
Astrocytes in Amyloid Generation and Alcohol Metabolism: Implications of Alcohol Use in Neurological Disorder(s)

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Culture
2.3. siRNA Transfection
2.4. Quantitative Real-Time PCR (qPCR)
2.5. Western Blot
2.6. Immunocytochemistry
2.7. Statistical Analysis
3. Results
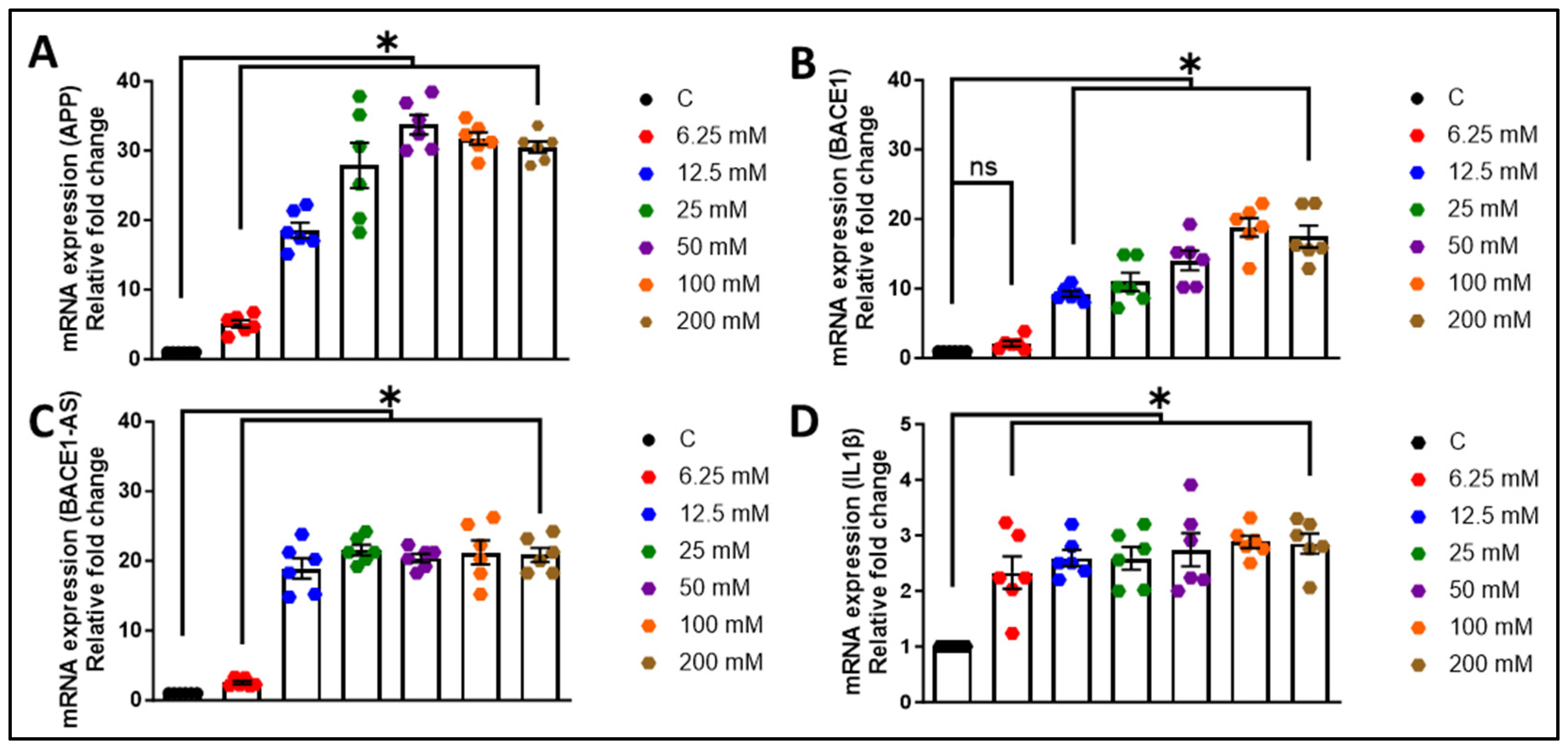
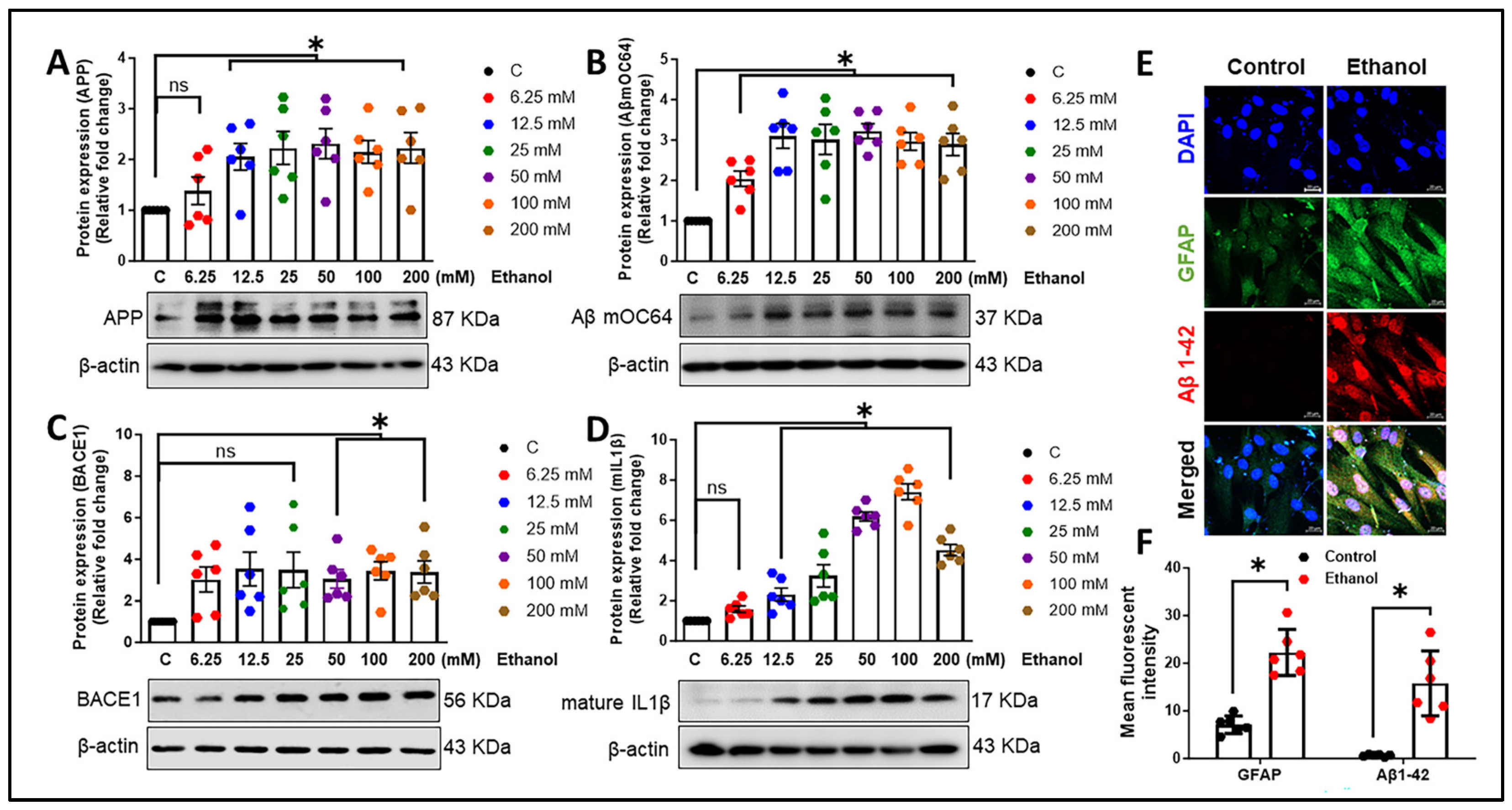
3.1. Alcohol Exposure Induces Astrocytic Amyloid Generation and Inflammation
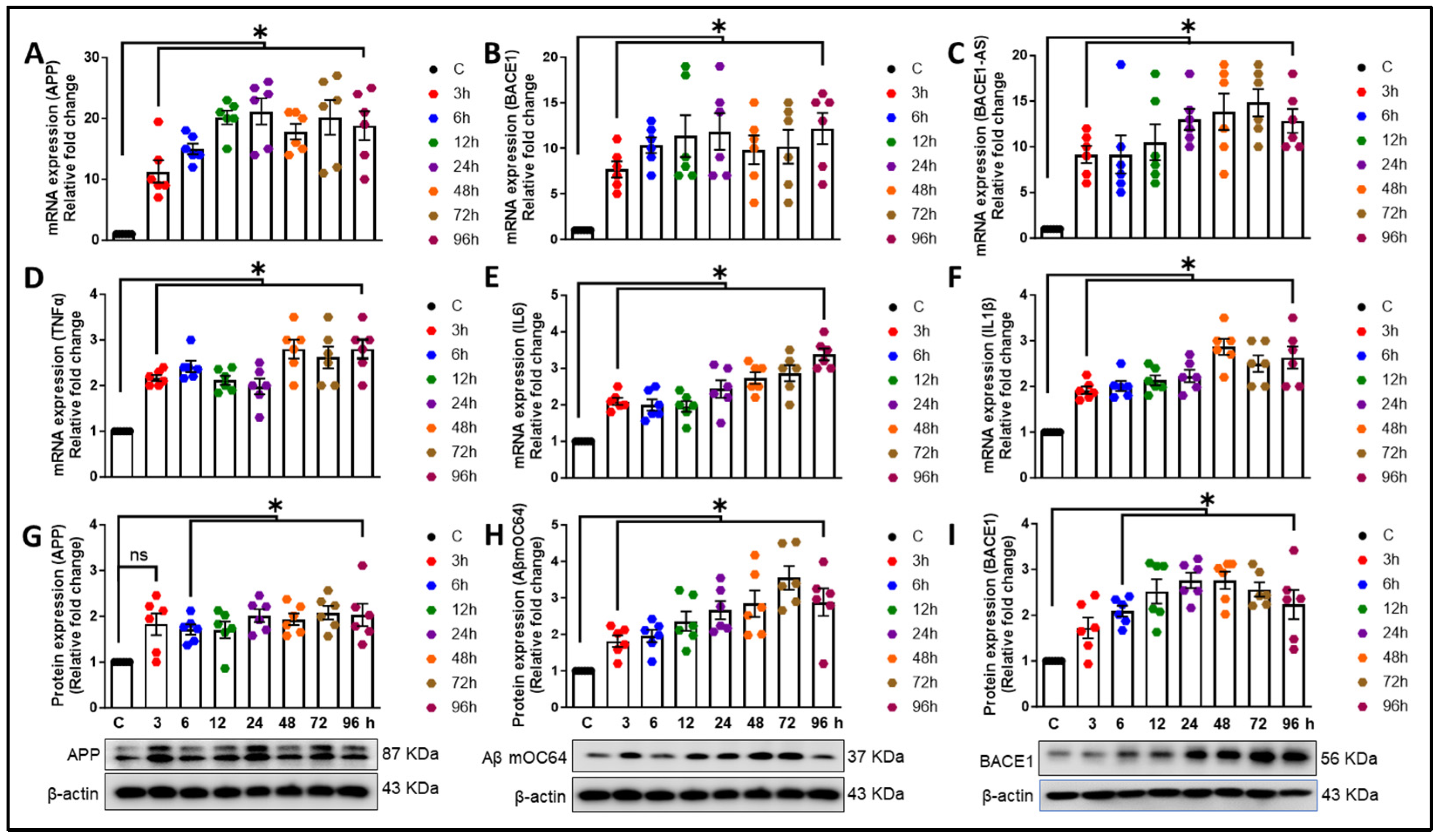
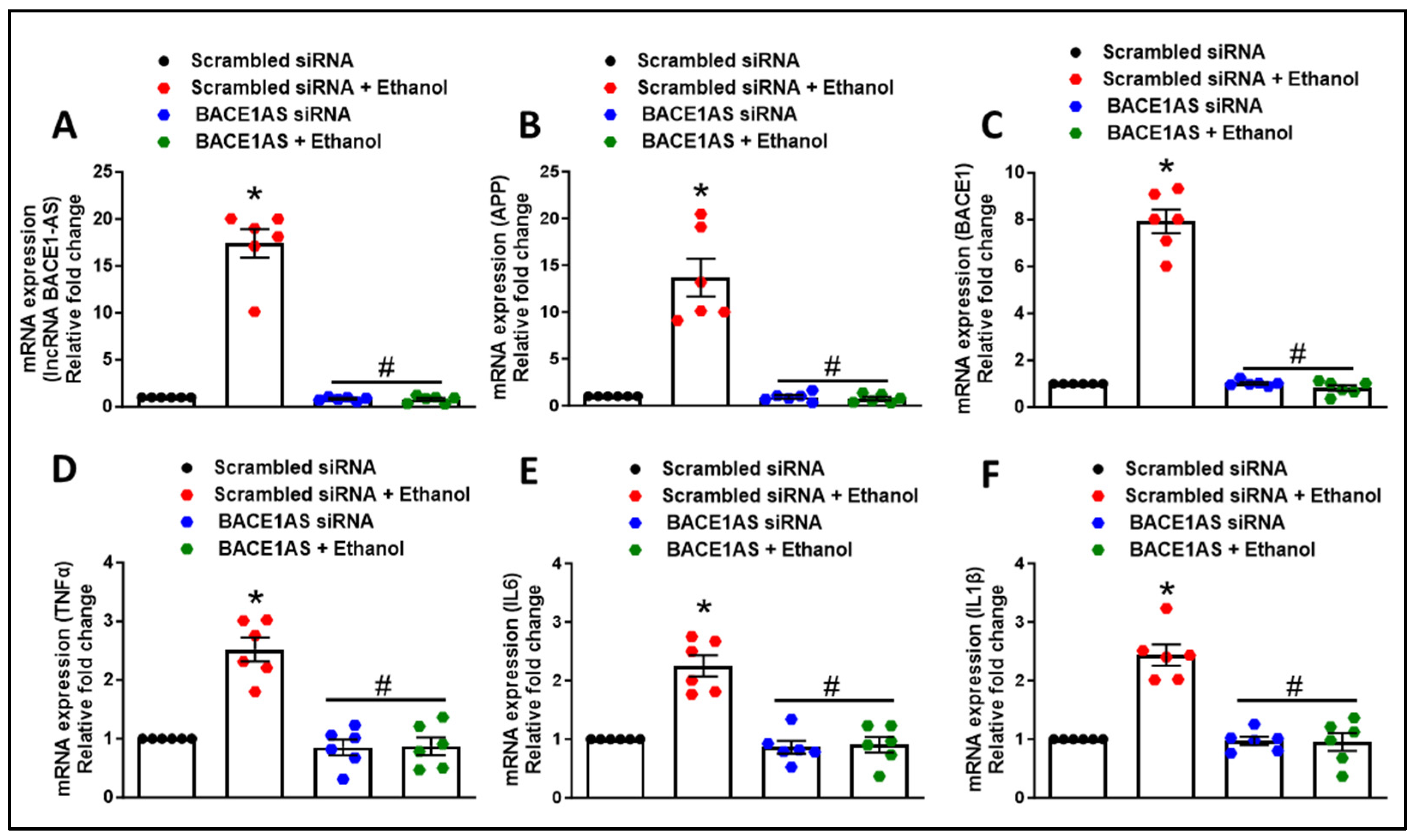
3.2. Long Noncoding RNA BACE1-AS Regulates Ethanol-Mediated Amyloid Generation and Inflammation in Astrocytes
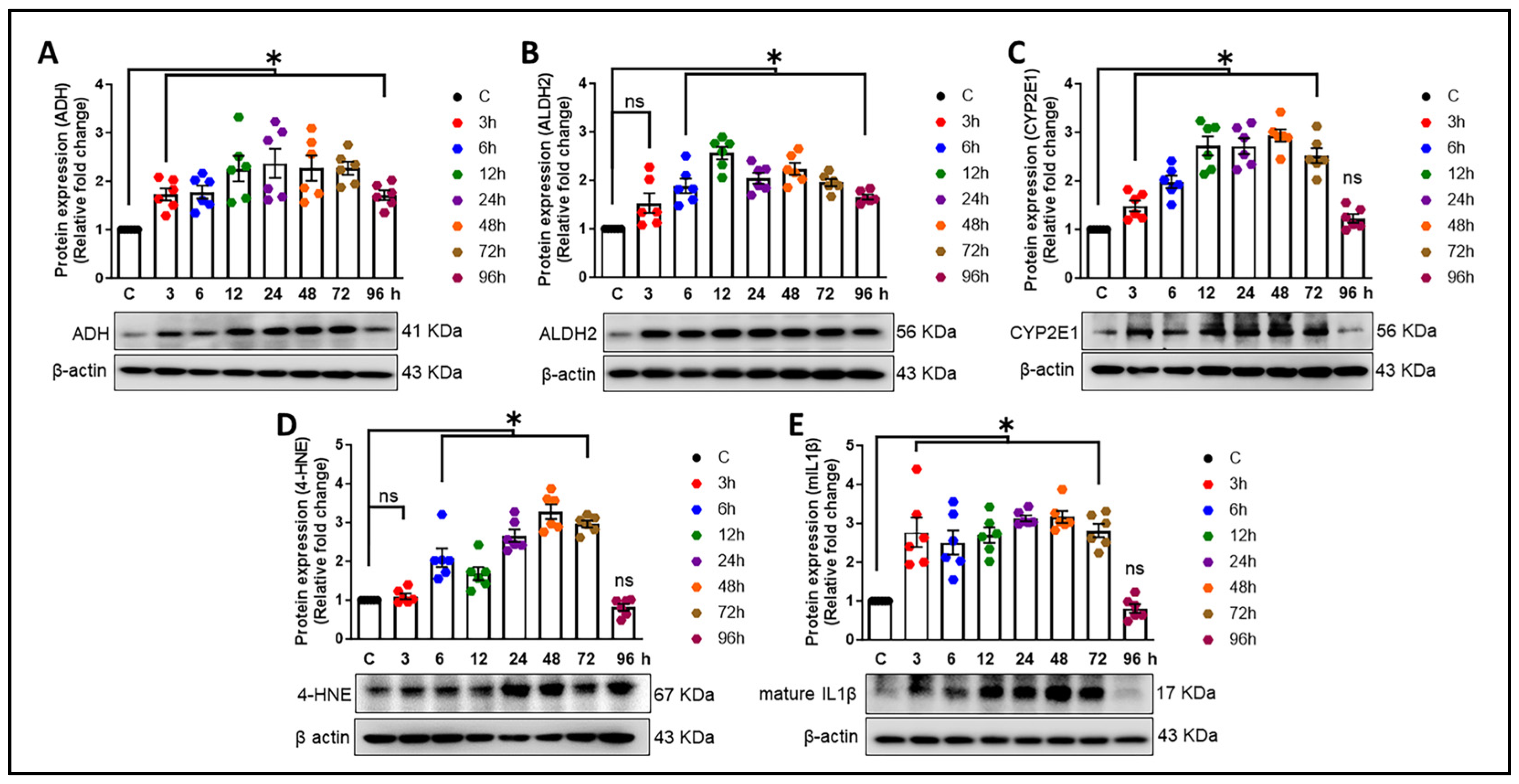
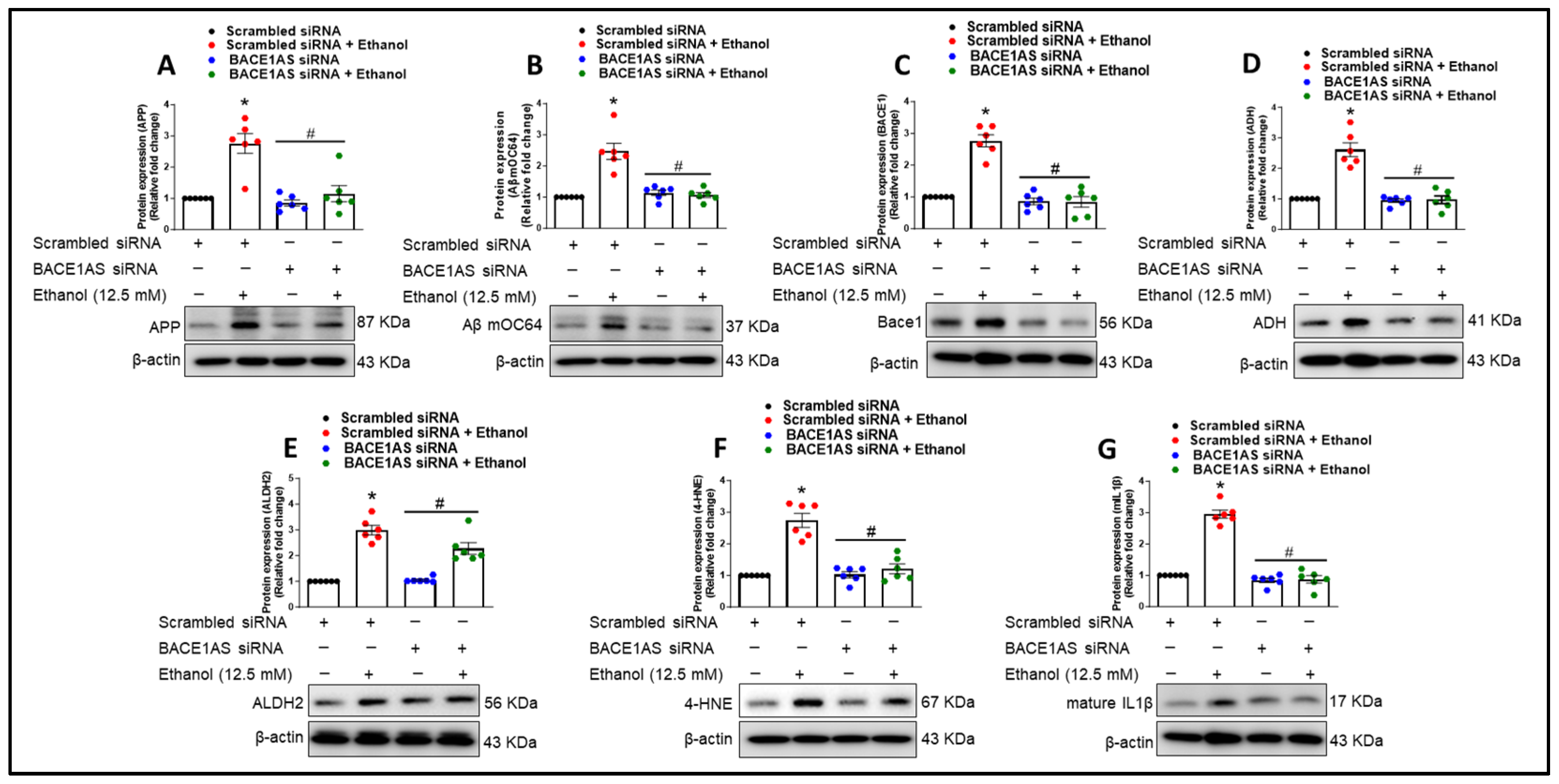
3.3. Alcohol-Induced Astrocytic Amyloid Generation Leading to Oxidative Stress and Neuroinflammation Involves lncRNA BACE1-AS
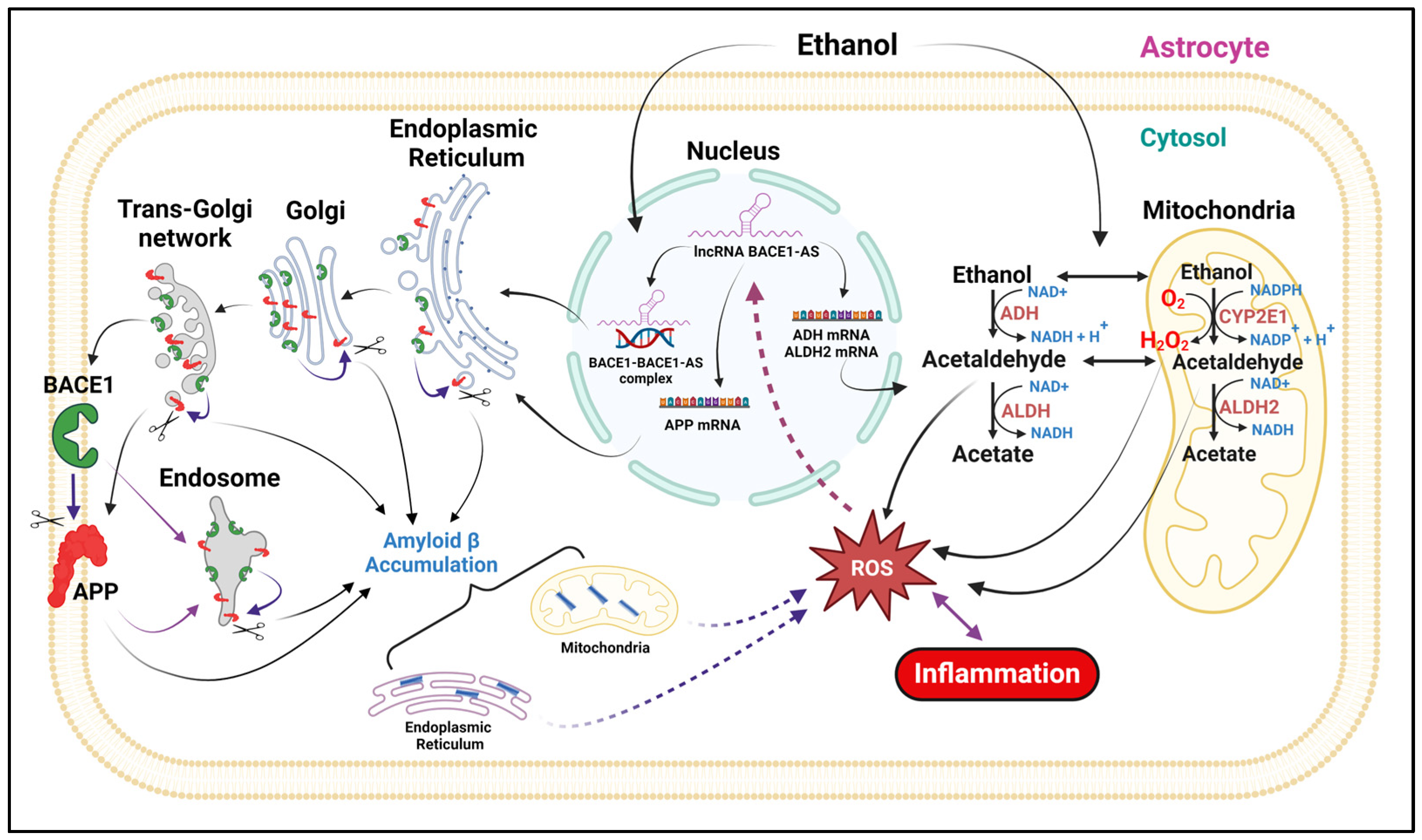
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hugo, J.; Ganguli, M. Dementia and cognitive impairment: Epidemiology, diagnosis, and treatment. Clin. Geriatr. Med. 2014, 30, 421–442. [Google Scholar] [CrossRef]
- Sabia, S.; Fayosse, A.; Dumurgier, J.; Dugravot, A.; Akbaraly, T.; Britton, A.; Kivimaki, M.; Singh-Manoux, A. Alcohol consumption and risk of dementia: 23 year follow-up of Whitehall II cohort study. BMJ 2018, 362, k2927. [Google Scholar] [CrossRef] [PubMed]
- Hart, C.L.; Marvin, C.B.; Silver, R.; Smith, E.E. Is cognitive functioning impaired in methamphetamine users? A critical review. Neuropsychopharmacology 2012, 37, 586–608. [Google Scholar] [CrossRef]
- Dublin, S.; Walker, R.L.; Gray, S.L.; Hubbard, R.A.; Anderson, M.L.; Yu, O.; Montine, T.J.; Crane, P.K.; Sonnen, J.A.; Larson, E.B. Use of Analgesics (Opioids and Nonsteroidal Anti-Inflammatory Drugs) and Dementia-Related Neuropathology in a Community-Based Autopsy Cohort. J. Alzheimers Dis. 2017, 58, 435–448. [Google Scholar] [CrossRef] [PubMed]
- Wiegmann, C.; Mick, I.; Brandl, E.J.; Heinz, A.; Gutwinski, S. Alcohol and Dementia—What is the Link? A Systematic Review. Neuropsychiatr. Dis. Treat. 2020, 16, 87–99. [Google Scholar] [CrossRef]
- Jeon, K.H.; Han, K.; Jeong, S.M.; Park, J.; Yoo, J.E.; Yoo, J.; Lee, J.; Kim, S.; Shin, D.W. Changes in Alcohol Consumption and Risk of Dementia in a Nationwide Cohort in South Korea. JAMA Netw. Open 2023, 6, e2254771. [Google Scholar] [CrossRef]
- Shimizu, Y.; Sawada, N.; Ihira, H.; Abe, S.K.; Inoue, M.; Yasuda, N.; Yamagishi, K.; Iwasaki, M.; Tsugane, S. Alcohol consumption from midlife and risk of disabling dementia in a large population-based cohort study in Japan. Int. J. Geriatr. Psychiatry 2023, 38, e5896. [Google Scholar] [CrossRef]
- Mechtcheriakov, S.; Brenneis, C.; Egger, K.; Koppelstaetter, F.; Schocke, M.; Marksteiner, J. A widespread distinct pattern of cerebral atrophy in patients with alcohol addiction revealed by voxel-based morphometry. J. Neurol. Neurosurg. Psychiatry 2007, 78, 610–614. [Google Scholar] [CrossRef]
- Grace, S.; Rossetti, M.G.; Allen, N.; Batalla, A.; Bellani, M.; Brambilla, P.; Chye, Y.; Cousijn, J.; Goudriaan, A.E.; Hester, R.; et al. Sex differences in the neuroanatomy of alcohol dependence: Hippocampus and amygdala subregions in a sample of 966 people from the ENIGMA Addiction Working Group. Transl. Psychiatry 2021, 11, 156. [Google Scholar] [CrossRef] [PubMed]
- Salinas, A.G.; Mateo, Y.; Carlson, V.C.C.; Stinnett, G.S.; Luo, G.; Seasholtz, A.F.; Grant, K.A.; Lovinger, D.M. Long-term alcohol consumption alters dorsal striatal dopamine release and regulation by D2 dopamine receptors in rhesus macaques. Neuropsychopharmacology 2021, 46, 1432–1441. [Google Scholar] [CrossRef]
- Lees, B.; Meredith, L.R.; Kirkland, A.E.; Bryant, B.E.; Squeglia, L.M. Effect of alcohol use on the adolescent brain and behavior. Pharmacol. Biochem. Behav. 2020, 192, 172906. [Google Scholar] [CrossRef] [PubMed]
- Zuccala, G.; Onder, G.; Pedone, C.; Cesari, M.; Landi, F.; Bernabei, R.; Cocchi, A.; Gruppo Italiano di Farmacoepidemiologia nell’Anziano Investigators. Dose-related impact of alcohol consumption on cognitive function in advanced age: Results of a multicenter survey. Alcohol. Clin. Exp. Res. 2001, 25, 1743–1748. [Google Scholar] [CrossRef] [PubMed]
- Kuhns, L.; Kroon, E.; Lesscher, H.; Mies, G.; Cousijn, J. Age-related differences in the effect of chronic alcohol on cognition and the brain: A systematic review. Transl. Psychiatry 2022, 12, 345. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Yingjie, Z.; Na, A.; Qi, Q.; Wei, L.; Wenzheng, W.; Lin, S.; Shifu, X. The Effects of Light-to-Moderate Alcohol Consumption on the Cognitive Function of Community Nondemented Male Elderly: A Cohort Study. Behav. Neurol. 2021, 2021, 5681913. [Google Scholar] [CrossRef] [PubMed]
- Nguyen-Louie, T.T.; Castro, N.; Matt, G.E.; Squeglia, L.M.; Brumback, T.; Tapert, S.F. Effects of Emerging Alcohol and Marijuana Use Behaviors on Adolescents’ Neuropsychological Functioning Over Four Years. J. Stud. Alcohol. Drugs 2015, 76, 738–748. [Google Scholar] [CrossRef] [PubMed]
- Winward, J.L.; Hanson, K.L.; Tapert, S.F.; Brown, S.A. Heavy alcohol use, marijuana use, and concomitant use by adolescents are associated with unique and shared cognitive decrements. J. Int. Neuropsychol. Soc. 2014, 20, 784–795. [Google Scholar] [CrossRef] [PubMed]
- Nguyen-Louie, T.T.; Tracas, A.; Squeglia, L.M.; Matt, G.E.; Eberson-Shumate, S.; Tapert, S.F. Learning and Memory in Adolescent Moderate, Binge, and Extreme-Binge Drinkers. Alcohol. Clin. Exp. Res. 2016, 40, 1895–1904. [Google Scholar] [CrossRef] [PubMed]
- Holtzman, D.M.; Morris, J.C.; Goate, A.M. Alzheimer’s disease: The challenge of the second century. Sci. Transl. Med. 2011, 3, 77sr71. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Browne, A.; Divito, J.R.; Stevenson, J.A.; Romano, D.; Dong, Y.; Xie, Z.; Tanzi, R.E. Amyloid-beta production via cleavage of amyloid-beta protein precursor is modulated by cell density. J. Alzheimers Dis. 2010, 22, 683–984. [Google Scholar] [CrossRef]
- Sil, S.; Singh, S.; Chemparathy, D.T.; Chivero, E.T.; Gordon, L.; Buch, S. Astrocytes & Astrocyte derived Extracellular Vesicles in Morphine Induced Amyloidopathy: Implications for Cognitive Deficits in Opiate Abusers. Aging Dis. 2021, 12, 1389–1408. [Google Scholar] [CrossRef]
- Cole, S.L.; Vassar, R. The Alzheimer’s disease beta-secretase enzyme, BACE1. Mol. Neurodegener. 2007, 2, 22. [Google Scholar] [CrossRef] [PubMed]
- Sayad, A.; Najafi, S.; Hussen, B.M.; Abdullah, S.T.; Movahedpour, A.; Taheri, M.; Hajiesmaeili, M. The Emerging Roles of the beta-Secretase BACE1 and the Long Non-coding RNA BACE1-AS in Human Diseases: A Focus on Neurodegenerative Diseases and Cancer. Front. Aging Neurosci. 2022, 14, 853180. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Wang, Y.; Yang, H.; Xu, Y.; Zhou, X.; Zhang, X.; Xie, Z.; Bi, J. The effect of BACE1-AS on beta-amyloid generation by regulating BACE1 mRNA expression. BMC Mol. Biol. 2019, 20, 23. [Google Scholar] [CrossRef] [PubMed]
- Chandrashekar, D.V.; Steinberg, R.A.; Han, D.; Sumbria, R.K. Alcohol as a Modifiable Risk Factor for Alzheimer’s Disease-Evidence from Experimental Studies. Int. J. Mol. Sci. 2023, 24, 9492. [Google Scholar] [CrossRef] [PubMed]
- Tyas, S.L. Alcohol use and the risk of developing Alzheimer’s disease. Alcohol Res. Health 2001, 25, 299–306. [Google Scholar] [PubMed]
- Heymann, D.; Stern, Y.; Cosentino, S.; Tatarina-Nulman, O.; Dorrejo, J.N.; Gu, Y. The Association Between Alcohol Use and the Progression of Alzheimer’s Disease. Curr. Alzheimer Res. 2016, 13, 1356–1362. [Google Scholar] [CrossRef] [PubMed]
- Tucker, A.E.; Alicea Pauneto, C.D.M.; Barnett, A.M.; Coleman, L.G., Jr. Chronic Ethanol Causes Persistent Increases in Alzheimer’s Tau Pathology in Female 3xTg-AD Mice: A Potential Role for Lysosomal Impairment. Front. Behav. Neurosci. 2022, 16, 886634. [Google Scholar] [CrossRef] [PubMed]
- Kalinin, S.; Gonzalez-Prieto, M.; Scheiblich, H.; Lisi, L.; Kusumo, H.; Heneka, M.T.; Madrigal, J.L.M.; Pandey, S.C.; Feinstein, D.L. Transcriptome analysis of alcohol-treated microglia reveals downregulation of beta amyloid phagocytosis. J. Neuroinflamm. 2018, 15, 141. [Google Scholar] [CrossRef] [PubMed]
- Day, S.M.; Gironda, S.C.; Clarke, C.W.; Snipes, J.A.; Nicol, N.I.; Kamran, H.; Vaughan, W.; Weiner, J.L.; Macauley, S.L. Ethanol exposure alters Alzheimer’s-related pathology, behavior, and metabolism in APP/PS1 mice. Neurobiol. Dis. 2023, 177, 105967. [Google Scholar] [CrossRef]
- Gabr, A.A.; Lee, H.J.; Onphachanh, X.; Jung, Y.H.; Kim, J.S.; Chae, C.W.; Han, H.J. Ethanol-induced PGE(2) up-regulates Abeta production through PKA/CREB signaling pathway. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2942–2953. [Google Scholar] [CrossRef]
- Gonzalez-Reyes, R.E.; Nava-Mesa, M.O.; Vargas-Sanchez, K.; Ariza-Salamanca, D.; Mora-Munoz, L. Involvement of Astrocytes in Alzheimer’s Disease from a Neuroinflammatory and Oxidative Stress Perspective. Front. Mol. Neurosci. 2017, 10, 427. [Google Scholar] [CrossRef] [PubMed]
- Sil, S.; Hu, G.; Liao, K.; Niu, F.; Callen, S.; Periyasamy, P.; Fox, H.S.; Buch, S. HIV-1 Tat-mediated astrocytic amyloidosis involves the HIF-1alpha/lncRNA BACE1-AS axis. PLoS Biol. 2020, 18, e3000660. [Google Scholar] [CrossRef] [PubMed]
- Chemparathy, D.T.; Ray, S.; Ochs, C.; Ferguson, N.; Gawande, D.Y.; Dravid, S.M.; Callen, S.; Sil, S.; Buch, S. Neuropathogenic role of astrocyte-derived extracellular vesicles in HIV-associated neurocognitive disorders. J. Extracell. Vesicles 2024, 13, e12439. [Google Scholar] [CrossRef] [PubMed]
- Chivero, E.T.; Dagur, R.S.; Peeples, E.S.; Sil, S.; Liao, K.; Ma, R.; Chen, L.; Gurumurthy, C.B.; Buch, S.; Hu, G. Biogenesis, physiological functions and potential applications of extracellular vesicles in substance use disorders. Cell Mol. Life Sci. 2021, 78, 4849–4865. [Google Scholar] [CrossRef] [PubMed]
- Sil, S.; Niu, F.; Tom, E.; Liao, K.; Periyasamy, P.; Buch, S. Cocaine Mediated Neuroinflammation: Role of Dysregulated Autophagy in Pericytes. Mol. Neurobiol. 2019, 56, 3576–3590. [Google Scholar] [CrossRef] [PubMed]
- Sil, S.; Periyasamy, P.; Guo, M.L.; Callen, S.; Buch, S. Morphine-Mediated Brain Region-Specific Astrocytosis Involves the ER Stress-Autophagy Axis. Mol. Neurobiol. 2018, 55, 6713–6733. [Google Scholar] [CrossRef] [PubMed]
- Abrahao, K.P.; Salinas, A.G.; Lovinger, D.M. Alcohol and the Brain: Neuronal Molecular Targets, Synapses, and Circuits. Neuron 2017, 96, 1223–1238. [Google Scholar] [CrossRef] [PubMed]
- Stroud, J.C.; Liu, C.; Teng, P.K.; Eisenberg, D. Toxic fibrillar oligomers of amyloid-beta have cross-beta structure. Proc. Natl. Acad. Sci. USA 2012, 109, 7717–7722. [Google Scholar] [CrossRef] [PubMed]
- Vena, A.A.; Zandy, S.L.; Cofresi, R.U.; Gonzales, R.A. Behavioral, neurobiological, and neurochemical mechanisms of ethanol self-administration: A translational review. Pharmacol. Ther. 2020, 212, 107573. [Google Scholar] [CrossRef]
- Edenberg, H.J. The genetics of alcohol metabolism: Role of alcohol dehydrogenase and aldehyde dehydrogenase variants. Alcohol Res. Health 2007, 30, 5–13. [Google Scholar]
- Jin, M.; Ande, A.; Kumar, A.; Kumar, S. Regulation of cytochrome P450 2e1 expression by ethanol: Role of oxidative stress-mediated pkc/jnk/sp1 pathway. Cell Death Dis. 2013, 4, e554. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Zhang, T.; Kusumanchi, P.; Han, S.; Yang, Z.; Liangpunsakul, S. Alcohol Metabolizing Enzymes, Microsomal Ethanol Oxidizing System, Cytochrome P450 2E1, Catalase, and Aldehyde Dehydrogenase in Alcohol-Associated Liver Disease. Biomedicines 2020, 8, 50. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Suastegui, W.A.; Ramos-Chavez, L.A.; Rubio-Osornio, M.; Calvillo-Velasco, M.; Atzin-Mendez, J.A.; Guevara, J.; Silva-Adaya, D. The Role of CYP2E1 in the Drug Metabolism or Bioactivation in the Brain. Oxid. Med. Cell Longev. 2017, 2017, 4680732. [Google Scholar] [CrossRef]
- Chen, C.H.; Joshi, A.U.; Mochly-Rosen, D. The Role of Mitochondrial Aldehyde Dehydrogenase 2 (ALDH2) in Neuropathology and Neurodegeneration. Acta Neurol. Taiwan. 2016, 25, 111–123. [Google Scholar]
- Yan, R.; Han, P.; Miao, H.; Greengard, P.; Xu, H. The transmembrane domain of the Alzheimer’s beta-secretase (BACE1) determines its late Golgi localization and access to beta -amyloid precursor protein (APP) substrate. J. Biol. Chem. 2001, 276, 36788–36796. [Google Scholar] [CrossRef] [PubMed]
- Hansson Petersen, C.A.; Alikhani, N.; Behbahani, H.; Wiehager, B.; Pavlov, P.F.; Alafuzoff, I.; Leinonen, V.; Ito, A.; Winblad, B.; Glaser, E.; et al. The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc. Natl. Acad. Sci. USA 2008, 105, 13145–13150. [Google Scholar] [CrossRef] [PubMed]
- Greenfield, J.P.; Tsai, J.; Gouras, G.K.; Hai, B.; Thinakaran, G.; Checler, F.; Sisodia, S.S.; Greengard, P.; Xu, H. Endoplasmic reticulum and trans-Golgi network generate distinct populations of Alzheimer beta-amyloid peptides. Proc. Natl. Acad. Sci. USA 1999, 96, 742–747. [Google Scholar] [CrossRef] [PubMed]
- Cook, D.G.; Forman, M.S.; Sung, J.C.; Leight, S.; Kolson, D.L.; Iwatsubo, T.; Lee, V.M.; Doms, R.W. Alzheimer’s A beta(1-42) is generated in the endoplasmic reticulum/intermediate compartment of NT2N cells. Nat. Med. 1997, 3, 1021–1023. [Google Scholar] [CrossRef]
- Hartmann, T.; Bieger, S.C.; Bruhl, B.; Tienari, P.J.; Ida, N.; Allsop, D.; Roberts, G.W.; Masters, C.L.; Dotti, C.G.; Unsicker, K.; et al. Distinct sites of intracellular production for Alzheimer’s disease A beta40/42 amyloid peptides. Nat. Med. 1997, 3, 1016–1020. [Google Scholar] [CrossRef]
- Xu, H.; Sweeney, D.; Wang, R.; Thinakaran, G.; Lo, A.C.; Sisodia, S.S.; Greengard, P.; Gandy, S. Generation of Alzheimer beta-amyloid protein in the trans-Golgi network in the apparent absence of vesicle formation. Proc. Natl. Acad. Sci. USA 1997, 94, 3748–3752. [Google Scholar] [CrossRef]
- Cataldo, A.M.; Petanceska, S.; Terio, N.B.; Peterhoff, C.M.; Durham, R.; Mercken, M.; Mehta, P.D.; Buxbaum, J.; Haroutunian, V.; Nixon, R.A. Abeta localization in abnormal endosomes: Association with earliest Abeta elevations in AD and Down syndrome. Neurobiol. Aging 2004, 25, 1263–1272. [Google Scholar] [CrossRef] [PubMed]
- Pasternak, S.H.; Callahan, J.W.; Mahuran, D.J. The role of the endosomal/lysosomal system in amyloid-beta production and the pathophysiology of Alzheimer’s disease: Reexamining the spatial paradox from a lysosomal perspective. J. Alzheimers Dis. 2004, 6, 53–65. [Google Scholar] [CrossRef]
- Takahashi, R.H.; Almeida, C.G.; Kearney, P.F.; Yu, F.; Lin, M.T.; Milner, T.A.; Gouras, G.K. Oligomerization of Alzheimer’s beta-amyloid within processes and synapses of cultured neurons and brain. J. Neurosci. 2004, 24, 3592–3599. [Google Scholar] [CrossRef]
- Lee, E.K.; Park, Y.W.; Shin, D.Y.; Mook-Jung, I.; Yoo, Y.J. Cytosolic amyloid-beta peptide 42 escaping from degradation induces cell death. Biochem. Biophys. Res. Commun. 2006, 344, 471–477. [Google Scholar] [CrossRef]
- Buckig, A.; Tikkanen, R.; Herzog, V.; Schmitz, A. Cytosolic and nuclear aggregation of the amyloid beta-peptide following its expression in the endoplasmic reticulum. Histochem. Cell Biol. 2002, 118, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Snezhkina, A.V.; Kudryavtseva, A.V.; Kardymon, O.L.; Savvateeva, M.V.; Melnikova, N.V.; Krasnov, G.S.; Dmitriev, A.A. ROS Generation and Antioxidant Defense Systems in Normal and Malignant Cells. Oxid. Med. Cell Longev. 2019, 2019, 6175804. [Google Scholar] [CrossRef] [PubMed]
- Dumont, M.; Ho, D.J.; Calingasan, N.Y.; Xu, H.; Gibson, G.; Beal, M.F. Mitochondrial dihydrolipoyl succinyltransferase deficiency accelerates amyloid pathology and memory deficit in a transgenic mouse model of amyloid deposition. Free Radic. Biol. Med. 2009, 47, 1019–1027. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Xu, H.; Yu, H.; Zhang, N.; Ye, Y.; Estevez, A.G.; Deng, H.; Gibson, G.E. Inactivation and reactivation of the mitochondrial alpha-ketoglutarate dehydrogenase complex. J. Biol. Chem. 2011, 286, 17640–17648. [Google Scholar] [CrossRef]
- Rehm, J.; Gmel, G.; Sempos, C.T.; Trevisan, M. Alcohol-related morbidity and mortality. Alcohol Res. Health 2003, 27, 39–51. [Google Scholar]
- Schwarzinger, M.; Pollock, B.G.; Hasan, O.S.M.; Dufouil, C.; Rehm, J.; QalyDays Study, G. Contribution of alcohol use disorders to the burden of dementia in France 2008-13: A nationwide retrospective cohort study. Lancet Public Health 2018, 3, e124–e132. [Google Scholar] [CrossRef]
- Ruitenberg, A.; van Swieten, J.C.; Witteman, J.C.; Mehta, K.M.; van Duijn, C.M.; Hofman, A.; Breteler, M.M. Alcohol consumption and risk of dementia: The Rotterdam Study. Lancet 2002, 359, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Neafsey, E.J.; Collins, M.A. Moderate alcohol consumption and cognitive risk. Neuropsychiatr. Dis. Treat. 2011, 7, 465–484. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Tong, J.; Liu, J.; Wang, Y. Downregulation of ROCK2 attenuates alcohol-induced inflammation and oxidative stress in astrocytes. Int. J. Neurosci. 2020, 132, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Brenner, E.; Tiwari, G.R.; Kapoor, M.; Liu, Y.; Brock, A.; Mayfield, R.D. Single cell transcriptome profiling of the human alcohol-dependent brain. Hum. Mol. Genet. 2020, 29, 1144–1153. [Google Scholar] [CrossRef] [PubMed]
- Leuba, G.; Wernli, G.; Vernay, A.; Kraftsik, R.; Mohajeri, M.H.; Saini, K.D. Neuronal and nonneuronal quantitative BACE immunocytochemical expression in the entorhinohippocampal and frontal regions in Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 2005, 19, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.F.; Wang, B.J.; Cheng, H.T.; Kuo, L.H.; Wolfe, M.S. Tumor necrosis factor-alpha, interleukin-1beta, and interferon-gamma stimulate gamma-secretase-mediated cleavage of amyloid precursor protein through a JNK-dependent MAPK pathway. J. Biol. Chem. 2004, 279, 49523–49532. [Google Scholar] [CrossRef] [PubMed]
- Orre, M.; Kamphuis, W.; Osborn, L.M.; Jansen, A.H.P.; Kooijman, L.; Bossers, K.; Hol, E.M. Isolation of glia from Alzheimer’s mice reveals inflammation and dysfunction. Neurobiol. Aging 2014, 35, 2746–2760. [Google Scholar] [CrossRef]
- Barucker, C.; Harmeier, A.; Weiske, J.; Fauler, B.; Albring, K.F.; Prokop, S.; Hildebrand, P.; Lurz, R.; Heppner, F.L.; Huber, O.; et al. Nuclear translocation uncovers the amyloid peptide Abeta42 as a regulator of gene transcription. J. Biol. Chem. 2014, 289, 20182–20191. [Google Scholar] [CrossRef]
- Gezen-Ak, D.; Atasoy, I.L.; Candas, E.; Alaylioglu, M.; Dursun, E. The Transcriptional Regulatory Properties of Amyloid Beta 1-42 may Include Regulation of Genes Related to Neurodegeneration. Neuromol. Med. 2018, 20, 363–375. [Google Scholar] [CrossRef]
- Bhat, R.; Crowe, E.P.; Bitto, A.; Moh, M.; Katsetos, C.D.; Garcia, F.U.; Johnson, F.B.; Trojanowski, J.Q.; Sell, C.; Torres, C. Astrocyte senescence as a component of Alzheimer’s disease. PLoS ONE 2012, 7, e45069. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Yarishkin, O.; Hwang, Y.J.; Chun, Y.E.; Park, M.; Woo, D.H.; Bae, J.Y.; Kim, T.; Lee, J.; Chun, H.; et al. GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease. Nat. Med. 2014, 20, 886–896. [Google Scholar] [CrossRef] [PubMed]
- Wyss-Coray, T.; Loike, J.D.; Brionne, T.C.; Lu, E.; Anankov, R.; Yan, F.; Silverstein, S.C.; Husemann, J. Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat. Med. 2003, 9, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Umhau, J.C.; Schwandt, M.; Solomon, M.G.; Yuan, P.; Nugent, A.; Zarate, C.A.; Drevets, W.C.; Hall, S.D.; George, D.T.; Heilig, M. Cerebrospinal fluid monocyte chemoattractant protein-1 in alcoholics: Support for a neuroinflammatory model of chronic alcoholism. Alcohol. Clin. Exp. Res. 2014, 38, 1301–1306. [Google Scholar] [CrossRef] [PubMed]
- Karadayian, A.G.; Malanga, G.; Czerniczyniec, A.; Lombardi, P.; Bustamante, J.; Lores-Arnaiz, S. Free radical production and antioxidant status in brain cortex non-synaptic mitochondria and synaptosomes at alcohol hangover onset. Free Radic. Biol. Med. 2017, 108, 692–703. [Google Scholar] [CrossRef] [PubMed]
- Zabel, M.K.; Kirsch, W.M. From development to dysfunction: Microglia and the complement cascade in CNS homeostasis. Ageing Res. Rev. 2013, 12, 749–756. [Google Scholar] [CrossRef] [PubMed]
- Contreras-Zentella, M.L.; Villalobos-Garcia, D.; Hernandez-Munoz, R. Ethanol Metabolism in the Liver, the Induction of Oxidant Stress, and the Antioxidant Defense System. Antioxidants 2022, 11, 1258. [Google Scholar] [CrossRef] [PubMed]
- Zakhari, S. Overview: How is alcohol metabolized by the body? Alcohol. Res. Health 2006, 29, 245–254. [Google Scholar] [PubMed]
- Jin, S.; Cao, Q.; Yang, F.; Zhu, H.; Xu, S.; Chen, Q.; Wang, Z.; Lin, Y.; Cinar, R.; Pawlosky, R.J.; et al. Brain ethanol metabolism by astrocytic ALDH2 drives the behavioural effects of ethanol intoxication. Nat. Metab. 2021, 3, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Waddell, J.; McKenna, M.C.; Kristian, T. Brain ethanol metabolism and mitochondria. Curr. Top. Biochem. Res. 2022, 23, 1–13. [Google Scholar]
- Hampel, H.; Vassar, R.; De Strooper, B.; Hardy, J.; Willem, M.; Singh, N.; Zhou, J.; Yan, R.; Vanmechelen, E.; De Vos, A.; et al. The beta-Secretase BACE1 in Alzheimer’s Disease. Biol. Psychiatry 2021, 89, 745–756. [Google Scholar] [CrossRef] [PubMed]
- McDade, E.; Voytyuk, I.; Aisen, P.; Bateman, R.J.; Carrillo, M.C.; De Strooper, B.; Haass, C.; Reiman, E.M.; Sperling, R.; Tariot, P.N.; et al. The case for low-level BACE1 inhibition for the prevention of Alzheimer disease. Nat. Rev. Neurol. 2021, 17, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, Y.; Zhou, M.; Yu, L.; Si, Z. m6A modified BACE1-AS contributes to liver metastasis and stemness-like properties in colorectal cancer through TUFT1 dependent activation of Wnt signaling. J. Exp. Clin. Cancer Res. 2023, 42, 306. [Google Scholar] [CrossRef] [PubMed]
- Guglas, K.; Kolenda, T.; Teresiak, A.; Kopczynska, M.; Lasinska, I.; Mackiewicz, J.; Mackiewicz, A.; Lamperska, K. lncRNA Expression after Irradiation and Chemoexposure of HNSCC Cell Lines. Noncoding RNA 2018, 4, 33. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhang, T.; Yang, L.; Na, X.; Qu, Y. LncRNA BACE1-AS promotes the progression of osteosarcoma through miR-762/SOX7 axis. Mol. Biol. Rep. 2022, 49, 5853–5862. [Google Scholar] [CrossRef] [PubMed]
- Bampatsias, D.; Mavroeidis, I.; Tual-Chalot, S.; Vlachogiannis, N.I.; Bonini, F.; Sachse, M.; Mavraganis, G.; Mareti, A.; Kritsioti, C.; Laina, A.; et al. Beta-Secretase-1 Antisense RNA Is Associated with Vascular Ageing and Atherosclerotic Cardiovascular Disease. Thromb. Haemost. 2022, 122, 1932–1942. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wang, H.; Li, H.; Lu, X.; Gao, Y.; Guo, X. Long noncoding RNA BACE1-antisense transcript plays a critical role in Parkinson’s disease via microRNA-214-3p/Cell death-inducing p53-target protein 1 axis. Bioengineered 2022, 13, 10889–10901. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Fang, J.; Zhou, Z.; Zhou, Q.; Sun, S.; Jin, Z.; Xi, Z.; Wei, J. Downregulation of lncRNA BACE1-AS improves dopamine-dependent oxidative stress in rats with Parkinson’s disease by upregulating microRNA-34b-5p and downregulating BACE1. Cell Cycle 2020, 19, 1158–1171. [Google Scholar] [CrossRef] [PubMed]
- Fotuhi, S.N.; Khalaj-Kondori, M.; Hoseinpour Feizi, M.A.; Talebi, M. Long Non-coding RNA BACE1-AS May Serve as an Alzheimer’s Disease Blood-Based Biomarker. J. Mol. Neurosci. 2019, 69, 351–359. [Google Scholar] [CrossRef]
- Karakas, D.; Ozpolat, B. The Role of LncRNAs in Translation. Noncoding RNA 2021, 7, 16. [Google Scholar] [CrossRef]
- Liu, T.; Huang, Y.; Chen, J.; Chi, H.; Yu, Z.; Wang, J.; Chen, C. Attenuated ability of BACE1 to cleave the amyloid precursor protein via silencing long noncoding RNA BACE1-AS expression. Mol. Med. Rep. 2014, 10, 1275–1281. [Google Scholar] [CrossRef] [PubMed]
- Zeng, T.; Ni, H.; Yu, Y.; Zhang, M.; Wu, M.; Wang, Q.; Wang, L.; Xu, S.; Xu, Z.; Xu, C.; et al. BACE1-AS prevents BACE1 mRNA degradation through the sequestration of BACE1-targeting miRNAs. J. Chem. Neuroanat. 2019, 98, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Ge, Y.; Liu, Q.; Li, Y.X.; Chao, X.; Guan, J.J.; Diwu, Y.C.; Zhang, Q. LncRNA BACE1-AS Promotes Autophagy-Mediated Neuronal Damage Through The miR-214-3p/ATG5 Signalling Axis In Alzheimer’s Disease. Neuroscience 2021, 455, 52–64. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, M.; Swanson, N.; Ray, S.; Buch, S.; Saraswathi, V.; Sil, S. Astrocytes in Amyloid Generation and Alcohol Metabolism: Implications of Alcohol Use in Neurological Disorder(s). Cells 2024, 13, 1173. https://doi.org/10.3390/cells13141173
Kumar M, Swanson N, Ray S, Buch S, Saraswathi V, Sil S. Astrocytes in Amyloid Generation and Alcohol Metabolism: Implications of Alcohol Use in Neurological Disorder(s). Cells. 2024; 13(14):1173. https://doi.org/10.3390/cells13141173
Chicago/Turabian StyleKumar, Mohit, Natalie Swanson, Sudipta Ray, Shilpa Buch, Viswanathan Saraswathi, and Susmita Sil. 2024. "Astrocytes in Amyloid Generation and Alcohol Metabolism: Implications of Alcohol Use in Neurological Disorder(s)" Cells 13, no. 14: 1173. https://doi.org/10.3390/cells13141173
APA StyleKumar, M., Swanson, N., Ray, S., Buch, S., Saraswathi, V., & Sil, S. (2024). Astrocytes in Amyloid Generation and Alcohol Metabolism: Implications of Alcohol Use in Neurological Disorder(s). Cells, 13(14), 1173. https://doi.org/10.3390/cells13141173