


Plasma-Derived Extracellular Vesicles and Non-Extracellular Vesicle Components from APCMin/+ Mice Promote Pro-Tumorigenic Activities and Activate Human Colonic Fibroblasts via the NF-κB Signaling Pathway



Abstract

1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Plasma Collection and Tissue Preparation for Tumor Counting
2.3. Cells and Reagents
2.4. Extracellular Vesicle Isolation from Mice Plasma Samples
2.5. Nanoparticles Tracking Analysis (NTA)
2.6. Scanning Electron Microscopy (SEM)
2.7. Protein Extraction from Cells LEVs and SEVs
2.8. Western Blotting
2.9. CCD-18Co Treatment with Plasma-Derived EVs
2.10. Wound Healing Assay
2.11. Cell Proliferation Assay
2.12. Human Cytokine Antibody Array
2.13. Preparation of Conditioned Media (CMs)
2.14. RNA Extraction and Real-Time PCR Analyses
2.15. Statistical Analysis
3. Results
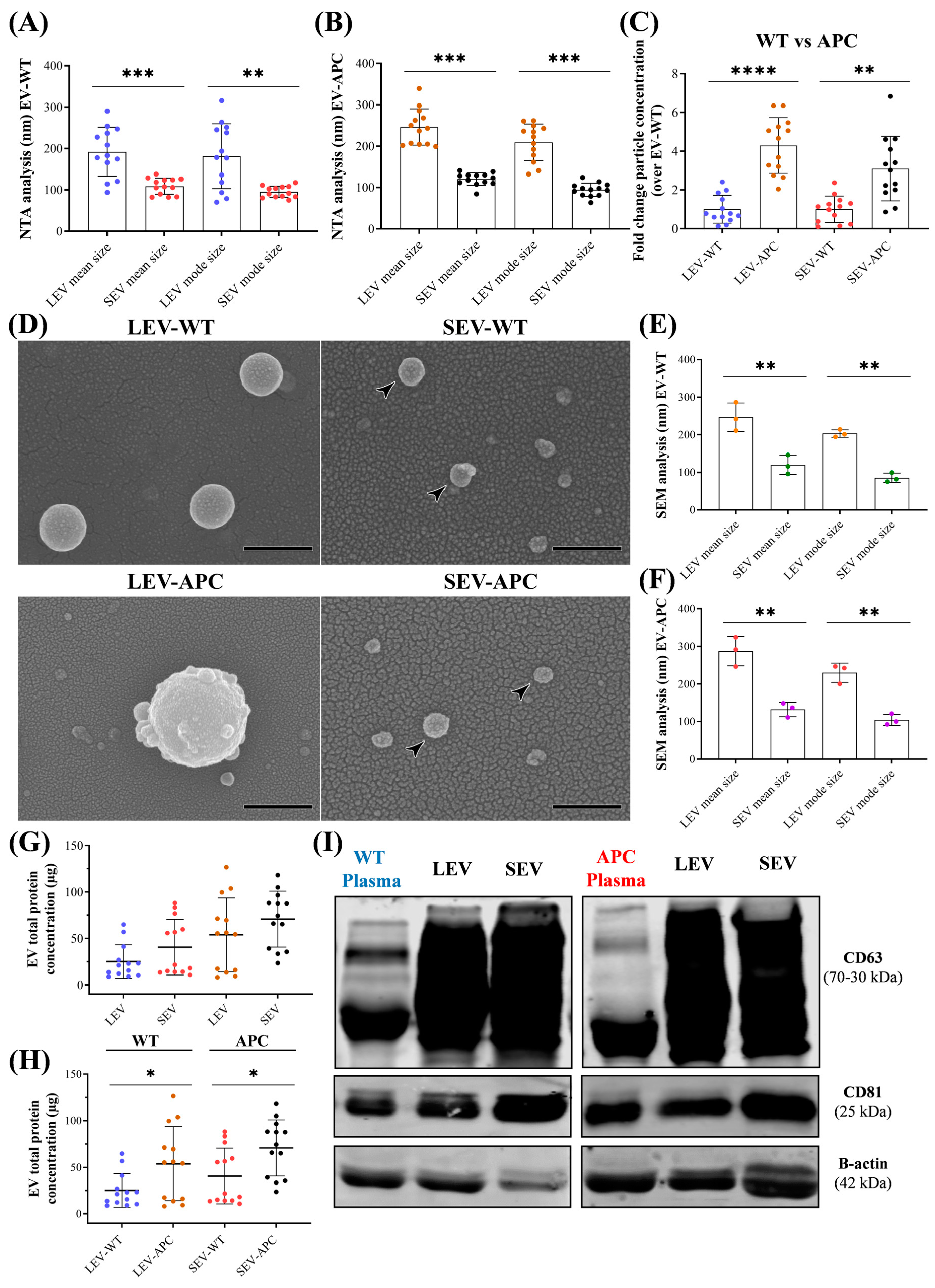
3.1. Characterization of Plasma-Derived EV Subtypes from WT and APCMin/+ Mice
3.2. Plasma-Derived EVs Increased Wound Healing in Human Colonic Fibroblasts
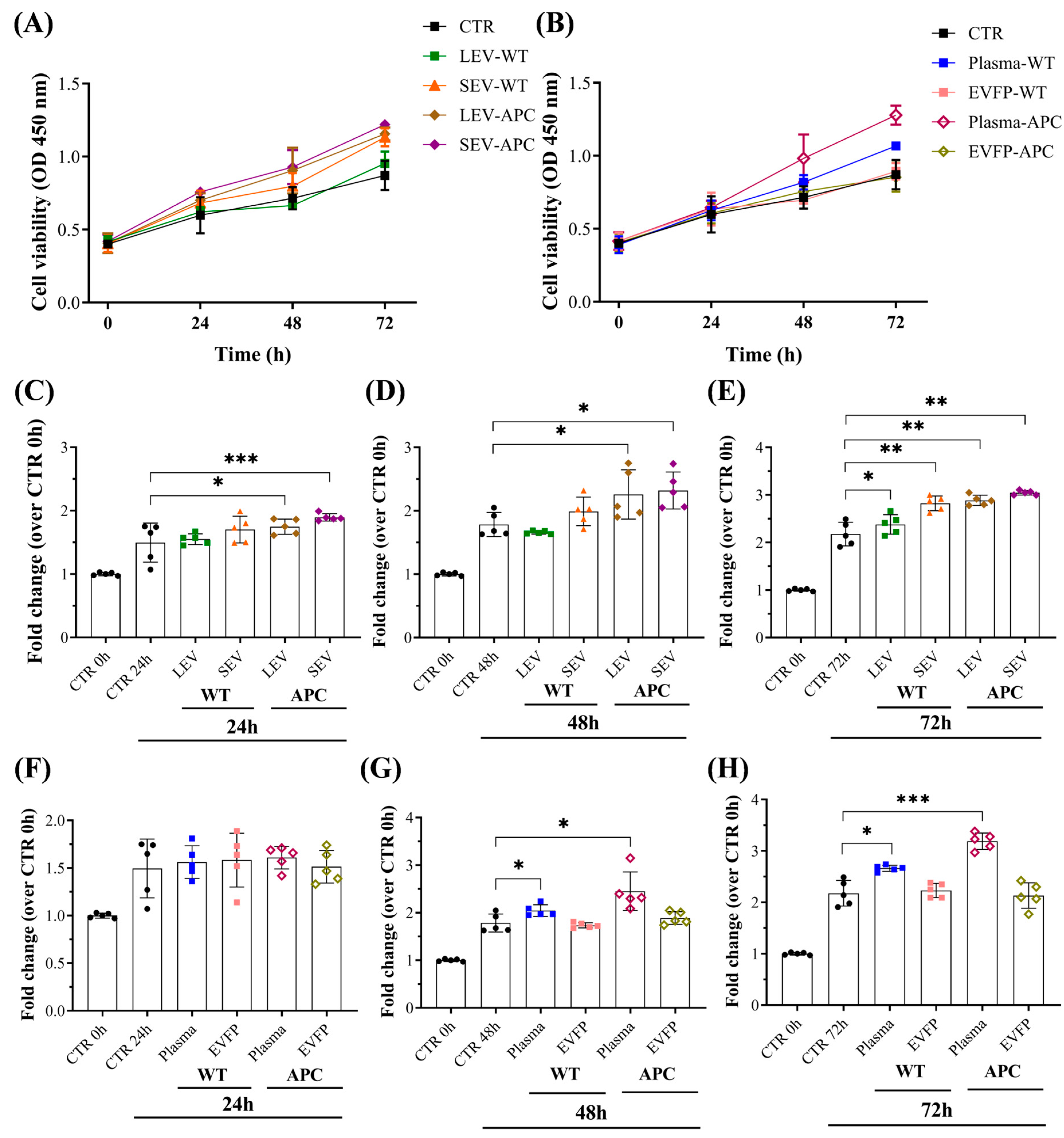
3.3. Plasma-Derived EVs Increased Cell Proliferation in Colon Fibroblasts
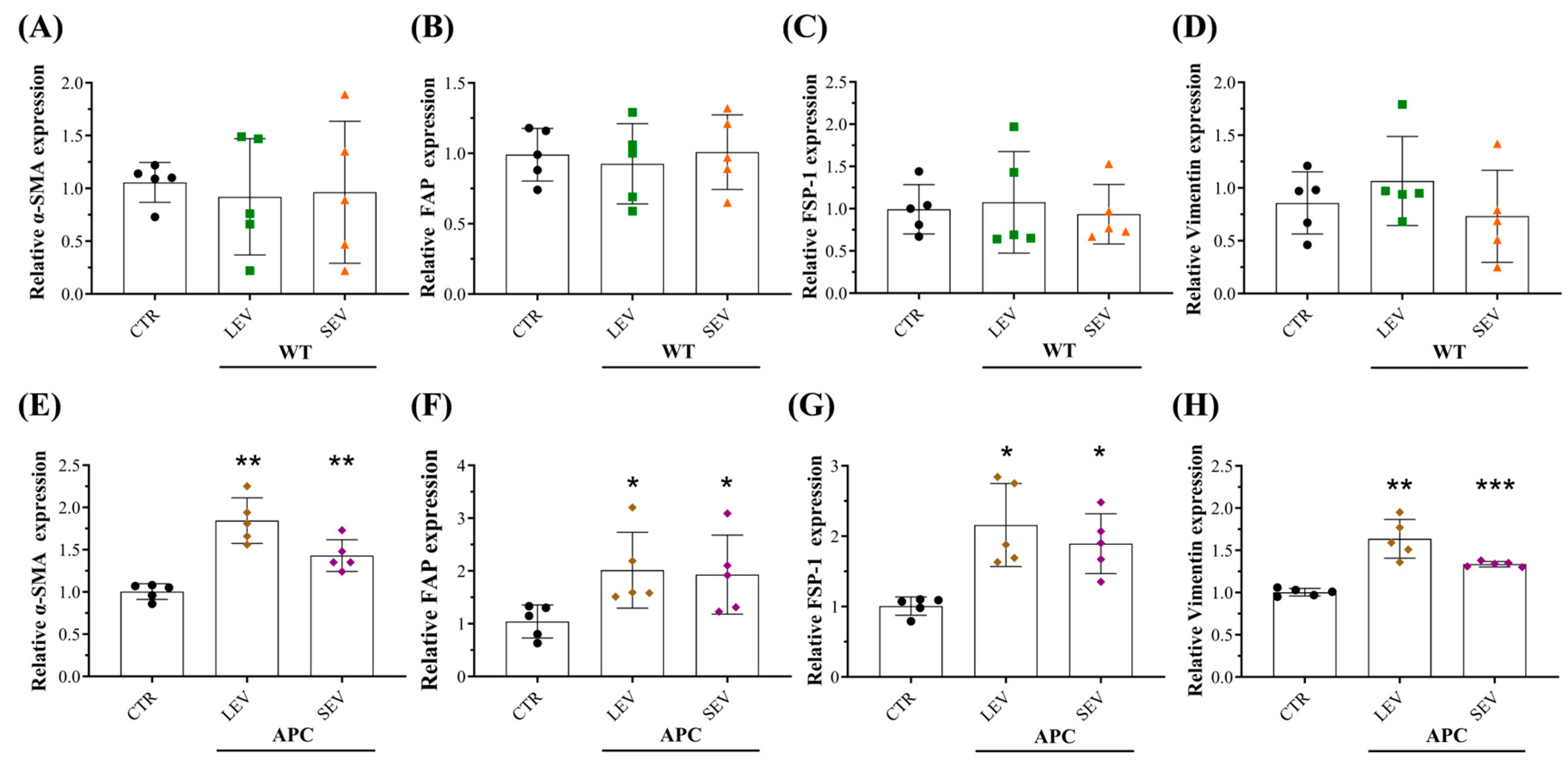
3.4. Plasma-Derived EVs from Tumor-Bearing Mice Increased Extracellular Matrix (ECM) Components in Fibroblasts
3.5. Plasma-Derived EVs from Tumor-Bearing Mice Reprogram Normal Colon Fibroblasts into CAFs
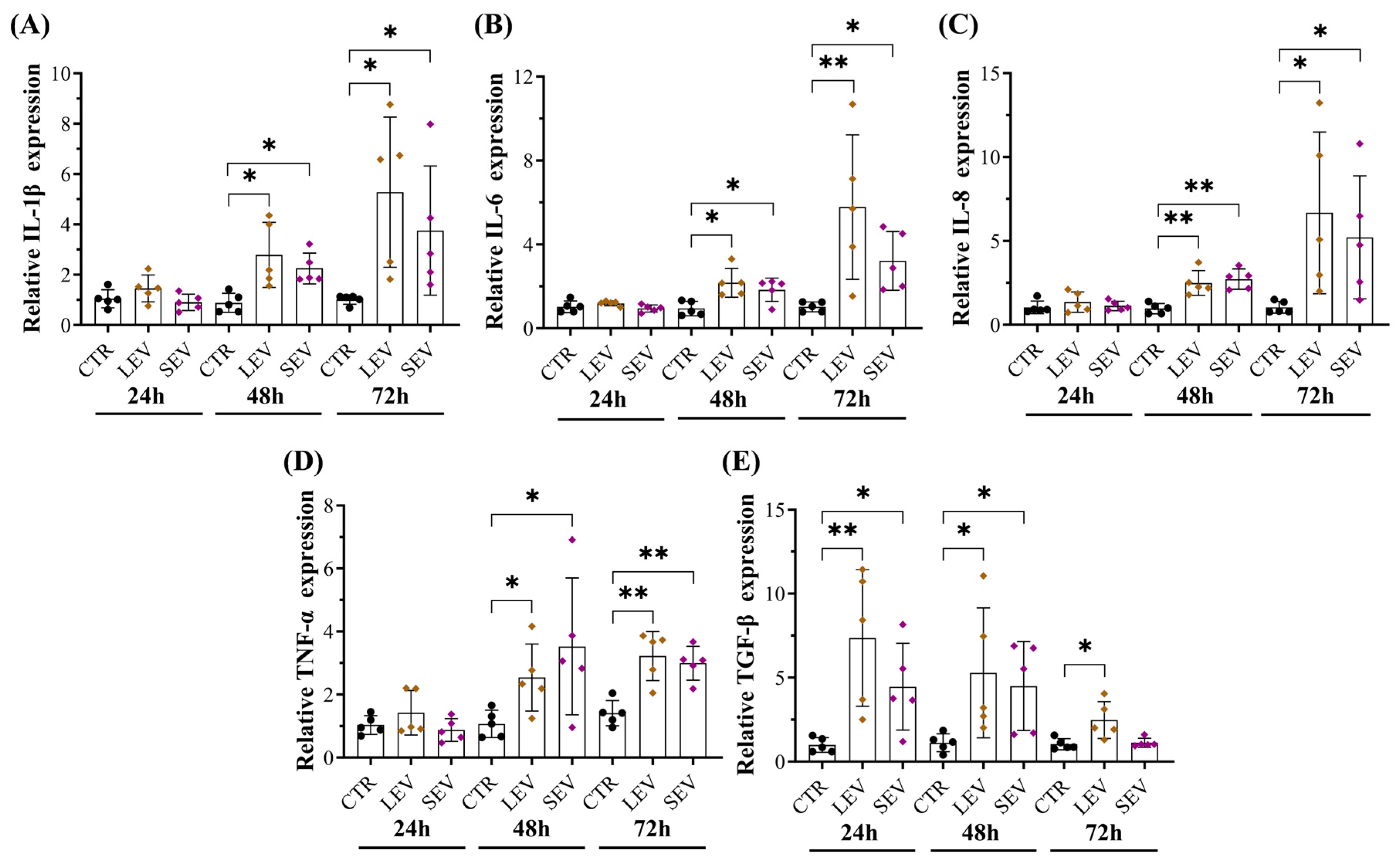
3.6. Tumor-Bearing Mice-Derived EVs Induced Pro-Inflammatory Responses in Colon Fibroblasts
3.7. APC-Derived EVs Increased Soluble Mediators in Fibroblasts Involved in CAF Transformation and Cancer Progression
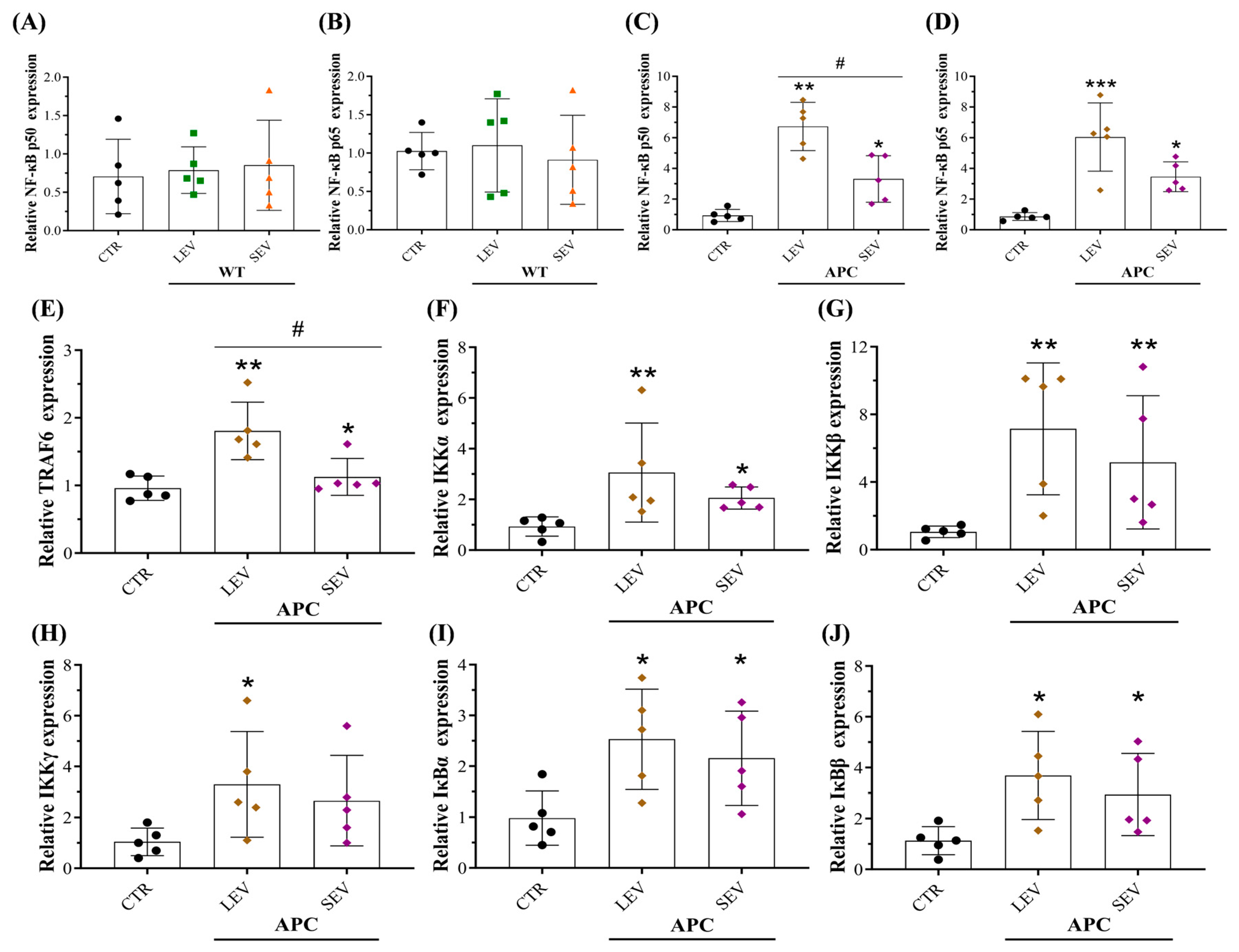
3.8. EVs from Tumor-Bearing Mice Promoted an Inflammatory CAF Phenotype via NF-κB Pathway Activation
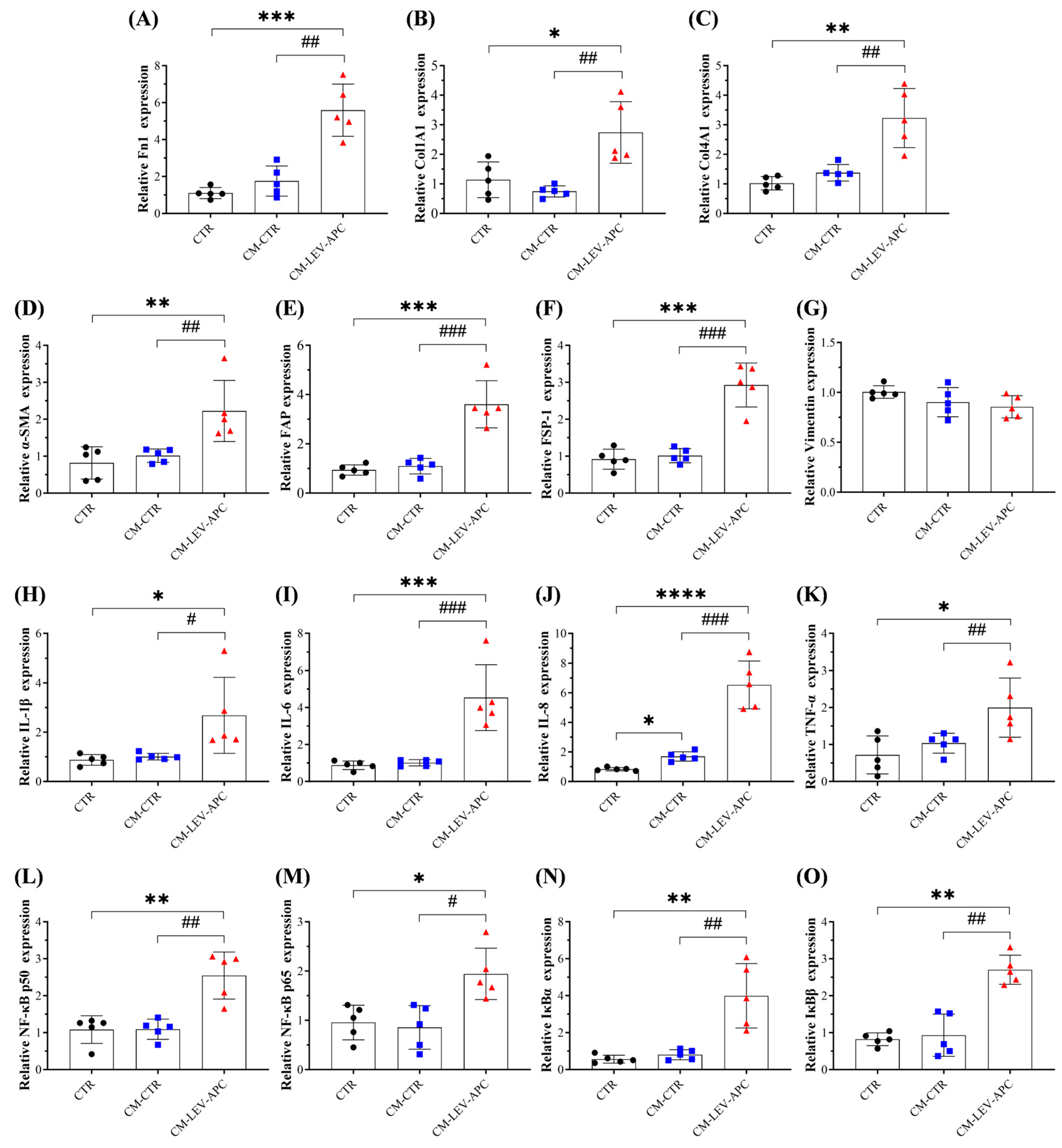
3.9. Fibroblasts Educated by APC-Derived EVs Released Soluble Factors with Malignant Behavior Effects on Surrounding Not-Activated Fibroblasts
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Wagle, N.S.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2023. Cancer J. Clin. 2023, 73, 233–254. [Google Scholar] [CrossRef] [PubMed]
- America Cancer Society. Colorectal Cancer Facts & Figures. Available online: https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/colorectal-cancer-facts-and-figures/colorectal-cancer-facts-and-figures-2023.pdf (accessed on 11 June 2024).
- Gmeiner, W.H. Recent Advances in Therapeutic Strategies to Improve Colorectal Cancer Treatment. Cancers 2024, 16, 1029. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Gautam, V.; Sandhu, A.; Rawat, K.; Sharma, A.; Saha, L. Current and emerging therapeutic approaches for colorectal cancer: A comprehensive review. World J. Gastro Surg. 2023, 15, 495–519. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.N. Genetic and epigenetic alterations of colorectal cancer. Intestig. Res. 2018, 16, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, D.; Shen, M. Tumor Microenvironment Shapes Colorectal Cancer Progression, Metastasis, and Treatment Responses. Front. Med. 2022, 9, 869010. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Enomoto, A.; Woods, S.L.; Burt, A.D.; Takahashi, M.; Worthley, D.L. Cancer-associated fibroblasts in gastrointestinal cancer. Nat. Rev. Gastro Hepat. 2019, 16, 282–295. [Google Scholar] [CrossRef] [PubMed]
- Buechler, M.B.; Fu, W.X.; Turley, S.J. Fibroblast-macrophage reciprocal interactions in health, fibrosis, and cancer. Immunity 2021, 54, 903–915. [Google Scholar] [CrossRef]
- Kazakova, A.; Sudarskikh, T.; Kovalev, O.; Kzhyshkowska, J.; Larionova, I. Interaction of tumor-associated macrophages with stromal and immune components in solid tumors: Research progress (Review). Int. J. Oncol. 2023, 62, 5480. [Google Scholar] [CrossRef] [PubMed]
- Boelens, M.C.; Wu, T.J.; Nabet, B.Y.; Xu, B.; Qiu, Y.; Yoon, T.; Azzam, D.J.; Victor, C.T.S.; Wiemann, B.Z.; Ishwaran, H.; et al. Exosome Transfer from Stromal to Breast Cancer Cells Regulates Therapy Resistance Pathways. Cell 2014, 159, 499–513. [Google Scholar] [CrossRef]
- Oszvald, A.; Szvicsek, Z.; Pápai, M.; Kelemen, A.; Varga, Z.; Tölgyes, T.; Dede, K.; Bursics, A.; Buzas, E.I.; Wiener, Z. Fibroblast-Derived Extracellular Vesicles Induce Colorectal Cancer Progression by Transmitting Amphiregulin. Front. Cell Dev. Biol. 2020, 8, 558. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Zhang, D.S.; Wang, T.; Ji, J.Z.; Jin, C.; Peng, C.F.; Tan, Y.Q.; Zhou, J.H.; Wang, L.; Feng, Y.F.; et al. CAF-derived exosomal WEE2-AS1 facilitates colorectal cancer progression via promoting degradation of MOB1A to inhibit the Hippo pathway. Cell Death Dis. 2022, 13, 796. [Google Scholar] [CrossRef] [PubMed]
- Fujita, Y.; Fujimoto, S.; Miyamoto, A.; Kaneko, R.; Kadota, T.; Watanabe, N.; Kizawa, R.; Kawamoto, H.; Watanabe, J.; Utsumi, H.; et al. Fibroblast-derived Extracellular Vesicles Induce Lung Cancer Progression in the Idiopathic Pulmonary Fibrosis Microenvironment. Am. J. Resp. Cell Mol. 2023, 69, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Li, T.T.; Tian, L.L.; Cao, J.; Liu, M. Cancer-associated fibroblasts secret extracellular vesicles to support cell proliferation and epithelial-mesenchymal transition in laryngeal squamous cell carcinoma. Mol. Cell Probes 2023, 72, 101934. [Google Scholar] [CrossRef] [PubMed]
- Xiong, L.J.; Wei, Y.M.; Jia, Q.; Chen, J.L.; Chen, T.; Yuan, J.Y.; Pi, C.; Liu, H.Y.; Tang, J.; Yin, S.Y.; et al. The application of extracellular vesicles in colorectal cancer metastasis and drug resistance: Recent advances and trends. J. Nanobiotechnol. 2023, 21, 143. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.X.; Huang, D.F.; Wu, J.Y.; Li, Z.Z.; Yi, X.M.; Zhong, T.Y. The Biological Effect of Small Extracellular Vesicles on Colorectal Cancer Metastasis. Cells 2022, 11, 4071. [Google Scholar] [CrossRef] [PubMed]
- Giusti, I.; Di Francesco, M.; Poppa, G.; Esposito, L.; D’Ascenzo, S.; Dolo, V. Tumor-Derived Extracellular Vesicles Activate Normal Human Fibroblasts to a Cancer-Associated Fibroblast-Like Phenotype, Sustaining a Pro-Tumorigenic Microenvironment. Front. Oncol. 2022, 12, 839880. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Lam, E.W.F.; Sun, Y. Extracellular vesicles in the tumor microenvironment: Old stories, but new tales. Mol. Cancer 2019, 18, 59. [Google Scholar] [CrossRef] [PubMed]
- Su, C.Y.; Zhang, J.Y.; Yarden, Y.; Fu, L.W. The key roles of cancer stem cell-derived extracellular vesicles. Signal Transduct. Ther. 2021, 6, 109. [Google Scholar] [CrossRef] [PubMed]
- Majood, M.; Rawat, S.; Mohanty, S. Delineating the role of extracellular vesicles in cancer metastasis: A comprehensive review. Front. Immunol. 2022, 13, 966661. [Google Scholar] [CrossRef] [PubMed]
- van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Cocucci, E.; Racchetti, G.; Podini, P.; Meldolesi, J. Enlargeosome traffic: Exocytosis triggered by various signals is followed by endocytosis, membrane shedding or both. Traffic 2007, 8, 742–757. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef]
- Tkach, M.; Kowal, J.; Théry, C. Why the need and how to approach the functional diversity of extracellular vesicles. Philos. Trans. R. Soc. B 2018, 372, 20160479. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, M.; Martin-Jaular, L.; Lavieu, G.; Théry, C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat. Cell Biol. 2019, 21, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Cheshomi, H.; Matin, M.M. Exosomes and their importance in metastasis, diagnosis, and therapy of colorectal cancer. J. Cell Biochem. 2019, 120, 2671–2686. [Google Scholar] [CrossRef] [PubMed]
- Glass, S.E.; Coffey, R.J. Recent Advances in the Study of Extracellular Vesicles in Colorectal Cancer. Gastroenterology 2022, 163, 1188–1197. [Google Scholar] [CrossRef] [PubMed]
- Fearon, E.R.; Vogelstein, B. A Genetic Model for Colorectal Tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Fearnhead, N.S.; Britton, M.P.; Bodmer, W.F. The ABC of APC. Hum. Mol. Genet. 2001, 10, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Moser, A.R.; Pitot, H.C.; Dove, W.F. A Dominant Mutation That Predisposes to Multiple Intestinal Neoplasia in the Mouse. Science 1990, 247, 322–324. [Google Scholar] [CrossRef] [PubMed]
- Su, L.K.; Kinzler, K.W.; Vogelstein, B.; Preisinger, A.C.; Moser, A.R.; Luongo, C.; Gould, K.A.; Dove, W.F. Multiple Intestinal Neoplasia Caused by a Mutation in the Murine Homolog of the Apc Gene. Science 1992, 256, 668–670. [Google Scholar] [CrossRef] [PubMed]
- Luongo, C.; Moser, A.R.; Gledhill, S.; Dove, W.F. Loss of Apc+ in Intestinal Adenomas from Min Mice. Cancer Res. 1994, 54, 5947–5952. [Google Scholar] [PubMed]
- Karimi, N.; Dalirfardouei, R.; Dias, T.; Lötvall, J.; Lässer, C. Tetraspanins distinguish separate extracellular vesicle subpopulations in human serum and plasma—Contributions of platelet extracellular vesicles in plasma samples. J. Extracell. Vesicles 2022, 11, e12213. [Google Scholar] [CrossRef] [PubMed]
- Bracht, J.W.P.; Los, M.; van Eijndhoven, M.A.J.; Bettin, B.; van der Pol, E.; Pegtel, D.M.; Nieuwland, R. Platelet removal from human blood plasma improves detection of extracellular vesicle-associated miRNA. J. Extracell. Vesicles 2023, 12, e12302. [Google Scholar] [CrossRef] [PubMed]
- Jeppesen, D.K.; Fenix, A.M.; Franklin, J.L.; Higginbotham, J.N.; Zhang, Q.; Zimmerman, L.J.; Liebler, D.C.; Ping, J.; Liu, Q.; Evans, R.; et al. Reassessment of Exosome Composition. Cell 2019, 177, 428. [Google Scholar] [CrossRef] [PubMed]
- Van Deun, J.; Mestdagh, P.; Agostinis, P.; Akay, Ö.; Anand, S.; Anckaert, J.; Martinez, Z.A.; Baetens, T.; Beghein, E.; Bertier, L.; et al. EV-TRACK: Transparent reporting and centralizing knowledge in extracellular vesicle research. Nat. Methods 2017, 14, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Arteaga-Blanco, L.A.; Mojoli, A.; Monteiro, R.Q.; Sandim, V.; Menna-Barreto, R.F.S.; Pereira-Dutra, F.S.; Bozza, P.T.; Resende, R.D.; Bou-Habib, D.C. Characterization and internalization of small extracellular vesicles released by human primary macrophages derived from circulating monocytes. PLoS ONE 2020, 15, 237795. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Ye, X.; Vishwakarma, V.; Preet, R.; Dixon, D.A. CRC-derived exosomes containing the RNA binding protein HuR promote lung cell proliferation by stabilizing c-Myc mRNA. Cancer Biol. Ther. 2022, 23, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Arnedo, A.; Torres Figueroa, F.; Clavijo, C.; Arbelaez, P.; Cruz, J.C.; Munoz-Camargo, C. An image J plugin for the high throughput image analysis of in vitro scratch wound healing assays. PLoS ONE 2020, 15, e0232565. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Morine, Y.; Tokuda, K.; Yamada, S.; Saito, Y.; Nishi, M.; Ikemoto, T.; Shimada, M. Cancer-associated fibroblast-induced M2-polarized macrophages promote hepatocellular carcinoma progression via the plasminogen activator inhibitor-1 pathway. Int. J. Oncol. 2021, 59, 20215239. [Google Scholar] [CrossRef] [PubMed]
- Welsh, J.A.; Goberdhan, D.C.I.; O’Driscoll, L.; Buzas, E.I.; Blenkiron, C.; Bussolati, B.; Cai, H.; Di Vizio, D.; Driedonks, T.A.P.; Erdbrugger, U.; et al. Minimal information for studies of extracellular vesicles (MISEV2023): From basic to advanced approaches. J. Extracell. Vesicles 2024, 13, e12404. [Google Scholar] [CrossRef] [PubMed]
- Clerici, S.P.; Peppelenbosch, M.; Fuhler, G.; Consonni, S.R.; Ferreira-Halder, C.V. Colorectal Cancer Cell-Derived Small Extracellular Vesicles Educate Human Fibroblasts to Stimulate Migratory Capacity. Front. Cell Dev. Biol. 2021, 9, 696373. [Google Scholar] [CrossRef] [PubMed]
- Nissen, N.I.; Karsdal, M.; Willumsen, N. Collagens and Cancer associated fibroblasts in the reactive stroma and its relation to Cancer biology. J. Exp. Clin. Cancer Res. 2019, 38, 115. [Google Scholar] [CrossRef] [PubMed]
- Lendahl, U.; Muhl, L.; Betsholtz, C. Identification, discrimination and heterogeneity of fibroblasts. Nat. Commun. 2022, 13, 3409. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.H.; Wu, A.T.H.; Bamodu, O.A.; Yadav, V.K.; Chao, T.Y.; Tzeng, Y.M.; Mukhopadhyay, D.; Hsiao, M.; Lee, J.C. Ovatodiolide Suppresses Oral Cancer Malignancy by Down-Regulating Exosomal Mir-21/STAT3/β-Catenin Cargo and Preventing Oncogenic Transformation of Normal Gingival Fibroblasts. Cancers 2020, 12, 56. [Google Scholar] [CrossRef] [PubMed]
- Arebro, J.; Towle, R.; Lee, C.M.; Bennewith, K.L.; Garnis, C. Extracellular vesicles promote activation of pro-inflammatory cancer-associated fibroblasts in oral cancer. Front. Cell Dev. Biol. 2023, 11, 1240159. [Google Scholar] [CrossRef] [PubMed]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [PubMed]
- Lahav, T.G.; Adler, O.; Zait, Y.; Shani, O.; Amer, M.; Doron, H.; Abramovitz, L.; Yofe, I.; Cohen, N.; Erez, N. Melanoma-derived extracellular vesicles instigate proinflammatory signaling in the metastatic microenvironment. Int. J. Cancer 2019, 145, 2521–2534. [Google Scholar] [CrossRef] [PubMed]
- Li, K.D.; Liu, T.; Chen, J.; Ni, H.Y.; Li, W.H. Survivin in breast cancer-derived exosomes activates fibroblasts by up-regulating SOD1, whose feedback promotes cancer proliferation and metastasis. J. Biol. Chem. 2020, 295, 13737–13752. [Google Scholar] [CrossRef]
- Goulet, C.R.; Bernard, G.; Tremblay, S.; Chabaud, S.; Bolduc, S.; Pouliot, F. Exosomes Induce Fibroblast Differentiation into Cancer-Associated Fibroblasts through TGFβ Signaling. Mol. Cancer Res. 2018, 16, 1196–1204. [Google Scholar] [CrossRef] [PubMed]
- Shelton, M.; Anene, C.A.; Nsengimana, J.; Roberts, W.; Newton-Bishop, J.; Boyne, J.R. The role of CAF derived exosomal microRNAs in the tumour microenvironment of melanoma. BBA-Rev. Cancer 2021, 1875, 188456. [Google Scholar] [CrossRef] [PubMed]
- Jahan, S.; Mukherjee, S.; Ali, S.; Bhardwaj, U.; Choudhary, R.K.; Balakrishnan, S.; Naseem, A.; Mir, S.A.; Banawas, S.; Alaidarous, M.; et al. Pioneer Role of Extracellular Vesicles as Modulators of Cancer Initiation in Progression, Drug Therapy, and Vaccine Prospects. Cells 2022, 11, 490. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.K.; Wu, L.; Yan, G.F.; Chen, Y.; Zhou, M.Y.; Wu, Y.Z.; Li, Y.S. Inflammation and tumor progression: Signaling pathways and targeted intervention. Signal Transduct. Target. Ther. 2021, 6, 263. [Google Scholar] [CrossRef]
- Wu, F.L.; Yang, J.; Liu, J.J.; Wang, Y.; Mu, J.T.; Zeng, Q.X.; Deng, S.Z.; Zhou, H.M. Signaling pathways in cancer-associated fibroblasts and targeted therapy for cancer. Signal Transduct. Target. Ther. 2021, 6, 218. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.Y.; Lee, J.J.; Yeh, C.Y.; Yang, C.J.; Kok, S.H.; Ko, J.Y.; Tsai, F.C.; Chia, J.S. Reciprocal activation of cancer-associated fibroblasts and oral squamous carcinoma cells through CXCL1. Oral. Oncol. 2019, 88, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Awaji, M.; Saxena, S.; Wu, L.Y.; Prajapati, D.R.; Purohit, A.; Varney, M.L.; Kumar, S.; Rachagani, S.; Ly, Q.P.; Jain, M.; et al. CXCR2 signaling promotes secretory cancer-associated fibroblasts in pancreatic ductal adenocarcinoma. Faseb J. 2020, 34, 9405–9418. [Google Scholar] [CrossRef]
- de Lope, L.R.; Sánchez-Herrero, E.; Serna-Blasco, R.; Provencio, M.; Romero, A. Cancer as an infective disease: The role of EVs in tumorigenesis. Mol. Oncol. 2023, 17, 390–406. [Google Scholar] [CrossRef] [PubMed]
- Shoucair, I.; Weber Mello, F.; Jabalee, J.; Maleki, S.; Garnis, C. The Role of Cancer-Associated Fibroblasts and Extracellular Vesicles in Tumorigenesis. Int. J. Mol. Sci. 2020, 21, 6837. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Liu, J.; Qian, H.; Zhuang, Q. Cancer-associated fibroblasts: From basic science to anticancer therapy. Exp. Mol. Med. 2023, 55, 1322–1332. [Google Scholar] [CrossRef]
- Nalbantoglu, I.; Blanc, V.; Davidson, N.O. Characterization of Colorectal Cancer Development in Apcmin/+ Mice. Methods Mol. Biol. 2016, 1422, 309–327. [Google Scholar] [CrossRef]
- Hosseinkhani, B.; Duran, G.; Hoeks, C.; Hermans, D.; Schepers, M.; Baeten, P.; Poelmans, J.; Coenen, B.; Bekar, K.; Pintelon, I.; et al. Cerebral microvascular endothelial cell-derived extracellular vesicles regulate blood—Brain barrier function. Fluids Barriers CNS 2023, 20, 95. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.; Garcia, V.; Rodriguez, M.; Compte, M.; Cisneros, E.; Veguillas, P.; Garcia, J.M.; Dominguez, G.; Campos-Martin, Y.; Cuevas, J.; et al. Analysis of exosome release and its prognostic value in human colorectal cancer. Genes Chromosomes Cancer 2012, 51, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Wei, P.; Wu, F.; Kang, B.; Sun, X.H.; Heskia, F.; Pachot, A.; Liang, J.; Li, D.W. Plasma extracellular vesicles detected by Single Molecule array technology as a liquid biopsy for colorectal cancer. J. Extracell. Vesicles 2020, 9, 1809765. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.F.; Zhang, X.L.; Wang, Y.J.; Fang, Y.; Li, M.L.; Liu, X.Y.; Luo, H.Y.; Tian, Y. The regulatory network of the chemokine CCL5 in colorectal cancer. Ann. Med. 2023, 55, 2205168. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, P.D.; Mendonça, S.; Martins, F.; Oliveira, M.J.; Velho, S. Modulation of Fibroblast Phenotype by Colorectal Cancer Cell-Secreted Factors Is Mostly Independent of Oncogenic KRAS. Cells 2022, 11, 2490. [Google Scholar] [CrossRef] [PubMed]
- Rai, A.; Greening, D.W.; Xu, R.; Suwakulsiri, W.; Simpson, R.J. Exosomes Derived from the Human Primary Colorectal Cancer Cell Line SW480 Orchestrate Fibroblast-Led Cancer Invasion. Proteomics 2020, 20, 2000016. [Google Scholar] [CrossRef] [PubMed]
- Hawinkels, L.J.A.C.; Paauwe, M.; Verspaget, H.W.; Wiercinska, E.; van der Zon, J.M.; van der Ploeg, K.; Koelink, P.J.; Lindeman, J.H.N.; Mesker, W.; ten Dijke, P.; et al. Interaction with colon cancer cells hyperactivates TGF-β signaling in cancer-associated fibroblasts. Oncogene 2014, 33, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Hassametto, A.; Tanomrat, R.; Muangthong, T.; Worawichawong, S.; Suwannalert, P. Role of Oxidative Stress-Dependent C/EBPbeta Expression on CAF Transformation Inducing HCT116 Colorectal Cancer Cell Progression; Migration and Invasion. Asian Pac. J. Cancer Prev. 2023, 24, 3825–3835. [Google Scholar] [CrossRef] [PubMed]
- Kennel, K.B.; Bozlar, M.; De Valk, A.F.; Greten, F.R. Cancer-Associated Fibroblasts in Inflammation and Antitumor Immunity. Clin. Cancer Res. 2023, 29, 1009–1016. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Sun, Y.; Peng, L.; Sun, J.; Sha, Z.; Lin, H.; Li, Y.; Li, C.; Li, H.; Shang, H.; et al. Extracellular vesicle-packaged ILK from mesothelial cells promotes fibroblast activation in peritoneal fibrosis. J. Extracell. Vesicles 2023, 12, e12334. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Kim, S.Y.; Song, S.J.; Hong, H.K.; Lee, Y.; Oh, B.Y.; Lee, W.Y.; Cho, Y.B. Crosstalk between CCL7 and CCR3 promotes metastasis of colon cancer cells via ERK-JNK signaling pathways. Oncotarget 2016, 7, 36842–36853. [Google Scholar] [CrossRef] [PubMed]
- Borowczak, J.; Szczerbowski, K.; Maniewski, M.; Kowalewski, A.; Janiczek-Polewska, M.; Szylberg, A.; Marszalek, A.; Szylberg, L. The Role of Inflammatory Cytokines in the Pathogenesis of Colorectal Carcinoma-Recent Findings and Review. Biomedicines 2022, 10, 1670. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Karin, M. NF–κB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef]
- Wu, X.; Zhou, Z.; Xu, S.; Liao, C.L.; Chen, X.; Li, B.; Peng, J.W.; Li, D.; Yang, L.F. Extracellular vesicle packaged LMP1-activated fibroblasts promote tumor progression via autophagy and stroma-tumor metabolism coupling. Cancer Lett. 2020, 478, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Vu, L.T.; Peng, B.; Zhang, D.X.; Ma, V.; Mathey-Andrews, C.A.; Lam, C.K.; Kiomourtzis, T.; Jin, J.; McReynolds, L.; Huang, L.; et al. Tumor-secreted extracellular vesicles promote the activation of cancer-associated fibroblasts via the transfer of microRNA-125b. J. Extracell. Vesicles 2019, 8, 1599680. [Google Scholar] [CrossRef]
- Naito, Y.; Yamamoto, Y.; Sakamoto, N.; Shimomura, I.; Kogure, A.; Kumazaki, M.; Yokoi, A.; Yashiro, M.; Kiyono, T.; Yanagihara, K.; et al. Cancer extracellular vesicles contribute to stromal heterogeneity by inducing chemokines in cancer-associated fibroblasts. Oncogene 2019, 38, 5566–5579. [Google Scholar] [CrossRef] [PubMed]
- Kong, J.; Tian, H.Z.; Zhang, F.Y.; Zhang, Z.B.; Li, J.; Liu, X.; Li, X.C.; Liu, J.; Li, X.J.; Jin, D.; et al. Extracellular vesicles of carcinoma-associated fibroblasts creates a pre-metastatic niche in the lung through activating fibroblasts. Mol. Cancer 2019, 18, 175. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibodies | Origin | Dilution | Supplier | Catalog Number# |
|---|---|---|---|---|
| CD81 | Rabbit | 1/1000 | Thermo Fisher | 10630D |
| CD63 | Rabbit | 1/1000 | Cell signaling (Danvers, MA, USA) | 52090 |
| α-SMA | Rabbit | 1/1000 | Cell signaling | 19245 |
| FAP | Rabbit | 1/1000 | Cell signaling | 66562 |
| Vimentin | Rabbit | 1/1000 | Cell signaling | 5741 |
| GAPDH | Rabbit | 1/1000 | Cell signaling | 5174 |
| Actin | Mouse | 1/5000 | MP Biomedicals (Irvine, CA, USA) | 691002 |
| NF-κB p65 | Rabbit | 1/1000 | Cell signaling | 8242 |
| Phospho-NF-κB p65 | Rabbit | 1/1000 | Cell signaling | 3033 |
| IκBα | Rabbit | 1/1000 | Cell signaling | 4812 |
| Phospho-IκBα | Rabbit | 1/1000 | Cell signaling | 2859 |
| Goat anti-Rabbit IgG | Goat | 1/10,000 | LI-COR | 926-68070 |
| Donkey anti-Mouse IgG | Donkey | 1/10,000 | LI-COR | 926-32212 |
| GENE | Forward | Reverse |
|---|---|---|
| ACTIN | TGGTTACAGGAAGTCCCTTGCC | ATGCTATCACCTCCCCTGTGTG |
| α-SMA | CCGACCGAATGCAGAAGGA | ACAGAGTATTTGCGCTCCGAA |
| FAP | GGAAGTGCCTGTTCCAGCAATG | TGTCTGCCAGTCTTCCCTGAAG |
| FSP-1 | CCTGGGGAAAAGGACAGATGAA | CATGGCAATGCAGGACAGGA |
| Vimentin | AGGCAAAGCAGGAGTCCACTGA | ATCTGGCGTTCCAGGGACTCAT |
| IL-1β | AGCTACGAATCTCCGACCAC | CGTTATCCCATGTGTCGAAGAA |
| IL-6 | GGTACATCCTCGACGGCATCT | GTGCCTCTTTGCTGCTTTCAC |
| IL-8 | ATGACTTCCAAGCTGGCC | TCTTCAAAAACTTCTCCACAACCC |
| TNF-α | GTGAGGAGGACGAACATC | GAGCCAGAAGAGGTTGAG |
| TGF-β | ACACCAACTATTGCTTCAG | TGTCCAGGCTCCAAATG |
| NF-κB p50 | ACACTGGAAGCACGAATGACAGA | CCTCCACCTTCTGCTTGCAA |
| NF-κB p65 | CAGGCGAGAGGAGCACAGATAC | TCCTTTCCTACAAGCTCGTGGG |
| IKKα | TGACAGCACAGAGATGGTGA | CTTCTGCTTACAGCCCAACA |
| IKKβ | ACAGCGAGCAAACCGAGTTTGG | CCTCTGTAAGTCCACAATGTCGG |
| IKKγ | AGCACCTGAAGAGATGCCAGCA | AGCCTGGCATTCCTTAGTGGCA |
| IκB-α | CACTCCATCCTGAAGGCTACCAAC | CACACTTCAACAGGAGTGACACCAG |
| IκB-β | GTACTCCCGACACCAACCAT | CGGACCATCTCCACATCTTT |
| TRAF6 | CTTTGGCAAATGTCATCTGTG | CTGAATGTGCATGGAATTGG |
| Fn1 | AGGCTTGAACCAACCTACGGATGA | GCCTAAGCACTGGCACAACAGTTT |
| Col1a1 | TCTGCGACAACGGCAAGGTG | GACGCCGGTGGTTTCTTGGT |
| Col4a1 | CTGCCTGGAGGAGTTTAGAAG | GAACATCTCGCTCCTCTCTATG |
| Cytokines/Chemokines | LEV-APC/Control Fibroblasts (Fold Change) | p-Value (LEV-APC/Fibroblasts) |
|---|---|---|
| Fold change > 1.5 | ||
| IL-6 | 2.45 | 0.025 |
| IL-8 # | 1.81 | 0.012 |
| CCL11 | 3.56 | 0.047 |
| CCL22 # | 2.78 | 0.029 |
| EGF | 2.78 | 0.046 |
| Angiogenin | 3.57 | 0.021 |
| OSM # | 2.12 | 0.043 |
| VEGF-A | 2.06 | 0.043 |
| FGF-4 | 2.09 | 0.042 |
| FLT-3 ligand # | 2.20 | 0.047 |
| IGFBP-3 # | 2.80 | 0.004 |
| CXCL-10 | 2.63 | 0.025 |
| Osteopontin # | 2.00 | 0.018 |
| TIMP-2 | 3.14 | 0.018 |
| Fold change > 5.5 | ||
| GRO-Alpha | 6.12 | 0.0451 |
| CCL5 | 11.2 | 0.0058 |
| IGF-1 # | 5.93 | 0.009 |
| Fold change > 20 | ||
| CXCL-5 (ENA-78) | 29.7 | 0.011 |
| GRO # | 21.8 | 0.0003 |
| CCL7 | 23.5 | 0.0285 |
| Cytokines/Chemokines | LEV-WT/Fibroblasts (Fold Change) | p-Value (LEV-WT/Fibroblasts) |
|---|---|---|
| IL-6 | 0.99 | 0.322 |
| IL-8 | 1.30 | 0.320 |
| CCL11 | 2.30 | 0.257 |
| CCL22 | 0.80 | 0.909 |
| EGF | 1.52 | 0.564 |
| Angiogenin | 2.33 | 0.158 |
| OSM | 0.83 | 0.995 |
| VEGF-A | 0.75 | 0.875 |
| FGF-4 | 0.89 | 0.967 |
| FLT-3 ligand | 0.70 | 0.745 |
| IGFBP-3 | 0.41 | 0.183 |
| CXCL-10 | 1.24 | 0.709 |
| Osteopontin | 0.84 | 0.708 |
| TIMP-2 | 1.68 | 0.569 |
| GRO-Alpha | 3.383 | 0.3044 |
| CCL5 | 2.793 | 0.5158 |
| IGF-1 | 2.522 | 0.2642 |
| CXCL-5 | 1.26 | 0.9987 |
| GRO | 2.327 | 0.6406 |
| CCL7 | 1.135 | 0.7315 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arteaga-Blanco, L.A.; Evans, A.E.; Dixon, D.A. Plasma-Derived Extracellular Vesicles and Non-Extracellular Vesicle Components from APCMin/+ Mice Promote Pro-Tumorigenic Activities and Activate Human Colonic Fibroblasts via the NF-κB Signaling Pathway. Cells 2024, 13, 1195. https://doi.org/10.3390/cells13141195
Arteaga-Blanco LA, Evans AE, Dixon DA. Plasma-Derived Extracellular Vesicles and Non-Extracellular Vesicle Components from APCMin/+ Mice Promote Pro-Tumorigenic Activities and Activate Human Colonic Fibroblasts via the NF-κB Signaling Pathway. Cells. 2024; 13(14):1195. https://doi.org/10.3390/cells13141195
Chicago/Turabian StyleArteaga-Blanco, Luis A., Andrew E. Evans, and Dan A. Dixon. 2024. "Plasma-Derived Extracellular Vesicles and Non-Extracellular Vesicle Components from APCMin/+ Mice Promote Pro-Tumorigenic Activities and Activate Human Colonic Fibroblasts via the NF-κB Signaling Pathway" Cells 13, no. 14: 1195. https://doi.org/10.3390/cells13141195
APA StyleArteaga-Blanco, L. A., Evans, A. E., & Dixon, D. A. (2024). Plasma-Derived Extracellular Vesicles and Non-Extracellular Vesicle Components from APCMin/+ Mice Promote Pro-Tumorigenic Activities and Activate Human Colonic Fibroblasts via the NF-κB Signaling Pathway. Cells, 13(14), 1195. https://doi.org/10.3390/cells13141195