characterization of the tumor microenvironment in central nervous system lymphoma reveals implications for immune-checkpoint therapy. Cancer Immunol. Immunother. 2020, 69, 1751–1766. [Google Scholar] [CrossRef] [PubMed]
- Katchi, T.; Liu, D. Diagnosis and treatment of CD20 negative B cell lymphomas. Biomark. Res. 2017, 5, 1–5. [Google Scholar] [CrossRef]
- Laurent, C.; Fabiani, B.; Do, C.; Tchernonog, E.; Cartron, G.; Gravelle, P.; Amara, N.; Malot, S.; Palisoc, M.M.; Copie-Bergman, C.; et al. Immune-checkpoint expression in Epstein-Barr virus positive and negative plasmablastic lymphoma: A clinical and pathological study in 82 patients. Haematologica 2016, 101, 976–984. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Type | Sub-Types | Major Role in TME | Function | Released Factors |
|---|---|---|---|---|
| TAMs | M1 | Anti-tumourous | Cytotoxicity | |
| M2 | Pro-tumourous | Suppression of CD8+ T cells | TGF-β ARG1 IL-10 IDO | |
| Angiogenesis | VEGF PDGF Angioprotein 2 CXCL1 FGF2 | |||
| Promoting dissemination | MMP2 MMP9 CCL18 Cathepsin | |||
| MDSCs | PNM-MDSCs | Pro-tumourous | Suppression of CD8+ T cells | TGF-β ARG1 IL-10 IDO |
| M-MDSCs | Pro-tumourous | Promoting of CD4+ Treg Suppression of NK cells | IDO1 | |
| Suppression of CD8+ T cells and B cells | iNOS promoting PGE2 | |||
| TANs | N1 | Anti-tumourous | Stimulation of NK cells | IL-8 |
| Cytotoxicity | ROS MPO H2O2 Proteases | |||
| N2 | Pro-tumourous | DNA damage | NOS ROS | |
| Suppression of CD8+ T cells | ARG1 | |||
| Angiogenesis | VEGF MMP9 | |||
| CAFs | mCAFs | Unspecified | Stromal matrix remodelling | MMP11 COL1A2 |
| iCAFs | Pro-tumourous | Promoting MDSCs | CCL2 IL-6 | |
| vCAFs | Pro-tumourous | Angiogenesis | ||
| tCAFs | Pro-tumourous | Promoting tumour growth | MME CAIX TMEM158 | |
| CD8+ T cells | Tc1 | Anti-tumourous | Cytotoxicity | IFN-y TNF-α Granzyme B Perforin |
| Tc2 | Pro-tumourous | Interactions in TME | IL-4 IL-5 IL-10 | |
| Tc9 | Pro-tumourous | Promoting tumour growth | IL-9 IL-4 | |
| Tc17 | Pro-tumourous | Promoting tumour growth Angiogenesis | IL-17 | |
| Promoting TANs | IL-17 | |||
| Suppression of NK cells | IL-22 | |||
| CD4+ Treg | Pro-tumourous | Suppression of CD8+ T cells | IL-2 IL-10 TGFb IL-35 CTLA-4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sinkarevs, S.; Strumfs, B.; Volkova, S.; Strumfa, I. Tumour Microenvironment: The General Principles of Pathogenesis and Implications in Diffuse Large B Cell Lymphoma. Cells 2024, 13, 1057. https://doi.org/10.3390/cells13121057
Sinkarevs S, Strumfs B, Volkova S, Strumfa I. Tumour Microenvironment: The General Principles of Pathogenesis and Implications in Diffuse Large B Cell Lymphoma. Cells. 2024; 13(12):1057. https://doi.org/10.3390/cells13121057
Chicago/Turabian StyleSinkarevs, Stanislavs, Boriss Strumfs, Svetlana Volkova, and Ilze Strumfa. 2024. "Tumour Microenvironment: The General Principles of Pathogenesis and Implications in Diffuse Large B Cell Lymphoma" Cells 13, no. 12: 1057. https://doi.org/10.3390/cells13121057