
Focal Adhesion Kinase and Colony Stimulating Factors: Intestinal Homeostasis and Innate Immunity Crosstalk


{kind=link}
{kind=link}
{kind=link}
Abstract
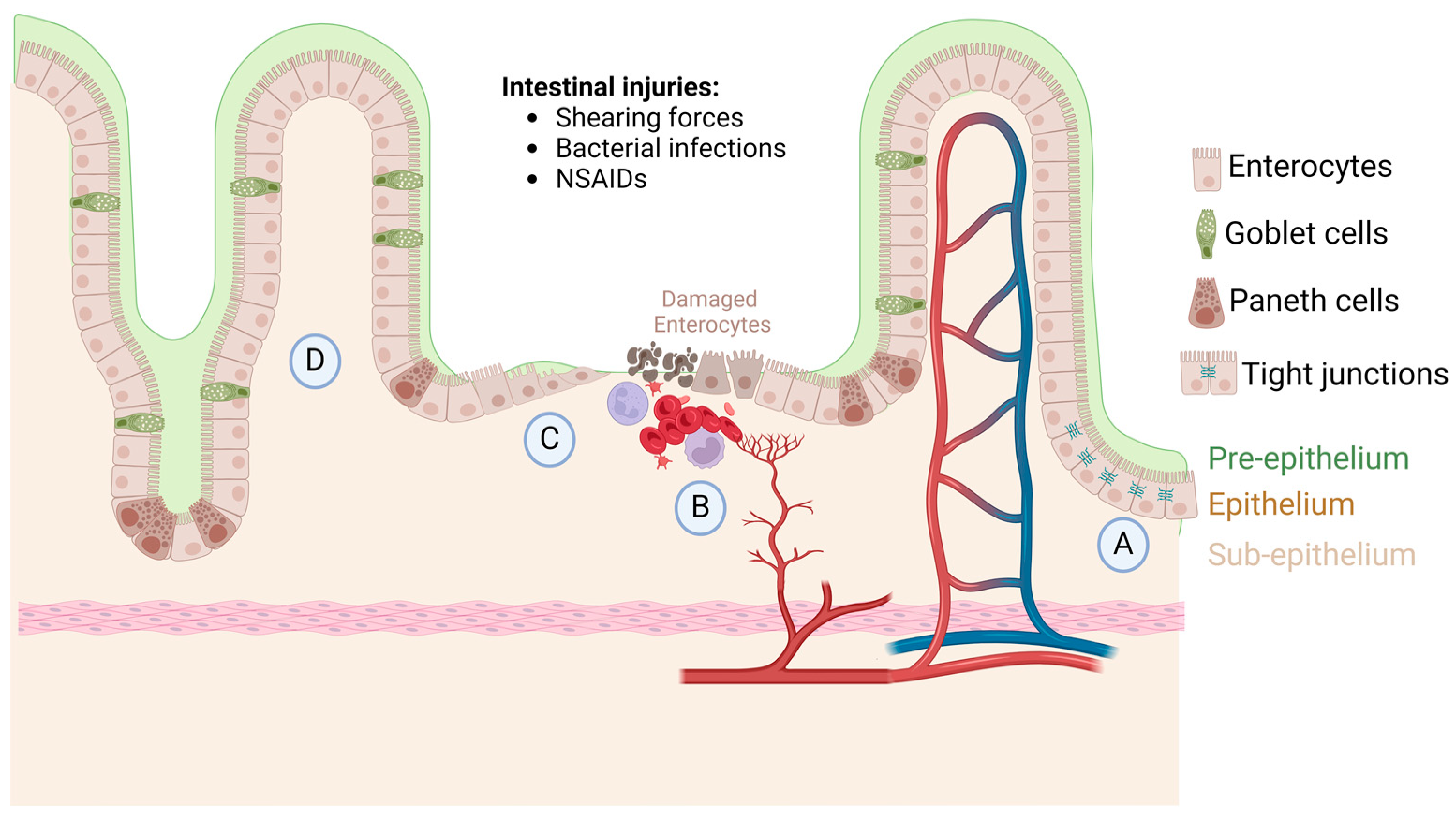
1. Intestinal Homeostasis, Injury, and Repair
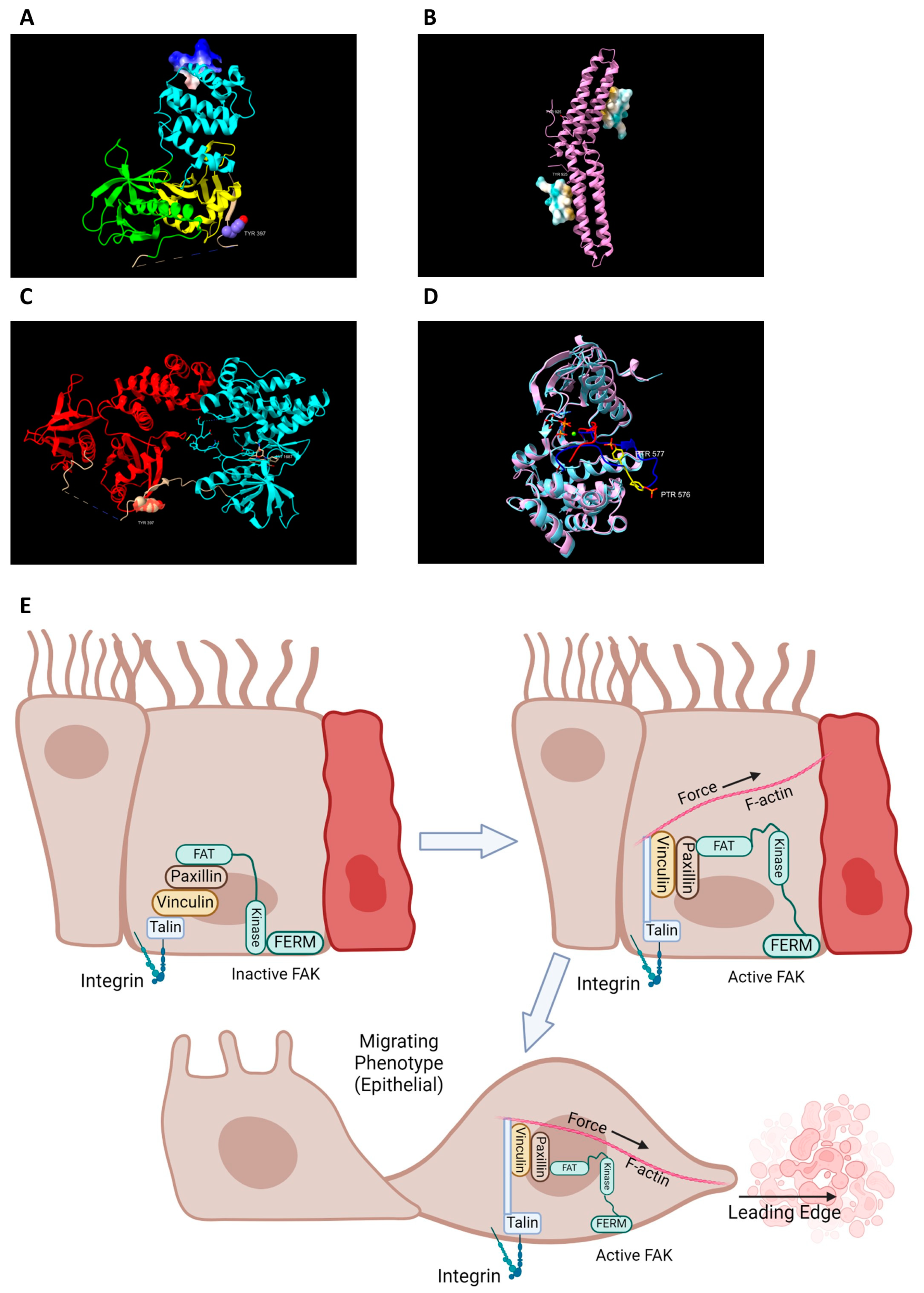
2. FAK Activation and Its Role in Intestinal Healing
3. Small FAK Activating Drugs
4. Role of FAK in Chemotaxis of Immune Cells
5. Macrophage Polarization and Response in Intestinal Healing
6. Waterfall Effect: Continuum of Macrophage Differentiation in Intestinal Healing
7. Resident Macrophages Function in Maintaining Epithelial Integrity
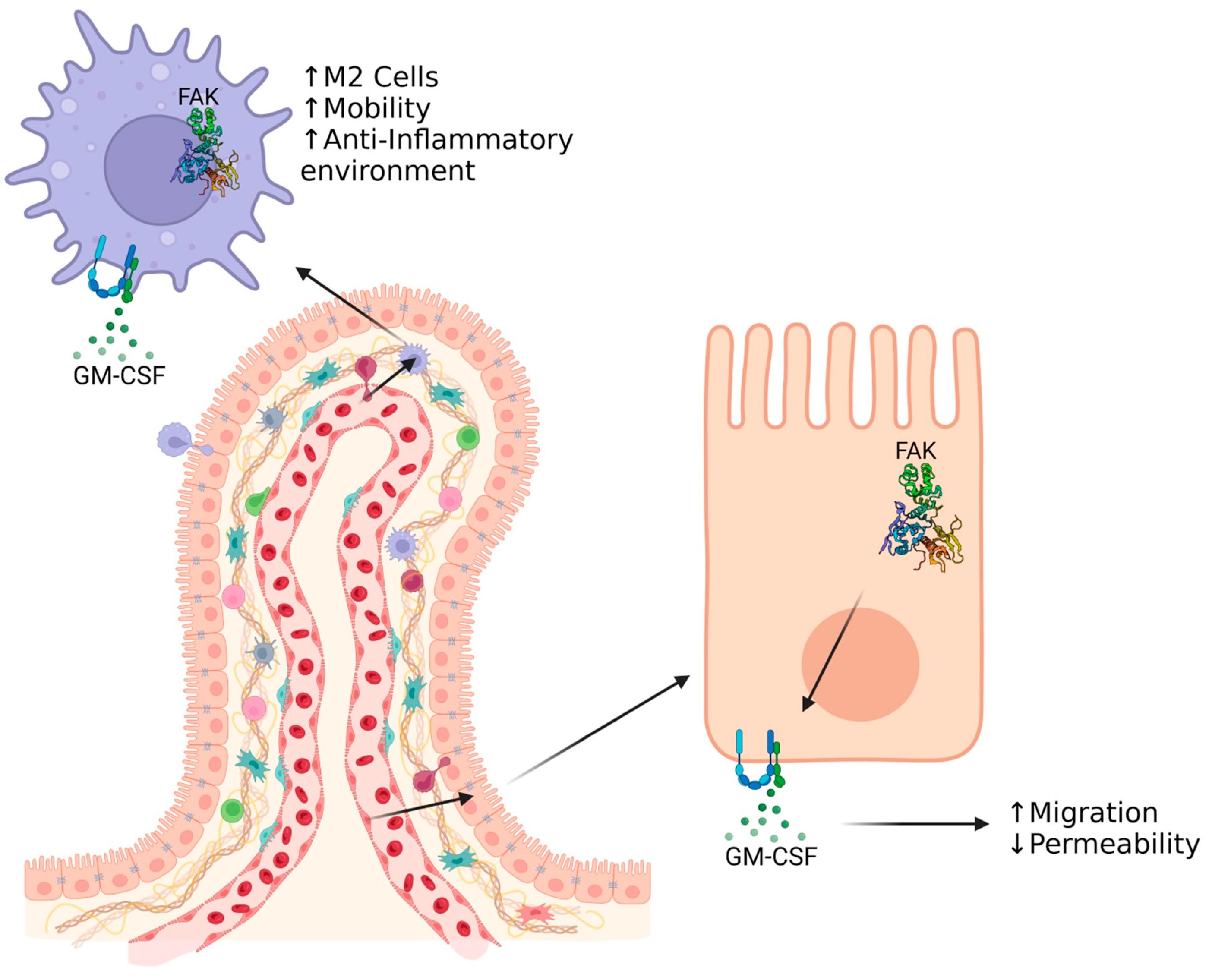
8. GM-CSF as a Factor for Intestinal Healing
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Oncel, S.; Basson, M.D. Gut homeostasis, injury, and healing: New therapeutic targets. World J. Gastroenterol. 2022, 28, 1725–1750. [Google Scholar] [CrossRef]
- Lichtenberger, L.M.; Trier, J.S. Changes in gastrin levels, food intake, and duodenal mucosal growth during lactation. Am. J. Physiol. 1979, 237, E98–E105. [Google Scholar] [CrossRef]
- Matuz, J. Role of mucus in mucosal protection through ethanol and pepsin damage models. Acta Physiol. Hung. 1992, 80, 189–194. [Google Scholar]
- Atuma, C.; Strugala, V.; Allen, A.; Holm, L. The adherent gastrointestinal mucus gel layer: Thickness and physical state in vivo. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 280, G922–G929. [Google Scholar] [CrossRef]
- Takezono, Y.; Joh, T.; Oshima, T.; Suzuki, H.; Seno, K.; Yokoyama, Y.; Alexander, J.S.; Itoh, M. Role of prostaglandins in maintaining gastric mucus-cell permeability against acid exposure. J. Lab. Clin. Med. 2004, 143, 52–58. [Google Scholar] [CrossRef]
- Wallace, J.L. Prostaglandins, NSAIDs, and gastric mucosal protection: Why doesn’t the stomach digest itself? Physiol. Rev. 2008, 88, 1547–1565. [Google Scholar] [CrossRef] [PubMed]
- Di Tommaso, N.; Gasbarrini, A.; Ponziani, F.R. Intestinal Barrier in Human Health and Disease. Int. J. Environ. Res. Public. Health 2021, 18, 12836. [Google Scholar] [CrossRef]
- Yibirin, M.; De Oliveira, D.; Valera, R.; Plitt, A.E.; Lutgen, S. Adverse Effects Associated with Proton Pump Inhibitor Use. Cureus 2021, 13, e12759. [Google Scholar] [CrossRef]
- Perry, I.E.; Sonu, I.; Scarpignato, C.; Akiyama, J.; Hongo, M.; Vega, K.J. Potential proton pump inhibitor-related adverse effects. Ann. N. Y. Acad. Sci. 2020, 1481, 43–58. [Google Scholar] [CrossRef]
- Watanabe, T.; Higuchi, K.; Kobata, A.; Nishio, H.; Tanigawa, T.; Shiba, M.; Tominaga, K.; Fujiwara, Y.; Oshitani, N.; Asahara, T.; et al. Non-steroidal anti-inflammatory drug-induced small intestinal damage is Toll-like receptor 4 dependent. Gut 2008, 57, 181–187. [Google Scholar] [CrossRef]
- Vilardi, A.; Przyborski, S.; Mobbs, C.; Rufini, A.; Tufarelli, C. Current understanding of the interplay between extracellular matrix remodelling and gut permeability in health and disease. Cell Death Discov. 2024, 10, 258. [Google Scholar] [CrossRef] [PubMed]
- Silva, F.A.; Rodrigues, B.L.; Ayrizono, M.L.; Leal, R.F. The Immunological Basis of Inflammatory Bowel Disease. Gastroenterol. Res. Pract. 2016, 2016, 2097274. [Google Scholar] [CrossRef] [PubMed]
- Fernando, E.H.; Gordon, M.H.; Beck, P.L.; MacNaughton, W.K. Inhibition of Intestinal Epithelial Wound Healing through Protease-Activated Receptor-2 Activation in Caco2 Cells. J. Pharmacol. Exp. Ther. 2018, 367, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Klenske, E.; Bojarski, C.; Waldner, M.; Rath, T.; Neurath, M.F.; Atreya, R. Targeting mucosal healing in Crohn’s disease: What the clinician needs to know. Therap Adv. Gastroenterol. 2019, 12, 1756284819856865. [Google Scholar] [CrossRef] [PubMed]
- Leoni, G.; Neumann, P.A.; Sumagin, R.; Denning, T.L.; Nusrat, A. Wound repair: Role of immune-epithelial interactions. Mucosal Immunol. 2015, 8, 959–968. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.M.; Duckworth, C.A.; Burkitt, M.D.; Watson, A.J.; Campbell, B.J.; Pritchard, D.M. Epithelial cell shedding and barrier function: A matter of life and death at the small intestinal villus tip. Vet. Pathol. 2015, 52, 445–455. [Google Scholar] [CrossRef]
- Landen, N.X.; Li, D.; Stahle, M. Transition from inflammation to proliferation: A critical step during wound healing. Cell Mol. Life Sci. 2016, 73, 3861–3885. [Google Scholar] [CrossRef]
- Xue, X.; Falcon, D.M. The Role of Immune Cells and Cytokines in Intestinal Wound Healing. Int. J. Mol. Sci. 2019, 20, 6097. [Google Scholar] [CrossRef] [PubMed]
- Patnaude, L.; Mayo, M.; Mario, R.; Wu, X.; Knight, H.; Creamer, K.; Wilson, S.; Pivorunas, V.; Karman, J.; Phillips, L.; et al. Mechanisms and regulation of IL-22-mediated intestinal epithelial homeostasis and repair. Life Sci. 2021, 271, 119195. [Google Scholar] [CrossRef]
- Pearson, M.A.; Reczek, D.; Bretscher, A.; Karplus, P.A. Structure of the ERM protein moesin reveals the FERM domain fold masked by an extended actin binding tail domain. Cell 2000, 101, 259–270. [Google Scholar] [CrossRef]
- Tapial Martinez, P.; Lopez Navajas, P.; Lietha, D. FAK Structure and Regulation by Membrane Interactions and Force in Focal Adhesions. Biomolecules 2020, 10, 179. [Google Scholar] [CrossRef]
- Wang, Y.; Zheng, J.; Han, Y.; Zhang, Y.; Su, L.; Hu, D.; Fu, X. JAM-A knockdown accelerates the proliferation and migration of human keratinocytes, and improves wound healing in rats via FAK/Erk signaling. Cell Death Dis. 2018, 9, 848. [Google Scholar] [CrossRef]
- Teranishi, S.; Kimura, K.; Nishida, T. Role of formation of an ERK-FAK-paxillin complex in migration of human corneal epithelial cells during wound closure in vitro. Investig. Ophthalmol. Vis. Sci. 2009, 50, 5646–5652. [Google Scholar] [CrossRef] [PubMed]
- Tanimura, S.; Takeda, K. ERK signalling as a regulator of cell motility. J. Biochem. 2017, 162, 145–154. [Google Scholar] [CrossRef]
- Owen, K.A.; Abshire, M.Y.; Tilghman, R.W.; Casanova, J.E.; Bouton, A.H. FAK regulates intestinal epithelial cell survival and proliferation during mucosal wound healing. PLoS ONE 2011, 6, e23123. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Colome, A.M.; Lee-Rivera, I.; Benavides-Hidalgo, R.; Lopez, E. Paxillin: A crossroad in pathological cell migration. J. Hematol. Oncol. 2017, 10, 50. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Mayer, B.J. Phosphorylation of p130Cas initiates Rac activation and membrane ruffling. BMC Cell Biol. 2008, 9, 50. [Google Scholar] [CrossRef]
- Sanders, M.A.; Basson, M.D. p130cas but not paxillin is essential for Caco-2 intestinal epithelial cell spreading and migration on collagen IV. J. Biol. Chem. 2005, 280, 23516–23522. [Google Scholar] [CrossRef]
- Geiger, B. A role for p130Cas in mechanotransduction. Cell 2006, 127, 879–881. [Google Scholar] [CrossRef]
- Chodniewicz, D.; Klemke, R.L. Regulation of integrin-mediated cellular responses through assembly of a CAS/Crk scaffold. Biochim. Biophys. Acta 2004, 1692, 63–76. [Google Scholar] [CrossRef]
- Lim, S.T.; Chen, X.L.; Lim, Y.; Hanson, D.A.; Vo, T.T.; Howerton, K.; Larocque, N.; Fisher, S.J.; Schlaepfer, D.D.; Ilic, D. Nuclear FAK promotes cell proliferation and survival through FERM-enhanced p53 degradation. Mol. Cell 2008, 29, 9–22. [Google Scholar] [CrossRef]
- Meng, E.C.; Goddard, T.D.; Pettersen, E.F.; Couch, G.S.; Pearson, Z.J.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Tools for structure building and analysis. Protein Sci. 2023, 32, e4792. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef]
- Goddard, T.D.; Huang, C.C.; Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 2018, 27, 14–25. [Google Scholar] [CrossRef]
- Mousson, A.; Sick, E.; Carl, P.; Dujardin, D.; De Mey, J.; Ronde, P. Targeting Focal Adhesion Kinase Using Inhibitors of Protein-Protein Interactions. Cancers 2018, 10, 278. [Google Scholar] [CrossRef]
- Cary, L.A.; Chang, J.F.; Guan, J.L. Stimulation of cell migration by overexpression of focal adhesion kinase and its association with Src and Fyn. J. Cell Sci. 1996, 109 Pt. 7, 1787–1794. [Google Scholar] [CrossRef]
- Oncel, S.; Basson, M.D. ZINC40099027 promotes monolayer circular defect closure by a novel pathway involving cytosolic activation of focal adhesion kinase and downstream paxillin and ERK1/2. Cell Tissue Res. 2022, 390, 261–279. [Google Scholar] [CrossRef]
- Colicelli, J. Human RAS superfamily proteins and related GTPases. Sci. STKE 2004, 2004, RE13. [Google Scholar] [CrossRef]
- Miller, N.L.; Lawson, C.; Chen, X.L.; Lim, S.T.; Schlaepfer, D.D. Rgnef (p190RhoGEF) knockout inhibits RhoA activity, focal adhesion establishment, and cell motility downstream of integrins. PLoS ONE 2012, 7, e37830. [Google Scholar] [CrossRef]
- Vleugel, M.M.; Greijer, A.E.; Bos, R.; van der Wall, E.; van Diest, P.J. c-Jun activation is associated with proliferation and angiogenesis in invasive breast cancer. Hum. Pathol. 2006, 37, 668–674. [Google Scholar] [CrossRef]
- Guller, M.; Toualbi-Abed, K.; Legrand, A.; Michel, L.; Mauviel, A.; Bernuau, D.; Daniel, F. c-Fos overexpression increases the proliferation of human hepatocytes by stabilizing nuclear Cyclin D1. World J. Gastroenterol. 2008, 14, 6339–6346. [Google Scholar] [CrossRef]
- Chaturvedi, L.S.; Marsh, H.M.; Basson, M.D. Role of RhoA and its effectors ROCK and mDia1 in the modulation of deformation-induced FAK, ERK, p38, and MLC motogenic signals in human Caco-2 intestinal epithelial cells. Am. J. Physiol. Cell Physiol. 2011, 301, C1224–C1238. [Google Scholar] [CrossRef]
- Wang, Q.; More, S.K.; Vomhof-DeKrey, E.E.; Golovko, M.Y.; Basson, M.D. Small molecule FAK activator promotes human intestinal epithelial monolayer wound closure and mouse ulcer healing. Sci. Rep. 2019, 9, 14669. [Google Scholar] [CrossRef]
- Wang, Q.; Gallardo-Macias, R.; Rashmi; Golovko, M.Y.; Elsayed, A.A.R.; More, S.K.; Oncel, S.; Gurvich, V.J.; Basson, M.D. Discovery of Novel Small-Molecule FAK Activators Promoting Mucosal Healing. ACS Med. Chem. Lett. 2021, 12, 356–364. [Google Scholar] [CrossRef]
- Wang, Q.; Gallardo-Macias, R.; Vomhof-DeKrey, E.E.; Gupta, R.; Golovko, S.A.; Golovko, M.Y.; Oncel, S.; Gurvich, V.J.; Basson, M.D. A novel drug-like water-soluble small molecule Focal Adhesion Kinase (FAK) activator promotes intestinal mucosal healing. Curr. Res. Pharmacol. Drug Discov. 2023, 4, 100147. [Google Scholar] [CrossRef]
- Maloy, K.J.; Powrie, F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature 2011, 474, 298–306. [Google Scholar] [CrossRef]
- Digiacomo, G.; Tusa, I.; Bacci, M.; Cipolleschi, M.G.; Dello Sbarba, P.; Rovida, E. Fibronectin induces macrophage migration through a SFK-FAK/CSF-1R pathway. Cell Adh Migr. 2017, 11, 327–337. [Google Scholar] [CrossRef]
- Maa, M.C.; Chang, M.Y.; Li, J.; Li, Y.Y.; Hsieh, M.Y.; Yang, C.J.; Chen, Y.J.; Li, Y.; Chen, H.C.; Cheng, W.E.; et al. The iNOS/Src/FAK axis is critical in Toll-like receptor-mediated cell motility in macrophages. Biochim. Biophys. Acta 2011, 1813, 136–147. [Google Scholar] [CrossRef]
- Abshire, M.Y.; Thomas, K.S.; Owen, K.A.; Bouton, A.H. Macrophage motility requires distinct alpha5beta1/FAK and alpha4beta1/paxillin signaling events. J. Leukoc. Biol. 2011, 89, 251–257. [Google Scholar] [CrossRef]
- Owen, K.A.; Pixley, F.J.; Thomas, K.S.; Vicente-Manzanares, M.; Ray, B.J.; Horwitz, A.F.; Parsons, J.T.; Beggs, H.E.; Stanley, E.R.; Bouton, A.H. Regulation of lamellipodial persistence, adhesion turnover, and motility in macrophages by focal adhesion kinase. J. Cell Biol. 2007, 179, 1275–1287. [Google Scholar] [CrossRef]
- Ghilas, S.; O’Keefe, R.; Mielke, L.A.; Raghu, D.; Buchert, M.; Ernst, M. Crosstalk between epithelium, myeloid and innate lymphoid cells during gut homeostasis and disease. Front. Immunol. 2022, 13, 944982. [Google Scholar] [CrossRef]
- Boutilier, A.J.; Elsawa, S.F. Macrophage Polarization States in the Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 6995. [Google Scholar] [CrossRef]
- Dijkgraaf, E.M.; Heusinkveld, M.; Tummers, B.; Vogelpoel, L.T.; Goedemans, R.; Jha, V.; Nortier, J.W.; Welters, M.J.; Kroep, J.R.; van der Burg, S.H. Chemotherapy alters monocyte differentiation to favor generation of cancer-supporting M2 macrophages in the tumor microenvironment. Cancer Res. 2013, 73, 2480–2492. [Google Scholar] [CrossRef]
- Fujiwara, N.; Kobayashi, K. Macrophages in inflammation. Curr. Drug Targets Inflamm. Allergy 2005, 4, 281–286. [Google Scholar] [CrossRef]
- Mily, A.; Kalsum, S.; Loreti, M.G.; Rekha, R.S.; Muvva, J.R.; Lourda, M.; Brighenti, S. Polarization of M1 and M2 Human Monocyte-Derived Cells and Analysis with Flow Cytometry upon Mycobacterium tuberculosis Infection. J. Vis. Exp. 2020, 163, e61807. [Google Scholar] [CrossRef]
- Fukao, T.; Koyasu, S. PI3K and negative regulation of TLR signaling. Trends Immunol. 2003, 24, 358–363. [Google Scholar] [CrossRef]
- Covarrubias, A.J.; Aksoylar, H.I.; Horng, T. Control of macrophage metabolism and activation by mTOR and Akt signaling. Semin. Immunol. 2015, 27, 286–296. [Google Scholar] [CrossRef]
- Krzyszczyk, P.; Schloss, R.; Palmer, A.; Berthiaume, F. The Role of Macrophages in Acute and Chronic Wound Healing and Interventions to Promote Pro-wound Healing Phenotypes. Front. Physiol. 2018, 9, 419. [Google Scholar] [CrossRef]
- Borisenko, G.G.; Matsura, T.; Liu, S.X.; Tyurin, V.A.; Jianfei, J.; Serinkan, F.B.; Kagan, V.E. Macrophage recognition of externalized phosphatidylserine and phagocytosis of apoptotic Jurkat cells—Existence of a threshold. Arch. Biochem. Biophys. 2003, 413, 41–52. [Google Scholar] [CrossRef]
- Van Dyken, S.J.; Locksley, R.M. Interleukin-4- and interleukin-13-mediated alternatively activated macrophages: Roles in homeostasis and disease. Annu. Rev. Immunol. 2013, 31, 317–343. [Google Scholar] [CrossRef]
- Yunna, C.; Mengru, H.; Lei, W.; Weidong, C. Macrophage M1/M2 polarization. Eur. J. Pharmacol. 2020, 877, 173090. [Google Scholar] [CrossRef]
- Brazil, J.C.; Quiros, M.; Nusrat, A.; Parkos, C.A. Innate immune cell-epithelial crosstalk during wound repair. J. Clin. Investig. 2019, 129, 2983–2993. [Google Scholar] [CrossRef]
- Quiros, M.; Nishio, H.; Neumann, P.A.; Siuda, D.; Brazil, J.C.; Azcutia, V.; Hilgarth, R.; O’Leary, M.N.; Garcia-Hernandez, V.; Leoni, G.; et al. Macrophage-derived IL-10 mediates mucosal repair by epithelial WISP-1 signaling. J. Clin. Investig. 2017, 127, 3510–3520. [Google Scholar] [CrossRef]
- Naqvi, S.; Martin, K.J.; Arthur, J.S. CREB phosphorylation at Ser133 regulates transcription via distinct mechanisms downstream of cAMP and MAPK signalling. Biochem. J. 2014, 458, 469–479. [Google Scholar] [CrossRef]
- Sharma, J.N.; Al-Omran, A.; Parvathy, S.S. Role of nitric oxide in inflammatory diseases. Inflammopharmacology 2007, 15, 252–259. [Google Scholar] [CrossRef]
- Bain, C.C.; Mowat, A.M. Macrophages in intestinal homeostasis and inflammation. Immunol. Rev. 2014, 260, 102–117. [Google Scholar] [CrossRef]
- Honda, M.; Surewaard, B.G.J.; Watanabe, M.; Hedrick, C.C.; Lee, W.Y.; Brown, K.; McCoy, K.D.; Kubes, P. Perivascular localization of macrophages in the intestinal mucosa is regulated by Nr4a1 and the microbiome. Nat. Commun. 2020, 11, 1329. [Google Scholar] [CrossRef]
- Brancato, S.K.; Albina, J.E. Wound macrophages as key regulators of repair: Origin, phenotype, and function. Am. J. Pathol. 2011, 178, 19–25. [Google Scholar] [CrossRef]
- Yang, P.; Liu, L.; Sun, L.; Fang, P.; Snyder, N.; Saredy, J.; Ji, Y.; Shen, W.; Qin, X.; Wu, Q.; et al. Immunological Feature and Transcriptional Signaling of Ly6C Monocyte Subsets From Transcriptome Analysis in Control and Hyperhomocysteinemic Mice. Front. Immunol. 2021, 12, 632333. [Google Scholar] [CrossRef]
- Zhang, J.; Patel, L.; Pienta, K.J. Targeting chemokine (C-C motif) ligand 2 (CCL2) as an example of translation of cancer molecular biology to the clinic. Prog. Mol. Biol. Transl. Sci. 2010, 95, 31–53. [Google Scholar] [CrossRef] [PubMed]
- Kadomoto, S.; Izumi, K.; Mizokami, A. Roles of CCL2-CCR2 Axis in the Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 8530. [Google Scholar] [CrossRef]
- Abbas, A.K.; Lichtman, A.H.; Baker, D.L. Basic Immunology: Functions and Disorders of the Immune System, 2nd ed.; Saunders: Philadelphia, PA, USA, 2004; 322p. [Google Scholar]
- De Schepper, S.; Verheijden, S.; Aguilera-Lizarraga, J.; Viola, M.F.; Boesmans, W.; Stakenborg, N.; Voytyuk, I.; Schmidt, I.; Boeckx, B.; Dierckx de Casterle, I.; et al. Self-Maintaining Gut Macrophages Are Essential for Intestinal Homeostasis. Cell 2018, 175, 400–415 e413. [Google Scholar] [CrossRef]
- Al-Sadi, R.; Boivin, M.; Ma, T. Mechanism of cytokine modulation of epithelial tight junction barrier. Front. Biosci. 2009, 14, 2765–2778. [Google Scholar] [CrossRef]
- Kim, M.S.; Kim, J.Y. Intestinal anti-inflammatory effects of cinnamon extracts in a co-culture model of intestinal epithelial Caco-2 cells and RAW264.7 macrophages. Appl. Biol. Chem. 2017, 60, 553–561. [Google Scholar] [CrossRef]
- Burgess, A.W.; Metcalf, D. Characterization of a serum factor stimulating the differentiation of myelomonocytic leukemic cells. Int. J. Cancer 1980, 26, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Vadhan-Raj, S.; Buescher, S.; LeMaistre, A.; Keating, M.; Walters, R.; Ventura, C.; Hittelman, W.; Broxmeyer, H.E.; Gutterman, J.U. Stimulation of hematopoiesis in patients with bone marrow failure and in patients with malignancy by recombinant human granulocyte-macrophage colony-stimulating factor. Blood 1988, 72, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.M.; Cory, S. Donald Metcalf: The father of modern hematology. Proc. Natl. Acad. Sci. USA 2015, 112, 2628–2629. [Google Scholar] [CrossRef] [PubMed]
- Egea, L.; McAllister, C.S.; Lakhdari, O.; Minev, I.; Shenouda, S.; Kagnoff, M.F. GM-CSF produced by nonhematopoietic cells is required for early epithelial cell proliferation and repair of injured colonic mucosa. J. Immunol. 2013, 190, 1702–1713. [Google Scholar] [CrossRef]
- Guillemin, G.; Boussin, F.D.; Le Grand, R.; Croitoru, J.; Coffigny, H.; Dormont, D. Granulocyte macrophage colony stimulating factor stimulates in vitro proliferation of astrocytes derived from simian mature brains. Glia 1996, 16, 71–80. [Google Scholar] [CrossRef]
- McCormick, T.S.; Hejal, R.B.; Leal, L.O.; Ghannoum, M.A. GM-CSF: Orchestrating the Pulmonary Response to Infection. Front. Pharmacol. 2021, 12, 735443. [Google Scholar] [CrossRef]
- Hamilton, J.A. GM-CSF-Dependent Inflammatory Pathways. Front. Immunol. 2019, 10, 2055. [Google Scholar] [CrossRef] [PubMed]
- Hansen, G.; Hercus, T.R.; McClure, B.J.; Stomski, F.C.; Dottore, M.; Powell, J.; Ramshaw, H.; Woodcock, J.M.; Xu, Y.; Guthridge, M.; et al. The structure of the GM-CSF receptor complex reveals a distinct mode of cytokine receptor activation. Cell 2008, 134, 496–507. [Google Scholar] [CrossRef] [PubMed]
- Hercus, T.R.; Broughton, S.E.; Ekert, P.G.; Ramshaw, H.S.; Perugini, M.; Grimbaldeston, M.; Woodcock, J.M.; Thomas, D.; Pitson, S.; Hughes, T.; et al. The GM-CSF receptor family: Mechanism of activation and implications for disease. Growth Factors 2012, 30, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Castro-Dopico, T.; Fleming, A.; Dennison, T.W.; Ferdinand, J.R.; Harcourt, K.; Stewart, B.J.; Cader, Z.; Tuong, Z.K.; Jing, C.; Lok, L.S.C.; et al. GM-CSF Calibrates Macrophage Defense and Wound Healing Programs during Intestinal Infection and Inflammation. Cell Rep. 2020, 32, 107857. [Google Scholar] [CrossRef] [PubMed]
- Domingues, R.G.; Hepworth, M.R. Immunoregulatory Sensory Circuits in Group 3 Innate Lymphoid Cell (ILC3) Function and Tissue Homeostasis. Front. Immunol. 2020, 11, 116. [Google Scholar] [CrossRef] [PubMed]
- Ribechini, E.; Hutchinson, J.A.; Hergovits, S.; Heuer, M.; Lucas, J.; Schleicher, U.; Jordan Garrote, A.L.; Potter, S.J.; Riquelme, P.; Brackmann, H.; et al. Novel GM-CSF signals via IFN-gammaR/IRF-1 and AKT/mTOR license monocytes for suppressor function. Blood Adv. 2017, 1, 947–960. [Google Scholar] [CrossRef] [PubMed]
- Egea, L.; Hirata, Y.; Kagnoff, M.F. GM-CSF: A role in immune and inflammatory reactions in the intestine. Expert. Rev. Gastroenterol. Hepatol. 2010, 4, 723–731. [Google Scholar] [CrossRef] [PubMed]
- Oncel, S.; Gupta, R.; Wang, Q.; Basson, M.D. ZINC40099027 Promotes Gastric Mucosal Repair in Ongoing Aspirin-Associated Gastric Injury by Activating Focal Adhesion Kinase. Cells 2021, 10, 908. [Google Scholar] [CrossRef]
- Lewis, J.D.; Parlett, L.E.; Jonsson Funk, M.L.; Brensinger, C.; Pate, V.; Wu, Q.; Dawwas, G.K.; Weiss, A.; Constant, B.D.; McCauley, M.; et al. Incidence, Prevalence, and Racial and Ethnic Distribution of Inflammatory Bowel Disease in the United States. Gastroenterology 2023, 165, 1197–1205 e1192. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brown, N.D.; Vomhof-DeKrey, E.E. Focal Adhesion Kinase and Colony Stimulating Factors: Intestinal Homeostasis and Innate Immunity Crosstalk. Cells 2024, 13, 1178. https://doi.org/10.3390/cells13141178
Brown ND, Vomhof-DeKrey EE. Focal Adhesion Kinase and Colony Stimulating Factors: Intestinal Homeostasis and Innate Immunity Crosstalk. Cells. 2024; 13(14):1178. https://doi.org/10.3390/cells13141178
Chicago/Turabian StyleBrown, Nicholas D., and Emilie E. Vomhof-DeKrey. 2024. "Focal Adhesion Kinase and Colony Stimulating Factors: Intestinal Homeostasis and Innate Immunity Crosstalk" Cells 13, no. 14: 1178. https://doi.org/10.3390/cells13141178
APA StyleBrown, N. D., & Vomhof-DeKrey, E. E. (2024). Focal Adhesion Kinase and Colony Stimulating Factors: Intestinal Homeostasis and Innate Immunity Crosstalk. Cells, 13(14), 1178. https://doi.org/10.3390/cells13141178