
Unravelling the Cerebellar Involvement in Autism Spectrum Disorders: Insights into Genetic Mechanisms and Developmental Pathways


Abstract
1. Introduction
2. Syndromic and Non-Syndromic ASD
3. Genomic Complexity in ASD
4. Neuropathological Mechanism of ASD
5. Cerebellar Involvement in ASD
5.1. Morphological and Functional Cerebellar Alterations in ASD
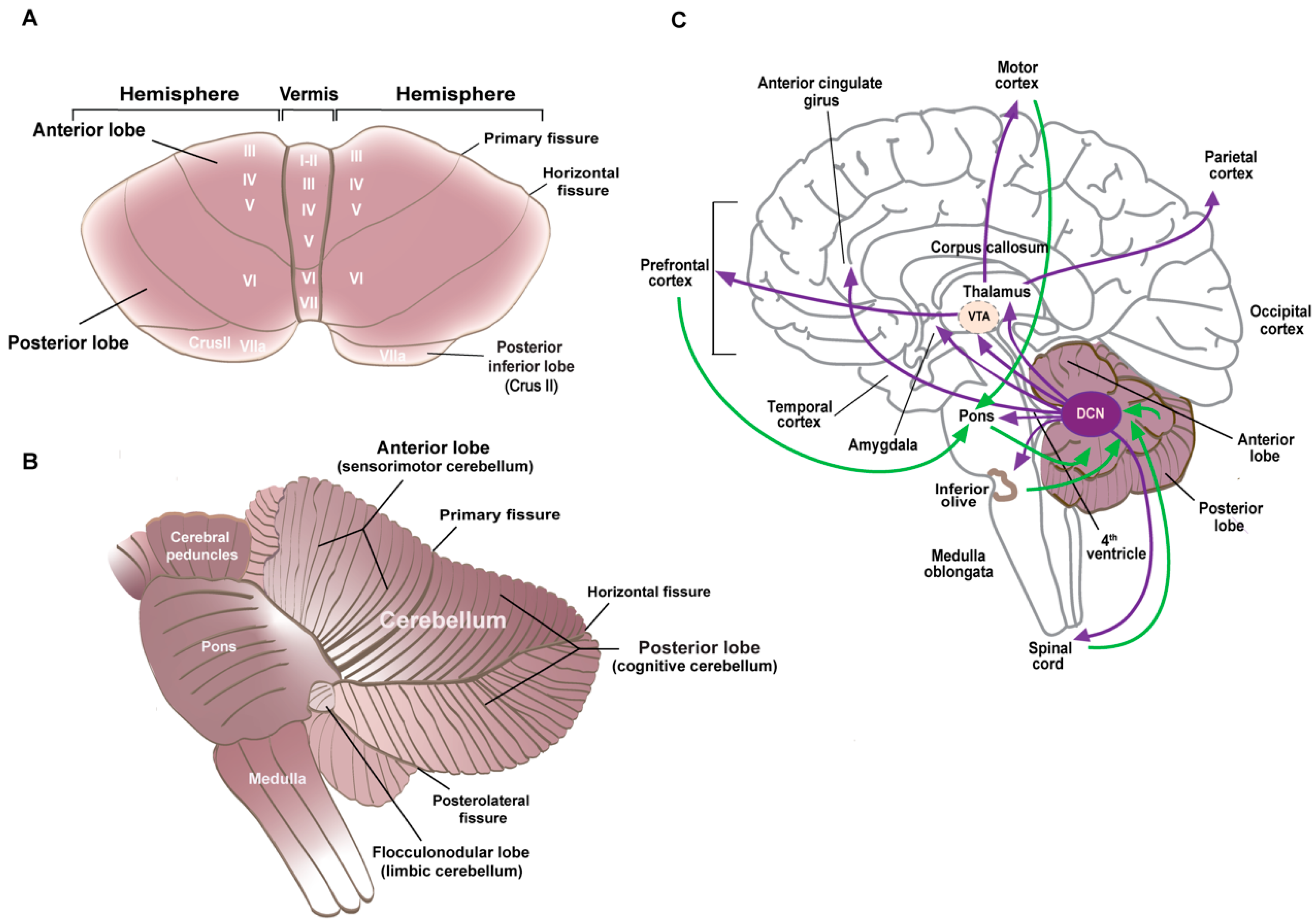
5.1.1. Cerebellar Anatomy and Neuronal Specification
5.1.2. Cerebellar Morphology and Connectivity Abnormalities in ASD
5.2. ASD Susceptibility Genes and Cerebellar Dysregulation
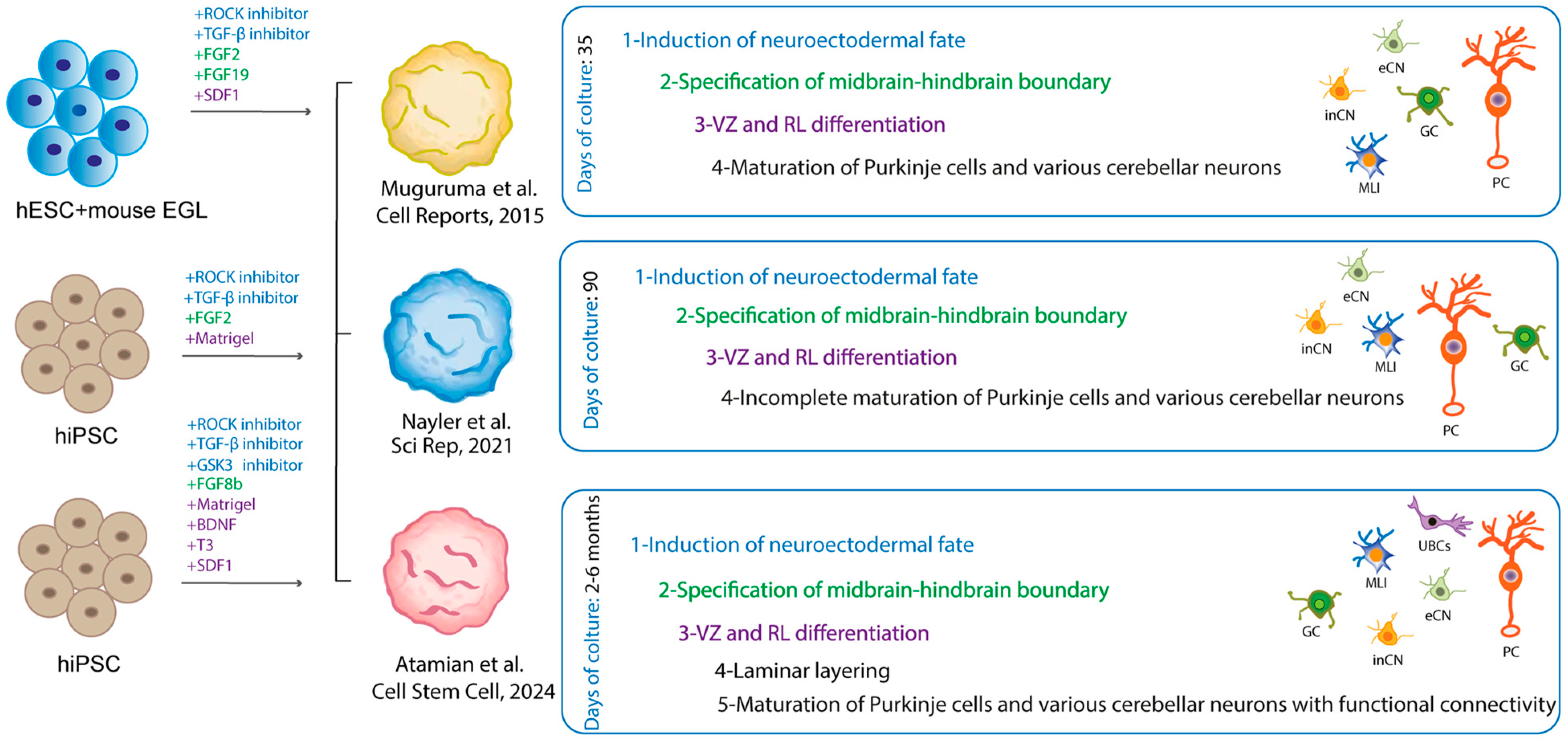
6. ASD Models to Highlight Pathology in the Cerebellum
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Glossary
References
- First, M.B. Diagnostic and statistical manual of mental disorders, 5th edition, and clinical utility. J. Nerv. Ment. Dis. 2013, 201, 727–729. [Google Scholar] [CrossRef] [PubMed]
- Sandin, S.; Lichtenstein, P.; Kuja-Halkola, R.; Hultman, C.; Larsson, H.; Reichenberg, A. The Heritability of Autism Spectrum Disorder. JAMA 2017, 318, 1182–1184. [Google Scholar] [CrossRef]
- Christensen, J.; Gronborg, T.K.; Sorensen, M.J.; Schendel, D.; Parner, E.T.; Pedersen, L.H.; Vestergaard, M. Prenatal valproate exposure and risk of autism spectrum disorders and childhood autism. JAMA 2013, 309, 1696–1703. [Google Scholar] [CrossRef]
- Castelbaum, L.; Sylvester, C.M.; Zhang, Y.; Yu, Q.; Constantino, J.N. On the Nature of Monozygotic Twin Concordance and Discordance for Autistic Trait Severity: A Quantitative Analysis. Behav. Genet. 2020, 50, 263–272. [Google Scholar] [CrossRef]
- Dworzynski, K.; Happé, F.; Bolton, P.; Ronald, A. Relationship between Symptom Domains in Autism Spectrum Disorders: A Population Based Twin Study. J. Autism Dev. Disord. 2009, 39, 1197–1210. [Google Scholar] [CrossRef] [PubMed]
- Willsey, A.J.; Sanders, S.J.; Li, M.; Dong, S.; Tebbenkamp, A.T.; Muhle, R.A.; Reilly, S.K.; Lin, L.; Fertuzinhos, S.; Miller, J.A.; et al. Coexpression Networks Implicate Human Midfetal Deep Cortical Projection Neurons in the Pathogenesis of Autism. Cell 2013, 155, 997–1007. [Google Scholar] [CrossRef]
- Wang, S.S.; Kloth, A.D.; Badura, A. The cerebellum, sensitive periods, and autism. Neuron 2014, 83, 518–532. [Google Scholar] [CrossRef] [PubMed]
- Stoodley, C.J.; Limperopoulos, C. Structure-function relationships in the developing cerebellum: Evidence from early-life cerebellar injury and neurodevelopmental disorders. Semin. Fetal Neonatal Med. 2016, 21, 356–364. [Google Scholar] [CrossRef] [PubMed]
- Vorstman, J.A.S.; Scherer, S.W. Contemplating syndromic autism. Genet. Med. 2023, 25, 100919. [Google Scholar] [CrossRef]
- Ziats, C.A.; Patterson, W.G.; Friez, M. Syndromic Autism Revisited: Review of the Literature and Lessons Learned. Pediatr. Neurol. 2021, 114, 21–25. [Google Scholar] [CrossRef]
- Sztainberg, Y.; Zoghbi, H.Y. Lessons learned from studying syndromic autism spectrum disorders. Nat. Neurosci. 2016, 19, 1408–1417. [Google Scholar] [CrossRef]
- Vyas, Y.; Cheyne, J.E.; Lee, K.; Jung, Y.; Cheung, P.Y.; Montgomery, J.M. Shankopathies in the Developing Brain in Autism Spectrum Disorders. Front. Neurosci. 2021, 15, 775431. [Google Scholar] [CrossRef]
- Phelan, K.; McDermid, H.E. The 22q13.3 Deletion Syndrome (Phelan-McDermid Syndrome). Mol. Syndromol. 2012, 2, 186–201. [Google Scholar] [CrossRef] [PubMed]
- Kloth, A.D.; Badura, A.; Li, A.; Cherskov, A.; Connolly, S.G.; Giovannucci, A.; Bangash, M.A.; Grasselli, G.; Penagarikano, O.; Piochon, C.; et al. Cerebellar associative sensory learning defects in five mouse autism models. eLife 2015, 4, e06085. [Google Scholar] [CrossRef]
- Ta, D.; Downs, J.; Baynam, G.; Wilson, A.; Richmond, P.; Leonard, H. A brief history of MECP2 duplication syndrome: 20-years of clinical understanding. Orphanet J. Rare Dis. 2022, 17, 131. [Google Scholar] [CrossRef]
- Good, K.V.; Vincent, J.B.; Ausió, J. MeCP2: The Genetic Driver of Rett Syndrome Epigenetics. Front. Genet. 2021, 12, 620859. [Google Scholar] [CrossRef] [PubMed]
- Balmer, D.; Goldstine, J.; Rao, Y.M.; LaSalle, J.M. Elevated methyl-CpG-binding protein 2 expression is acquired during postnatal human brain development and is correlated with alternative polyadenylation. J. Mol. Med. 2003, 81, 61–68. [Google Scholar] [CrossRef]
- Tillotson, R.; Bird, A. The Molecular Basis of MeCP2 Function in the Brain. J. Mol. Biol. 2020, 432, 1602–1623. [Google Scholar] [CrossRef]
- Chen, R.Z.; Akbarian, S.; Tudor, M.; Jaenisch, R. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat. Genet. 2001, 27, 327–331. [Google Scholar] [CrossRef]
- Kanner, L. Autistic disturbances of affective contact. Nerv. Child 1943, 2, 217–250. [Google Scholar]
- De Rubeis, S.; He, X.; Goldberg, A.P.; Poultney, C.S.; Samocha, K.; Ercument Cicek, A.; Kou, Y.; Liu, L.; Fromer, M.; Walker, S.; et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014, 515, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Krumm, N.; Turner, T.N.; Baker, C.; Vives, L.; Mohajeri, K.; Witherspoon, K.; Raja, A.; Coe, B.P.; Stessman, H.A.; He, Z.X.; et al. Excess of rare, inherited truncating mutations in autism. Nat. Genet. 2015, 47, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Gaugler, T.; Klei, L.; Sanders, S.J.; Bodea, C.A.; Goldberg, A.P.; Lee, A.B.; Mahajan, M.; Manaa, D.; Pawitan, Y.; Reichert, J.; et al. Most genetic risk for autism resides with common variation. Nat. Genet. 2014, 46, 881–885. [Google Scholar] [CrossRef] [PubMed]
- Hogart, A.; Wu, D.; LaSalle, J.M.; Schanen, N.C. The comorbidity of autism with the genomic disorders of chromosome 15q11.2-q13. Neurobiol. Dis. 2010, 38, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Maranga, C.; Fernandes, T.G.; Bekman, E.; da Rocha, S.T. Angelman syndrome: A journey through the brain. FEBS J. 2020, 287, 2154–2175. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Liu, Y.; Moreno, S.; Baudry, M.; Bi, X. Imbalanced Mechanistic Target of Rapamycin C1 and C2 Activity in the Cerebellum of Angelman Syndrome Mice Impairs Motor Function. J. Neurosci. 2015, 35, 4706–4718. [Google Scholar] [CrossRef] [PubMed]
- Egawa, K.; Kitagawa, K.; Inoue, K.; Takayama, M.; Takayama, C.; Saitoh, S.; Kishino, T.; Kitagawa, M.; Fukuda, A. Decreased tonic inhibition in cerebellar granule cells causes motor dysfunction in a mouse model of Angelman syndrome. Sci. Transl. Med. 2012, 4, 163ra157. [Google Scholar] [CrossRef] [PubMed]
- Vicari, S.; Napoli, E.; Cordeddu, V.; Menghini, D.; Alesi, V.; Loddo, S.; Novelli, A.; Tartaglia, M. Copy number variants in autism spectrum disorders. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2019, 92, 421–427. [Google Scholar] [CrossRef]
- Grove, J.; Ripke, S.; Als, T.D.; Mattheisen, M.; Walters, R.K.; Won, H.; Pallesen, J.; Agerbo, E.; Andreassen, O.A.; Anney, R.; et al. Identification of common genetic risk variants for autism spectrum disorder. Nat. Genet. 2019, 51, 431–444. [Google Scholar] [CrossRef]
- Matoba, N.; Liang, D.; Sun, H.; Aygün, N.; McAfee, J.C.; Davis, J.E.; Raffield, L.M.; Qian, H.; Piven, J.; Li, Y.; et al. Common genetic risk variants identified in the SPARK cohort support DDHD2 as a candidate risk gene for autism. Transl. Psychiatry 2020, 10, 265. [Google Scholar] [CrossRef]
- Visel, A.; Rubin, E.M.; Pennacchio, L.A. Genomic views of distant-acting enhancers. Nature 2009, 461, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, M.; Badayeva, Y.; Yeung, J.; Wu, J.; Abdalla-Wyse, A.; Yang, E.; Consortium, F.; Trost, B.; Scherer, S.W.; Goldowitz, D. Temporal analysis of enhancers during mouse cerebellar development reveals dynamic and novel regulatory functions. eLife 2022, 11, e74207. [Google Scholar] [CrossRef] [PubMed]
- Yeung, J.; Ha, T.J.; Swanson, D.J.; Goldowitz, D. A Novel and Multivalent Role of Pax6 in Cerebellar Development. J. Neurosci. Off. J. Soc. Neurosci. 2016, 36, 9057–9069. [Google Scholar] [CrossRef] [PubMed]
- Iossifov, I.; O’Roak, B.J.; Sanders, S.J.; Ronemus, M.; Krumm, N.; Levy, D.; Stessman, H.A.; Witherspoon, K.T.; Vives, L.; Patterson, K.E.; et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 2014, 515, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Ruzzo, E.K.; Perez-Cano, L.; Jung, J.Y.; Wang, L.K.; Kashef-Haghighi, D.; Hartl, C.; Singh, C.; Xu, J.; Hoekstra, J.N.; Leventhal, O.; et al. Inherited and De Novo Genetic Risk for Autism Impacts Shared Networks. Cell 2019, 178, 850–866.e26. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Sanders, S.J.; Liu, L.; De Rubeis, S.; Lim, E.T.; Sutcliffe, J.S.; Schellenberg, G.D.; Gibbs, R.A.; Daly, M.J.; Buxbaum, J.D.; et al. Integrated model of de novo and inherited genetic variants yields greater power to identify risk genes. PLoS Genet. 2013, 9, e1003671. [Google Scholar] [CrossRef]
- Sanders, S.J.; He, X.; Willsey, A.J.; Ercan-Sencicek, A.G.; Samocha, K.E.; Cicek, A.E.; Murtha, M.T.; Bal, V.H.; Bishop, S.L.; Dong, S.; et al. Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci. Neuron 2015, 87, 1215–1233. [Google Scholar] [CrossRef] [PubMed]
- Buxbaum, J.D.; Daly, M.J.; Devlin, B.; Lehner, T.; Roeder, K.; State, M.W. The autism sequencing consortium: Large-scale, high-throughput sequencing in autism spectrum disorders. Neuron 2012, 76, 1052–1056. [Google Scholar] [CrossRef] [PubMed]
- Satterstrom, F.K.; Kosmicki, J.A.; Wang, J.; Breen, M.S.; De Rubeis, S.; An, J.Y.; Peng, M.; Collins, R.; Grove, J.; Klei, L.; et al. Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell 2020, 180, 568–584.e23. [Google Scholar] [CrossRef]
- Zhou, X.; Feliciano, P.; Shu, C.; Wang, T.; Astrovskaya, I.; Hall, J.B.; Obiajulu, J.U.; Wright, J.R.; Murali, S.C.; Xu, S.X.; et al. Integrating de novo and inherited variants in 42,607 autism cases identifies mutations in new moderate-risk genes. Nat. Genet. 2022, 54, 1305–1319. [Google Scholar] [CrossRef]
- Fu, J.M.; Satterstrom, F.K.; Peng, M.; Brand, H.; Collins, R.L.; Dong, S.; Wamsley, B.; Klei, L.; Wang, L.; Hao, S.P.; et al. Rare coding variation provides insight into the genetic architecture and phenotypic context of autism. Nat. Genet. 2022, 54, 1320–1331. [Google Scholar] [CrossRef]
- Trost, B.; Thiruvahindrapuram, B.; Chan, A.J.S.; Engchuan, W.; Higginbotham, E.J.; Howe, J.L.; Loureiro, L.O.; Reuter, M.S.; Roshandel, D.; Whitney, J.; et al. Genomic architecture of autism from comprehensive whole-genome sequence annotation. Cell 2022, 185, 4409–4427.e18. [Google Scholar] [CrossRef]
- Parikshak, N.N.; Swarup, V.; Belgard, T.G.; Irimia, M.; Ramaswami, G.; Gandal, M.J.; Hartl, C.; Leppa, V.; Ubieta, L.T.; Huang, J.; et al. Genome-wide changes in lncRNA, splicing, and regional gene expression patterns in autism. Nature 2016, 540, 423–427. [Google Scholar] [CrossRef]
- Hagerman, R.J.; Berry-Kravis, E.; Hazlett, H.C.; Bailey, D.B., Jr.; Moine, H.; Kooy, R.F.; Tassone, F.; Gantois, I.; Sonenberg, N.; Mandel, J.L.; et al. Fragile X syndrome. Nat. Rev. Dis. Primers 2017, 3, 17065. [Google Scholar] [CrossRef]
- Milla, L.A.; Corral, L.; Rivera, J.; Zuñiga, N.; Pino, G.; Nunez-Parra, A.; Cea-Del Rio, C.A. Neurodevelopment and early pharmacological interventions in Fragile X Syndrome. Front. Neurosci. 2023, 17, 1213410. [Google Scholar] [CrossRef]
- Richter, J.D.; Zhao, X. The molecular biology of FMRP: New insights into fragile X syndrome. Nat. Rev. Neurosci. 2021, 22, 209–222. [Google Scholar] [CrossRef]
- Chen, Y.S.; Guo, L.; Han, M.; Zhang, S.M.; Chen, Y.Q.; Zou, J.; Bai, S.Y.; Cheng, G.R.; Zeng, Y. Cerebellum neuropathology and motor skill deficits in fragile X syndrome. Int. J. Dev. Neurosci. Off. J. Int. Soc. Dev. Neurosci. 2022, 82, 557–568. [Google Scholar] [CrossRef]
- Koekkoek, S.K.; Yamaguchi, K.; Milojkovic, B.A.; Dortland, B.R.; Ruigrok, T.J.; Maex, R.; De Graaf, W.; Smit, A.E.; VanderWerf, F.; Bakker, C.E.; et al. Deletion of FMR1 in Purkinje cells enhances parallel fiber LTD, enlarges spines, and attenuates cerebellar eyelid conditioning in Fragile X syndrome. Neuron 2005, 47, 339–352. [Google Scholar] [CrossRef]
- Gibson, J.M.; Vazquez, A.H.; Yamashiro, K.; Jakkamsetti, V.; Ren, C.; Lei, K.; Dentel, B.; Pascual, J.M.; Tsai, P.T. Cerebellar contribution to autism-relevant behaviors in fragile X syndrome models. Cell Rep. 2023, 42, 113533. [Google Scholar] [CrossRef] [PubMed]
- Rolland, T.; Cliquet, F.; Anney, R.J.L.; Moreau, C.; Traut, N.; Mathieu, A.; Huguet, G.; Duan, J.; Warrier, V.; Portalier, S.; et al. Phenotypic effects of genetic variants associated with autism. Nat. Med. 2023, 29, 1671–1680. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Ellis, S.E.; Ashar, F.N.; Moes, A.; Bader, J.S.; Zhan, J.; West, A.B.; Arking, D.E. Transcriptome analysis reveals dysregulation of innate immune response genes and neuronal activity-dependent genes in autism. Nat. Commun. 2014, 5, 5748. [Google Scholar] [CrossRef] [PubMed]
- Voineagu, I.; Wang, X.; Johnston, P.; Lowe, J.K.; Tian, Y.; Horvath, S.; Mill, J.; Cantor, R.M.; Blencowe, B.J.; Geschwind, D.H. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 2011, 474, 380–384. [Google Scholar] [CrossRef]
- Zerbo, O.; Leong, A.; Barcellos, L.; Bernal, P.; Fireman, B.; Croen, L.A. Immune mediated conditions in autism spectrum disorders. Brain Behav. Immun. 2015, 46, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Arenella, M.; Fanelli, G.; Kiemeney, L.A.; McAlonan, G.; Murphy, D.G.; Bralten, J. Genetic relationship between the immune system and autism. Brain Behav. Immun. Health 2023, 34, 100698. [Google Scholar] [CrossRef]
- Murtaza, N.; Uy, J.; Singh, K.K. Emerging proteomic approaches to identify the underlying pathophysiology of neurodevelopmental and neurodegenerative disorders. Mol. Autism 2020, 11, 27. [Google Scholar] [CrossRef]
- Broek, J.A.C.; Guest, P.C.; Rahmoune, H.; Bahn, S. Proteomic analysis of post mortem brain tissue from autism patients: Evidence for opposite changes in prefrontal cortex and cerebellum in synaptic connectivity-related proteins. Mol. Autism 2014, 5, 41. [Google Scholar] [CrossRef]
- Abraham, J.R.; Szoko, N.; Barnard, J.; Rubin, R.A.; Schlatzer, D.; Lundberg, K.; Li, X.; Natowicz, M.R. Proteomic Investigations of Autism Brain Identify Known and Novel Pathogenetic Processes. Sci. Rep. 2019, 9, 13118. [Google Scholar] [CrossRef] [PubMed]
- Pintacuda, G.; Hsu, Y.-H.H.; Tsafou, K.; Li, K.W.; Martín, J.M.; Riseman, J.; Biagini, J.C.; Ching, J.K.T.; Mena, D.; Gonzalez-Lozano, M.A.; et al. Protein interaction studies in human induced neurons indicate convergent biology underlying autism spectrum disorders. Cell Genom. 2023, 3, 100250. [Google Scholar] [CrossRef]
- Ramnani, N. The primate cortico-cerebellar system: Anatomy and function. Nat. Rev. Neurosci. 2006, 7, 511–522. [Google Scholar] [CrossRef]
- Van Overwalle, F.; Manto, M.; Cattaneo, Z.; Clausi, S.; Ferrari, C.; Gabrieli, J.D.E.; Guell, X.; Heleven, E.; Lupo, M.; Ma, Q.; et al. Consensus Paper: Cerebellum and Social Cognition. Cerebellum 2020, 19, 833–868. [Google Scholar] [CrossRef]
- Stoodley, C.J.; Schmahmann, J.D. Evidence for topographic organization in the cerebellum of motor control versus cognitive and affective processing. Cortex 2010, 46, 831–844. [Google Scholar] [CrossRef]
- Stoodley, C.J.; Tsai, P.T. Adaptive Prediction for Social Contexts: The Cerebellar Contribution to Typical and Atypical Social Behaviors. Annu. Rev. Neurosci. 2021, 44, 475–493. [Google Scholar] [CrossRef]
- Mapelli, L.; Soda, T.; D’Angelo, E.; Prestori, F. The Cerebellar Involvement in Autism Spectrum Disorders: From the Social Brain to Mouse Models. Int. J. Mol. Sci. 2022, 23, 3894. [Google Scholar] [CrossRef]
- Fetit, R.; Hillary, R.F.; Price, D.J.; Lawrie, S.M. The neuropathology of autism: A systematic review of post-mortem studies of autism and related disorders. Neurosci. Biobehav. Rev. 2021, 129, 35–62. [Google Scholar] [CrossRef]
- De Zeeuw, C.I.; Lisberger, S.G.; Raymond, J.L. Diversity and dynamism in the cerebellum. Nat. Neurosci. 2021, 24, 160–167. [Google Scholar] [CrossRef]
- Hull, C.; Regehr, W.G. The Cerebellar Cortex. Annu. Rev. Neurosci. 2022, 45, 151–175. [Google Scholar] [CrossRef]
- Haldipur, P.; Bharti, U.; Alberti, C.; Sarkar, C.; Gulati, G.; Iyengar, S.; Gressens, P.; Mani, S. Preterm delivery disrupts the developmental program of the cerebellum. PLoS ONE 2011, 6, e23449. [Google Scholar] [CrossRef]
- Aldinger, K.A.; Thomson, Z.; Phelps, I.G.; Haldipur, P.; Deng, M.; Timms, A.E.; Hirano, M.; Santpere, G.; Roco, C.; Rosenberg, A.B.; et al. Spatial and cell type transcriptional landscape of human cerebellar development. Nat. Neurosci. 2021, 24, 1163–1175. [Google Scholar] [CrossRef]
- Zhong, S.; Wang, M.; Huang, L.; Chen, Y.; Ge, Y.; Zhang, J.; Shi, Y.; Dong, H.; Zhou, X.; Wang, B.; et al. Single-cell epigenomics and spatiotemporal transcriptomics reveal human cerebellar development. Nat. Commun. 2023, 14, 7613. [Google Scholar] [CrossRef] [PubMed]
- Haldipur, P.; Millen, K.J.; Aldinger, K.A. Human Cerebellar Development and Transcriptomics: Implications for Neurodevelopmental Disorders. Annu. Rev. Neurosci. 2022, 45, 515–531. [Google Scholar] [CrossRef]
- Haldipur, P.; Aldinger, K.A.; Bernardo, S.; Deng, M.; Timms, A.E.; Overman, L.M.; Winter, C.; Lisgo, S.N.; Razavi, F.; Silvestri, E.; et al. Spatiotemporal expansion of primary progenitor zones in the developing human cerebellum. Science 2019, 366, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Courchesne, E.; Hesselink, J.R.; Jernigan, T.L.; Yeung-Courchesne, R. Abnormal neuroanatomy in a nonretarded person with autism. Unusual findings with magnetic resonance imaging. Arch. Neurol. 1987, 44, 335–341. [Google Scholar] [CrossRef]
- Scott, J.A.; Schumann, C.M.; Goodlin-Jones, B.L.; Amaral, D.G. A comprehensive volumetric analysis of the cerebellum in children and adolescents with autism spectrum disorder. Autism Res. Off. J. Int. Soc. Autism Res. 2009, 2, 246–257. [Google Scholar] [CrossRef]
- Webb, S.J.; Sparks, B.F.; Friedman, S.D.; Shaw, D.W.; Giedd, J.; Dawson, G.; Dager, S.R. Cerebellar vermal volumes and behavioral correlates in children with autism spectrum disorder. Psychiatry Res. 2009, 172, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Hiremath, C.; Khokhar, S.K.; Bansal, E.; Sagar, K.J.V.; Padmanabha, H.; Girimaji, A.S.; Narayan, S.; Kishore, M.T.; Yamini, B.K.; et al. Altered cerebellar lobular volumes correlate with clinical deficits in siblings and children with ASD: Evidence from toddlers. J. Transl. Med. 2023, 21, 246. [Google Scholar] [CrossRef] [PubMed]
- D’Mello, A.M.; Crocetti, D.; Mostofsky, S.H.; Stoodley, C.J. Cerebellar gray matter and lobular volumes correlate with core autism symptoms. NeuroImage Clin. 2015, 7, 631–639. [Google Scholar] [CrossRef]
- Laidi, C.; Floris, D.L.; Tillmann, J.; Elandaloussi, Y.; Zabihi, M.; Charman, T.; Wolfers, T.; Durston, S.; Moessnang, C.; Dell’Acqua, F.; et al. Cerebellar Atypicalities in Autism? Biol. Psychiatry 2022, 92, 674–682. [Google Scholar] [CrossRef]
- Limperopoulos, C.; Bassan, H.; Gauvreau, K.; Robertson, R.L., Jr.; Sullivan, N.R.; Benson, C.B.; Avery, L.; Stewart, J.; Soul, J.S.; Ringer, S.A.; et al. Does cerebellar injury in premature infants contribute to the high prevalence of long-term cognitive, learning, and behavioral disability in survivors? Pediatrics 2007, 120, 584–593. [Google Scholar] [CrossRef]
- Limperopoulos, C.; Chilingaryan, G.; Guizard, N.; Robertson, R.L.; Du Plessis, A.J. Cerebellar injury in the premature infant is associated with impaired growth of specific cerebral regions. Pediatr. Res. 2010, 68, 145–150. [Google Scholar] [CrossRef]
- Fatemi, S.H.; Halt, A.R.; Realmuto, G.; Earle, J.; Kist, D.A.; Thuras, P.; Merz, A. Purkinje cell size is reduced in cerebellum of patients with autism. Cell. Mol. Neurobiol. 2002, 22, 171–175. [Google Scholar] [CrossRef]
- Skefos, J.; Cummings, C.; Enzer, K.; Holiday, J.; Weed, K.; Levy, E.; Yuce, T.; Kemper, T.; Bauman, M. Regional alterations in purkinje cell density in patients with autism. PLoS ONE 2014, 9, e81255. [Google Scholar] [CrossRef]
- Fatemi, S.H.; Folsom, T.D.; Reutiman, T.J.; Thuras, P.D. Expression of GABA(B) receptors is altered in brains of subjects with autism. Cerebellum 2009, 8, 64–69. [Google Scholar] [CrossRef]
- Nicolini, C.; Fahnestock, M. The valproic acid-induced rodent model of autism. Exp. Neurol. 2018, 299, 217–227. [Google Scholar] [CrossRef]
- Guerra, M.; Medici, V.; Weatheritt, R.; Corvino, V.; Palacios, D.; Geloso, M.C.; Farini, D.; Sette, C. Fetal exposure to valproic acid dysregulates the expression of autism-linked genes in the developing cerebellum. Transl. Psychiatry 2023, 13, 114. [Google Scholar] [CrossRef]
- Roux, S.; Bailly, Y.; Bossu, J.L. Regional and sex-dependent alterations in Purkinje cell density in the valproate mouse model of autism. Neuroreport 2019, 30, 82–88. [Google Scholar] [CrossRef]
- Meyza, K.Z.; Blanchard, D.C. The BTBR mouse model of idiopathic autism—Current view on mechanisms. Neurosci. Biobehav. Rev. 2017, 76, 99–110. [Google Scholar] [CrossRef]
- Kiffmeyer, E.A.; Cosgrove, J.A.; Siganos, J.K.; Bien, H.E.; Vipond, J.E.; Vogt, K.R.; Kloth, A.D. Deficits in cerebellum-dependent learning and cerebellar morphology in male and female BTBR autism model mice. NeuroSci 2022, 3, 624–644. [Google Scholar] [CrossRef]
- Xiao, R.; Zhong, H.; Li, X.; Ma, Y.; Zhang, R.; Wang, L.; Zang, Z.; Fan, X. Abnormal Cerebellar Development Is Involved in Dystonia-Like Behaviors and Motor Dysfunction of Autistic BTBR Mice. Front. Cell Dev. Biol. 2020, 8, 231. [Google Scholar] [CrossRef]
- Greco, C.M.; Navarro, C.S.; Hunsaker, M.R.; Maezawa, I.; Shuler, J.F.; Tassone, F.; Delany, M.; Au, J.W.; Berman, R.F.; Jin, L.W.; et al. Neuropathologic features in the hippocampus and cerebellum of three older men with fragile X syndrome. Mol. Autism 2011, 2, 2. [Google Scholar] [CrossRef] [PubMed]
- Huber, K.M. The fragile X-cerebellum connection. Trends Neurosci. 2006, 29, 183–185. [Google Scholar] [CrossRef] [PubMed]
- Ertan, G.; Arulrajah, S.; Tekes, A.; Jordan, L.; Huisman, T.A. Cerebellar abnormality in children and young adults with tuberous sclerosis complex: MR and diffusion weighted imaging findings. J. Neuroradiol. 2010, 37, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Tsai, P.T.; Hull, C.; Chu, Y.; Greene-Colozzi, E.; Sadowski, A.R.; Leech, J.M.; Steinberg, J.; Crawley, J.N.; Regehr, W.G.; Sahin, M. Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature 2012, 488, 647–651. [Google Scholar] [CrossRef] [PubMed]
- D’Mello, A.M.; Stoodley, C.J. Cerebro-cerebellar circuits in autism spectrum disorder. Front. Neurosci. 2015, 9, 408. [Google Scholar] [CrossRef] [PubMed]
- Mostofsky, S.H.; Powell, S.K.; Simmonds, D.J.; Goldberg, M.C.; Caffo, B.; Pekar, J.J. Decreased connectivity and cerebellar activity in autism during motor task performance. Brain J. Neurol. 2009, 132, 2413–2425. [Google Scholar] [CrossRef] [PubMed]
- Unruh, K.E.; Bartolotti, J.V.; McKinney, W.S.; Schmitt, L.M.; Sweeney, J.A.; Mosconi, M.W. Functional connectivity of cortical-cerebellar networks in relation to sensorimotor behavior and clinical features in autism spectrum disorder. Cereb. Cortex 2023, 33, 8990–9002. [Google Scholar] [CrossRef] [PubMed]
- Jack, A.; Pelphrey, K.A. Annual Research Review: Understudied populations within the autism spectrum—Current trends and future directions in neuroimaging research. J. Child Psychol. Psychiatry Allied Discip. 2017, 58, 411–435. [Google Scholar] [CrossRef] [PubMed]
- Kana, R.K.; Maximo, J.O.; Williams, D.L.; Keller, T.A.; Schipul, S.E.; Cherkassky, V.L.; Minshew, N.J.; Just, M.A. Aberrant functioning of the theory-of-mind network in children and adolescents with autism. Mol. Autism 2015, 6, 59. [Google Scholar] [CrossRef]
- Oldehinkel, M.; Mennes, M.; Marquand, A.; Charman, T.; Tillmann, J.; Ecker, C.; Dell’Acqua, F.; Brandeis, D.; Banaschewski, T.; Baumeister, S.; et al. Altered Connectivity Between Cerebellum, Visual, and Sensory-Motor Networks in Autism Spectrum Disorder: Results from the EU-AIMS Longitudinal European Autism Project. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 2019, 4, 260–270. [Google Scholar] [CrossRef] [PubMed]
- Cerliani, L.; Mennes, M.; Thomas, R.M.; Di Martino, A.; Thioux, M.; Keysers, C. Increased Functional Connectivity Between Subcortical and Cortical Resting-State Networks in Autism Spectrum Disorder. JAMA Psychiatry 2015, 72, 767–777. [Google Scholar] [CrossRef]
- Khan, A.J.; Nair, A.; Keown, C.L.; Datko, M.C.; Lincoln, A.J.; Müller, R.-A. Cerebro-cerebellar Resting-State Functional Connectivity in Children and Adolescents with Autism Spectrum Disorder. Biol. Psychiatry 2015, 78, 625–634. [Google Scholar] [CrossRef]
- Verly, M.; Verhoeven, J.; Zink, I.; Mantini, D.; Peeters, R.; Deprez, S.; Emsell, L.; Boets, B.; Noens, I.; Steyaert, J.; et al. Altered functional connectivity of the language network in ASD: Role of classical language areas and cerebellum. NeuroImage Clin. 2014, 4, 374–382. [Google Scholar] [CrossRef] [PubMed]
- Okada, N.J.; Liu, J.; Tsang, T.; Nosco, E.; McDonald, N.M.; Cummings, K.K.; Jung, J.; Patterson, G.; Bookheimer, S.Y.; Green, S.A.; et al. Atypical cerebellar functional connectivity at 9 months of age predicts delayed socio-communicative profiles in infants at high and low risk for autism. J. Child Psychol. Psychiatry Allied Discip. 2022, 63, 1002–1016. [Google Scholar] [CrossRef] [PubMed]
- Marek, S.; Siegel, J.S.; Gordon, E.M.; Raut, R.V.; Gratton, C.; Newbold, D.J.; Ortega, M.; Laumann, T.O.; Adeyemo, B.; Miller, D.B.; et al. Spatial and Temporal Organization of the Individual Human Cerebellum. Neuron 2018, 100, 977–993.e7. [Google Scholar] [CrossRef] [PubMed]
- Clifford, H.; Dulneva, A.; Ponting, C.P.; Haerty, W.; Becker, E.B.E. A gene expression signature in developing Purkinje cells predicts autism and intellectual disability co-morbidity status. Sci. Rep. 2019, 9, 485. [Google Scholar] [CrossRef] [PubMed]
- Sepp, M.; Leiss, K.; Murat, F.; Okonechnikov, K.; Joshi, P.; Leushkin, E.; Spanig, L.; Mbengue, N.; Schneider, C.; Schmidt, J.; et al. Cellular development and evolution of the mammalian cerebellum. Nature 2024, 625, 788–796. [Google Scholar] [CrossRef] [PubMed]
- Sydnor, L.M.; Aldinger, K.A. Structure, Function, and Genetics of the Cerebellum in Autism. J. Psychiatry Brain Sci. 2022, 7, e220008. [Google Scholar] [CrossRef]
- Brandenburg, C.; Griswold, A.J.; Van Booven, D.J.; Kilander, M.B.C.; Frei, J.A.; Nestor, M.W.; Dykxhoorn, D.M.; Pericak-Vance, M.A.; Blatt, G.J. Transcriptomic analysis of isolated and pooled human postmortem cerebellar Purkinje cells in autism spectrum disorders. Front. Genet. 2022, 13, 944837. [Google Scholar] [CrossRef] [PubMed]
- Ament, S.A.; Cortes-Gutierrez, M.; Herb, B.R.; Mocci, E.; Colantuoni, C.; McCarthy, M.M. A single-cell genomic atlas for maturation of the human cerebellum during early childhood. Sci. Transl. Med. 2023, 15, eade1283. [Google Scholar] [CrossRef] [PubMed]
- Prata, J.; Machado, A.S.; von Doellinger, O.; Almeida, M.I.; Barbosa, M.A.; Coelho, R.; Santos, S.G. The Contribution of Inflammation to Autism Spectrum Disorders: Recent Clinical Evidence. Methods Mol. Biol. 2019, 2011, 493–510. [Google Scholar] [CrossRef]
- Ha, S.; Lee, D.; Cho, Y.S.; Chung, C.; Yoo, Y.E.; Kim, J.; Lee, J.; Kim, W.; Kim, H.; Bae, Y.C.; et al. Cerebellar Shank2 Regulates Excitatory Synapse Density, Motor Coordination, and Specific Repetitive and Anxiety-Like Behaviors. J. Neurosci. Off. J. Soc. Neurosci. 2016, 36, 12129–12143. [Google Scholar] [CrossRef]
- Peter, S.; ten Brinke, M.M.; Stedehouder, J.; Reinelt, C.M.; Wu, B.; Zhou, H.; Zhou, K.; Boele, H.-J.; Kushner, S.A.; Lee, M.G.; et al. Dysfunctional cerebellar Purkinje cells contribute to autism-like behaviour in Shank2-deficient mice. Nat. Commun. 2016, 7, 12627. [Google Scholar] [CrossRef] [PubMed]
- Watson, L.M.; Wong, M.M.K.; Vowles, J.; Cowley, S.A.; Becker, E.B.E. A Simplified Method for Generating Purkinje Cells from Human-Induced Pluripotent Stem Cells. Cerebellum 2018, 17, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Behesti, H.; Kocabas, A.; Buchholz, D.E.; Carroll, T.S.; Hatten, M.E. Altered temporal sequence of transcriptional regulators in the generation of human cerebellar granule cells. eLife 2021, 10, e67074. [Google Scholar] [CrossRef] [PubMed]
- Sundberg, M.; Tochitsky, I.; Buchholz, D.E.; Winden, K.; Kujala, V.; Kapur, K.; Cataltepe, D.; Turner, D.; Han, M.-J.; Woolf, C.J.; et al. Purkinje cells derived from TSC patients display hypoexcitability and synaptic deficits associated with reduced FMRP levels and reversed by rapamycin. Mol. Psychiatry 2018, 23, 2167–2183. [Google Scholar] [CrossRef] [PubMed]
- Muguruma, K.; Nishiyama, A.; Kawakami, H.; Hashimoto, K.; Sasai, Y. Self-Organization of Polarized Cerebellar Tissue in 3D Culture of Human Pluripotent Stem Cells. Cell Rep. 2015, 10, 537–550. [Google Scholar] [CrossRef] [PubMed]
- Nayler, S.; Agarwal, D.; Curion, F.; Bowden, R.; Becker, E.B.E. High-resolution transcriptional landscape of xeno-free human induced pluripotent stem cell-derived cerebellar organoids. Sci. Rep. 2021, 11, 12959. [Google Scholar] [CrossRef] [PubMed]
- Atamian, A.; Birtele, M.; Hosseini, N.; Nguyen, T.; Seth, A.; Del Dosso, A.; Paul, S.; Tedeschi, N.; Taylor, R.; Coba, M.P.; et al. Human cerebellar organoids with functional Purkinje cells. Cell Stem Cell 2024, 31, 39–51.e6. [Google Scholar] [CrossRef] [PubMed]
- Levy, R.J.; Paşca, S.P. What Have Organoids and Assembloids Taught Us About the Pathophysiology of Neuropsychiatric Disorders? Biol. Psychiatry 2023, 93, 632–641. [Google Scholar] [CrossRef]
- Schaaf, C.P.; Betancur, C.; Yuen, R.K.C.; Parr, J.R.; Skuse, D.H.; Gallagher, L.; Bernier, R.A.; Buchanan, J.A.; Buxbaum, J.D.; Chen, C.A.; et al. A framework for an evidence-based gene list relevant to autism spectrum disorder. Nat. Rev. Genet. 2020, 21, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Jacob, S.; Wolff, J.J.; Steinbach, M.S.; Doyle, C.B.; Kumar, V.; Elison, J.T. Neurodevelopmental heterogeneity and computational approaches for understanding autism. Transl. Psychiatry 2019, 9, 63–74. [Google Scholar] [CrossRef]
- Uddin, M.; Wang, Y.; Woodbury-Smith, M. Artificial intelligence for precision medicine in neurodevelopmental disorders. NPJ Digit. Med. 2019, 2, 112–121. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ADNP | Transcription factor that may mediate neuroprotective effects in normal growth and cancer |
| ANK2 | Member of ankyrin family of proteins that link the integral membrane proteins to cytoskeleton |
| ANKRD11 | Ankyrin repeat domain-containing protein. Inhibits ligand-dependent activation of transcription |
| ARID1B | Component of the SWI/SNF chromatin remodeling complex. May play a role in cell cycle activation |
| ASH1L | Member of the trithorax group of transcriptional activators |
| ASXL3 | Putative Polycomb group protein involved in the transcriptionally repressive state during development |
| CHD2 | DNA-binding helicase that binds to the promoter of target genes, leading to chromatin remodeling |
| CHD8 | DNA helicase that acts as a chromatin remodeling factor and regulates transcription |
| CTNNB1 | Part of a complex of proteins that constitute adherens junctions |
| DEAF1 | Transcription factor that binds to retinoic acid response element. Inhibitor of cell proliferation |
| DNMT3A | DNA methyltransferase involved in epigenetic modifications |
| DYNC1H1 | Dynein 1. Acts as a motor for the intracellular retrograde motility of vesicles and organelles along microtubules |
| DYRK1A | Kinase that promotes cell survival |
| FOXP1 | Transcriptional factor regulator of tissue- and cell type-specific gene transcription during development |
| GIGYF1 | Component of gyf family of adaptor protein involved in tyrosine kinase receptor signaling |
| GRIN2B | NMDA receptor family member involved in excitatory synaptic transmission in the CNS |
| IRF2BPL | E3 ubiquitin protein ligase involved in the degradation of target proteins in CNS development |
| KCNQ3 | Member of the family of potassium channel proteins |
| KDM6B | Lysine-specific demethylase and epigenetic modifier during cellular differentiation and development |
| MBD5 | Member of the methyl-CpG-binding domain (MBD) family. Is involved in cell division, growth, and differentiation |
| MED13L | Component of the mediator complex involved in the transcription of RNA polymerase II-dependent genes |
| MYT1L | Member of the zinc finger superfamily of transcription factors, found only in neuronal tissues |
| PHF21A | Component of the BHC complex that represses transcription of neuron-specific genes in non-neuronal cells |
| POGZ | Plays a role in mitotic cell cycle progression, kinetochore assembly, and mitotic sister chromatid cohesion |
| PTEN | Phosphatidylinositol-3,4,5-trisphosphate 3-phosphatase involved in tumor suppression |
| RAI1 | Transcriptional regulator of the circadian clock components involved development and neuronal differentiation |
| RFX3 | Transcriptional activator factor that binds DNA, required for cell differentiation in endocrine pancreas development |
| RORB | Protein of family of orphan nuclear receptors involved in cytoarchitectural patterning of neocortical neurons |
| SCN2A | Voltage-gated sodium channel subunit, responsible for action potential initiation and propagation in excitable cells |
| SETD5 | Regulator of chromatin and RNA elongation rate; crucial in NSC proliferation and synaptic transmission |
| SHANK2 | Synaptic protein that functions as a molecular scaffold in the postsynaptic density of excitatory synapses |
| SHANK3 | Synaptic protein that functions as a molecular scaffold in the postsynaptic density of excitatory synapses |
| SLC6A1 | Gamma-aminobutyric acid (GABA) transporter responsible for the reuptake of GABA from the synapse |
| SMARCC2 | Component of SWI/SNF neuronal progenitor-specific chromatin remodeling complexes |
| STXBP1 | Syntaxin-binding protein that participates in the regulation of synaptic vesicle docking and fusion |
| SYNGAP1 | Major constituent of the PSD essential for postsynaptic signaling and synaptic plasticity |
| TAOK1 | Serine/threonine-protein kinase involved in DNA damage response and regulation of cytoskeleton stability |
| TBR1 | Transcriptional repressor involved in cortical development, neuronal migration, and axonal projection |
| TCF20 | Transcriptional activator that binds to the regulatory region of MMP3 and thereby controls stromelysin expression |
| TCF4 | Transcription factor involved in the initiation of neuronal differentiation |
| TLK2 | Serine/threonine-protein kinase involved in DNA replication, transcription, repair, and chromosome segregation |
| TRIP12 | E3 ubiquitin-protein ligase involved in the ubiquitin fusion degradation (UFD) pathway and regulation of DNA repair |
| WAC | Signaling protein involved in cell-cycle checkpoint activation in response to DNA damage |
| FOXP1 | Transcriptional factor regulator of tissue- and cell type-specific gene transcription during development |
| FOXP2 | Transcriptional factor expressed in fetal and adult brains and involved in development |
| GABRB2 | Gamma-aminobutyric acid (GABA) A receptor mediating inhibitory synaptic transmission in the central nervous system |
| GRIN1 | Subunit of N-methyl-D-aspartate receptors, members of the glutamate receptor channel superfamily involved in synaptic plasticity |
| KCNB1 | Member of the family of potassium channel proteins |
| KCNQ3 | Member of the family of potassium channel proteins |
| MAGEL2 | Gene involved in the terminal differentiation of neurons |
| MYT1L | Member of the zinc finger superfamily of transcription factors, found only in neuronal tissues |
| NRXN1 | Cell-surface receptors involved in synapse formation in the central nervous system and required for efficient transmission |
| NRXN3 | Cell-surface receptors involved in synapse formation in the central nervous system and required for efficient transmission |
| RORB | Protein of family of orphan nuclear receptors involved in cytoarchitectural patterning of neocortical neurons |
| SCN2A | Voltage-gated sodium channel subunit, responsible for action potential initiation and propagation in excitable cells |
| SHANK2 | Synaptic protein that functions as a molecular scaffold in the postsynaptic density of excitatory synapses |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guerra, M.; Medici, V.; La Sala, G.; Farini, D. Unravelling the Cerebellar Involvement in Autism Spectrum Disorders: Insights into Genetic Mechanisms and Developmental Pathways. Cells 2024, 13, 1176. https://doi.org/10.3390/cells13141176
Guerra M, Medici V, La Sala G, Farini D. Unravelling the Cerebellar Involvement in Autism Spectrum Disorders: Insights into Genetic Mechanisms and Developmental Pathways. Cells. 2024; 13(14):1176. https://doi.org/10.3390/cells13141176
Chicago/Turabian StyleGuerra, Marika, Vanessa Medici, Gina La Sala, and Donatella Farini. 2024. "Unravelling the Cerebellar Involvement in Autism Spectrum Disorders: Insights into Genetic Mechanisms and Developmental Pathways" Cells 13, no. 14: 1176. https://doi.org/10.3390/cells13141176
APA StyleGuerra, M., Medici, V., La Sala, G., & Farini, D. (2024). Unravelling the Cerebellar Involvement in Autism Spectrum Disorders: Insights into Genetic Mechanisms and Developmental Pathways. Cells, 13(14), 1176. https://doi.org/10.3390/cells13141176