

A Novel Pyrazole Exhibits Potent Anticancer Cytotoxicity via Apoptosis, Cell Cycle Arrest, and the Inhibition of Tubulin Polymerization in Triple-Negative Breast Cancer Cells
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Cell Culture
2.3. Differential Nuclear Staining (DNS) Assay
2.4. Cytotoxic Concentration 50 (CC50) and Selective Cytotoxicity Index (SCI) Calculation
2.5. Annexin V-FITC and Propidium Iodide Assay
2.6. Caspase-3/7 Activation Assay
2.7. Cell Cycle Analysis
2.8. Transcriptome Analysis
2.9. Connectivity Map (CMap) Analysis
2.10. Computational Docking
2.11. Cytoskeleton Analysis with Fluorescent Microscopy
2.12. Tubulin Polymerization Assay
3. Results
3.1. Drug Screening Identified a Novel Pyrazole Derivative with Potent Cytotoxicity against a Panel of Different Cell Lines
3.2. PTA-1 Induces Apoptosis as Measured by Phosphatidylserine Externalization and Caspase-3/7 Activation
3.3. PTA-1 Interferes with Cell Cycle Progression
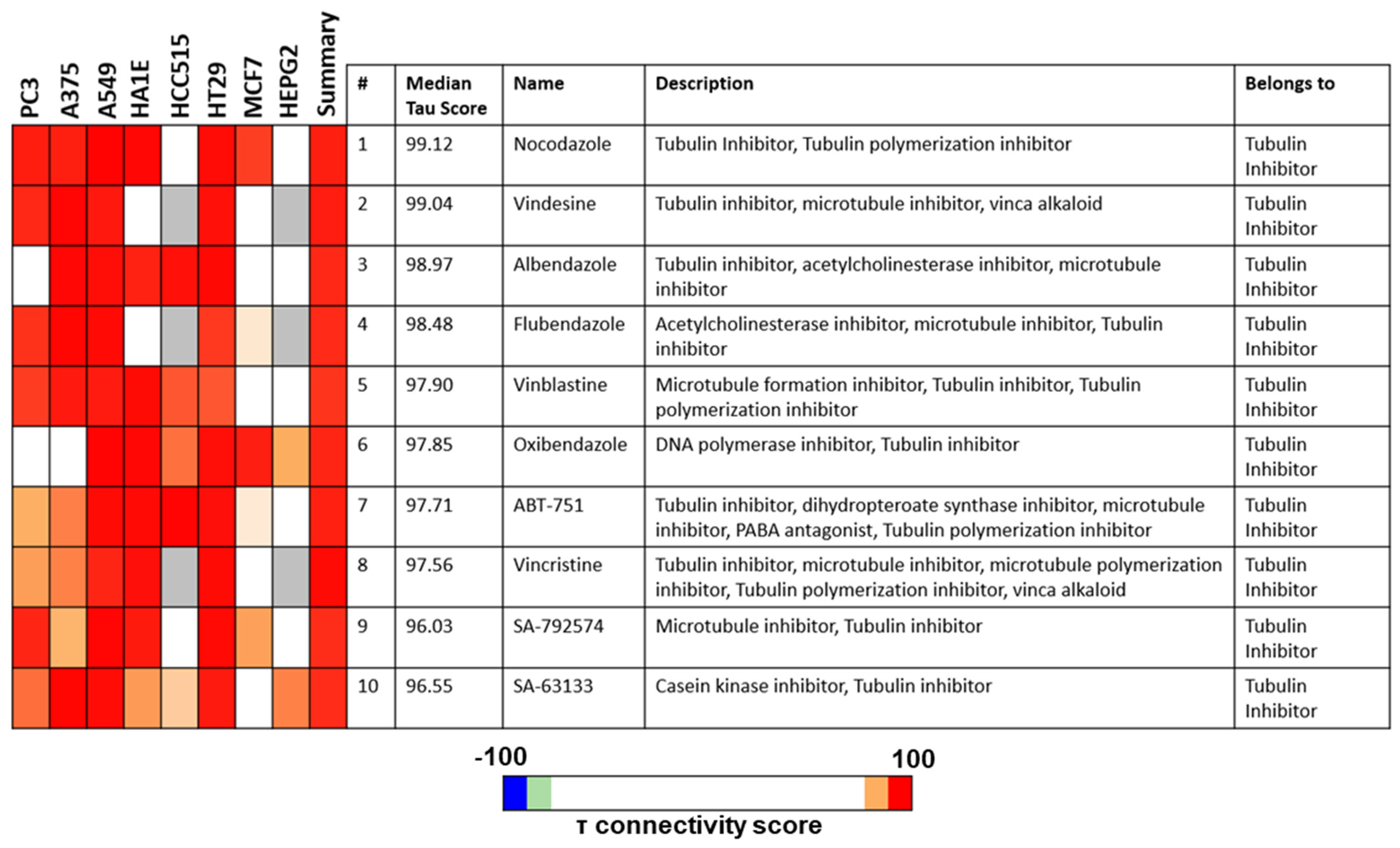
3.4. PTA-1-Treated MDA-MB-231 Cells Display a Transcriptome Profile Similar to Tubulin Polymerization Inhibitors
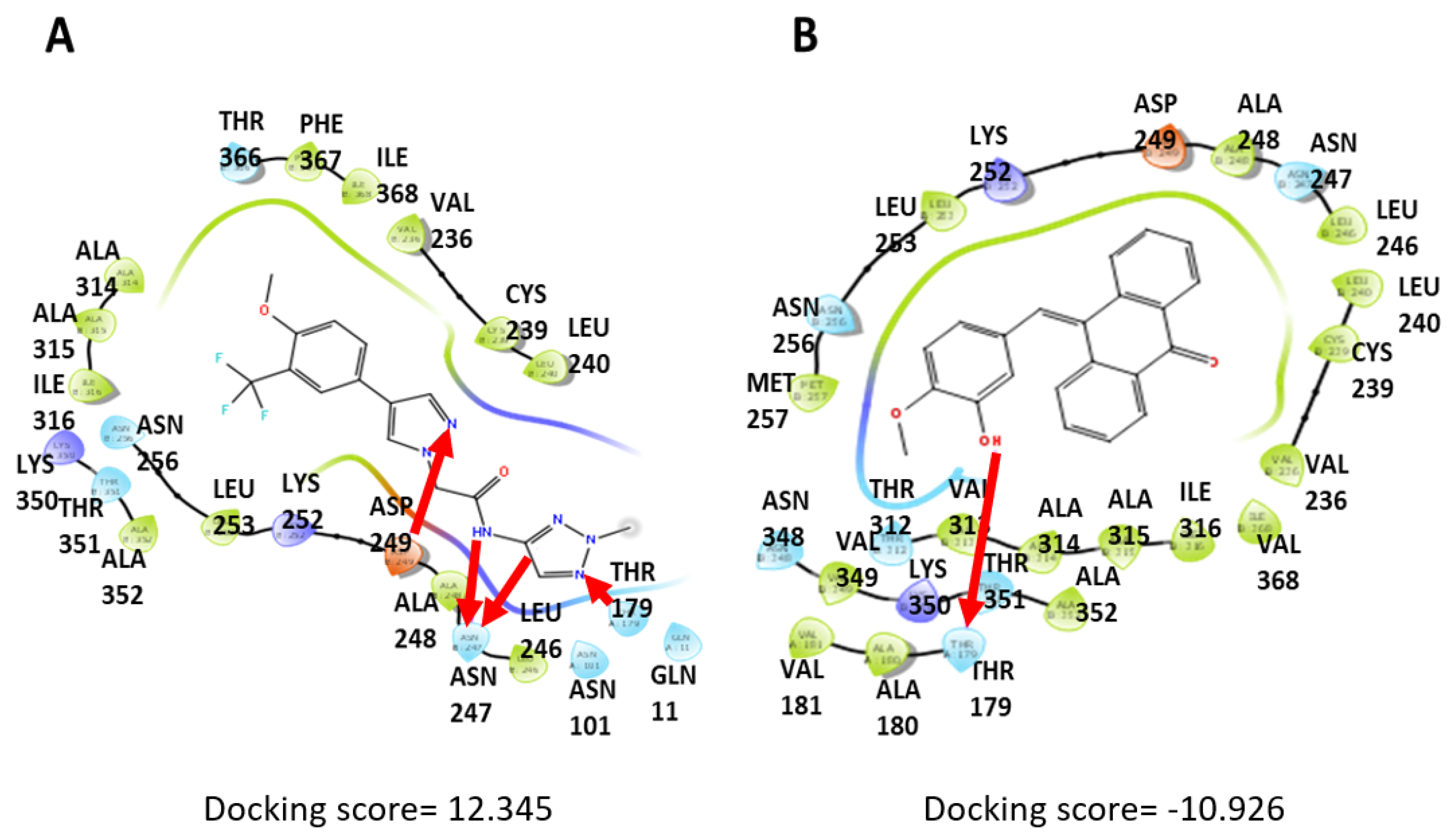
3.5. Computational Docking Shows That PTA-1 Interacts with Tubulin
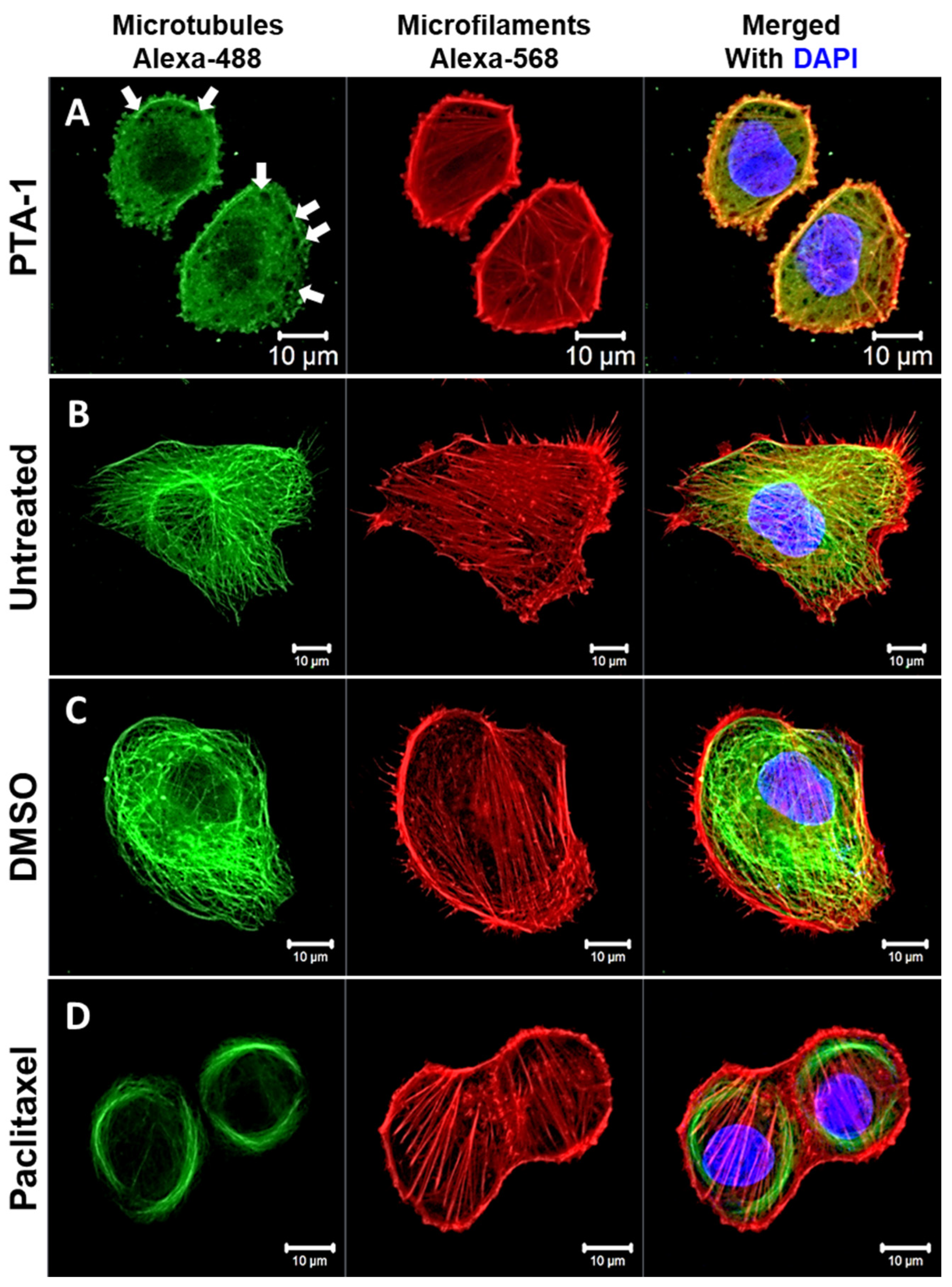
3.6. PTA-1 Disturbs Microtubule Organization in MDA-MB-231 Cells
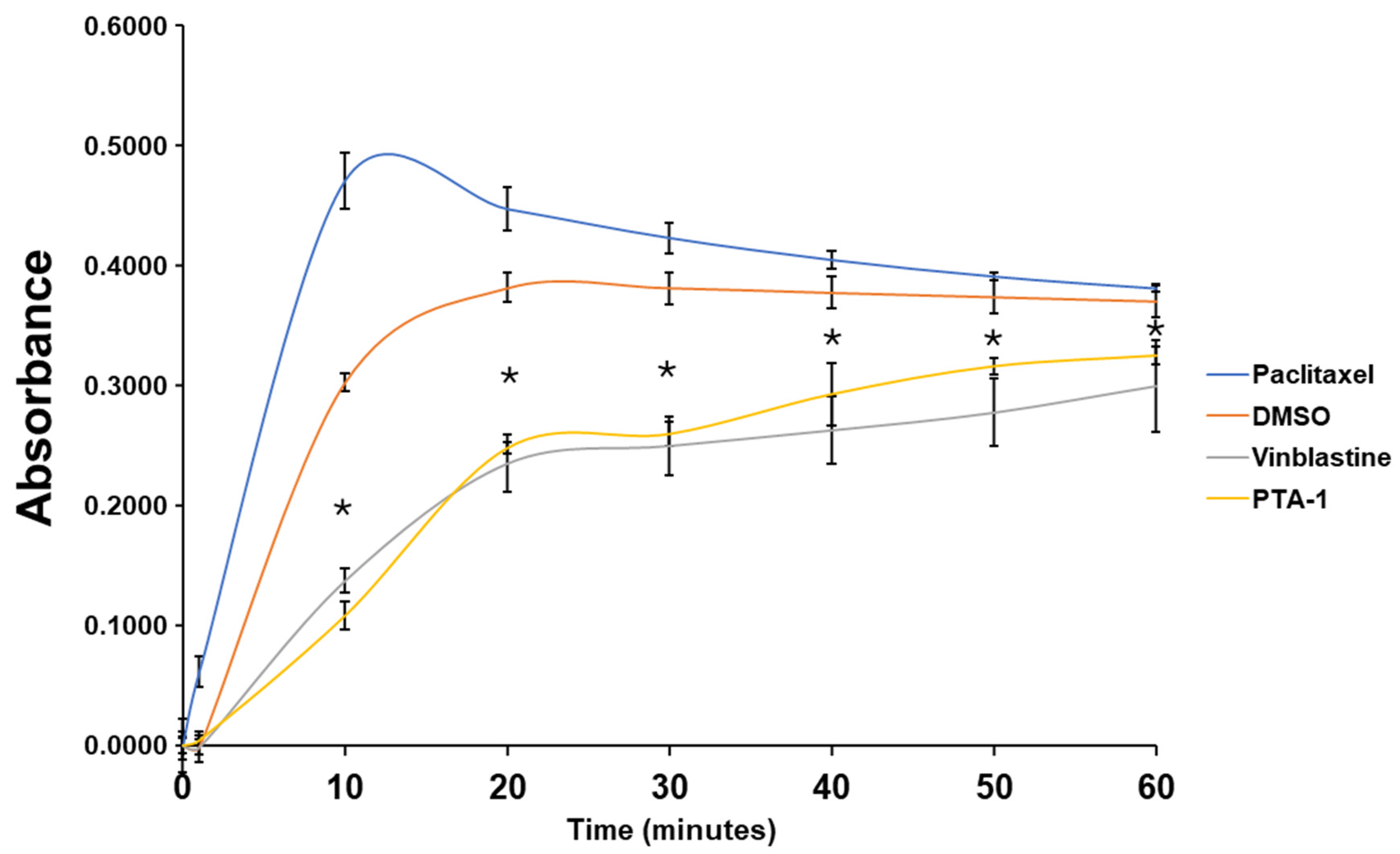
3.7. PTA-1 Inhibits Tubulin Polymerization
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- What Is Cancer?—National Cancer Institute. Available online: https://www.cancer.gov/about-cancer/understanding/what-is-cancer (accessed on 31 March 2022).
- Cancer—Symptoms and Causes—Mayo Clinic. Available online: https://www.mayoclinic.org/diseases-conditions/cancer/symptoms-causes/syc-20370588 (accessed on 31 March 2022).
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; He, F.; Hu, Y.; Wang, Q.; Wang, D.; Sha, Y.; Wu, J. Cancer Incidence and Mortality and Risk Factors in Member Countries of the “ Belt and Road “ Initiative. BMC Cancer 2022, 22, 582. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer Statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Debela, D.T.; Muzazu, S.G.; Heraro, K.D.; Ndalama, M.T.; Mesele, B.W.; Haile, D.C.; Kitui, S.K.; Manyazewal, T. New Approaches and Procedures for Cancer Treatment: Current Perspectives. SAGE Open Med. 2021, 9, 205031212110343. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A. Cancer Statistics, 2024. CA Cancer J. Clin. 2024, 74, 12–49. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.D.; Nogueira, L. Cancer Treatment and Survivorship Statistics, 2022. CA Cancer J. Clin. 2022, 72, 409–436. [Google Scholar] [CrossRef] [PubMed]
- Mc Erlain, T.; Burke, A.; Branco, C.M. Life after Cell Death-Survival and Survivorship Following Chemotherapy. Cancers 2021, 13, 2942. [Google Scholar] [CrossRef] [PubMed]
- Anand, U.; Dey, A.; Chandel, A.K.S.; Sanyal, R.; Mishra, A.; Pandey, D.K.; De Falco, V.; Upadhyay, A.; Kandimalla, R.; Chaudhary, A.; et al. Cancer Chemotherapy and beyond: Current Status, Drug Candidates, Associated Risks and Progress in Targeted Therapeutics. Genes Dis. 2023, 10, 1367–1401. [Google Scholar] [CrossRef]
- Khachatryan, H.; Olszowy, B.; Barrero, C.A.; Gordon, J.; Perez-Leal, O. Identification of Inhibitors of Tubulin Polymerization Using a CRISPR-Edited Cell Line with Endogenous Fluorescent Tagging of β-Tubulin and Histone H1. Biomolecules 2023, 13, 249. [Google Scholar] [CrossRef]
- Di Cesare, E.; Verrico, A.; Miele, A.; Giubettini, M.; Rovella, P.; Coluccia, A.; Famiglini, V.; La Regina, G.; Cundari, E.; Silvestri, R.; et al. Mitotic Cell Death Induction by Targeting the Mitotic Spindle with Tubulin-Inhibitory Indole Derivative Molecules. Oncotarget 2017, 8, 19738–19759. [Google Scholar] [CrossRef] [PubMed]
- Singh, H.; Kumar, M.; Nepali, K.; Gupta, M.K.; Saxena, A.K.; Sharma, S.; Bedi, P.M.S. Triazole Tethered C5-Curcuminoid-Coumarin Based Molecular Hybrids as Novel Antitubulin Agents: Design, Synthesis, Biological Investigation and Docking Studies. Eur. J. Med. Chem. 2016, 116, 102–115. [Google Scholar] [CrossRef] [PubMed]
- Hawash, M. Recent Advances of Tubulin Inhibitors Targeting the Colchicine Binding Site for Cancer Therapy. Biomolecules 2022, 12, 1843. [Google Scholar] [CrossRef] [PubMed]
- El-Brolsy, H.M.E.M.; Hanafy, N.A.N.; El-Kemary, M.A. Fighting Non-Small Lung Cancer Cells Using Optimal Functionalization of Targeted Carbon Quantum Dots Derived from Natural Sources Might Provide Potential Therapeutic and Cancer Bio Image Strategies. Int. J. Mol. Sci. 2022, 23, 13283. [Google Scholar] [CrossRef] [PubMed]
- Lema, C.; Varela-Ramirez, A.; Aguilera, R.J. Differential Nuclear Staining Assay for High-Throughput Screening to Identify Cytotoxic Compounds. Curr. Cell. Biochem. 2011, 1, 1–14. [Google Scholar] [PubMed]
- Varela-Ramirez, A.; Costanzo, M.; Carrasco, Y.P.; Pannell, K.H.; Aguilera, R.J. Cytotoxic Effects of Two Organotin Compounds and Their Mode of Inflicting Cell Death on Four Mammalian Cancer Cells. Cell Biol. Toxicol. 2011, 27, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Hess, J.D.; Macias, L.H.; Gutierrez, D.A.; Moran-Santibanez, K.; Contreras, L.; Medina, S.; Villanueva, P.J.; Kirken, R.A.; Varela-Ramirez, A.; Penichet, M.L.; et al. Identification of a Unique Cytotoxic Thieno[2,3-c]Pyrazole Derivative with Potent and Selective Anticancer Effects In Vitro. Biology 2022, 11, 930. [Google Scholar] [CrossRef] [PubMed]
- Robles-Escajeda, E.; Das, U.; Ortega, N.M.; Parra, K.; Francia, G.; Dimmock, J.R.; Varela-Ramirez, A.; Aguilera, R.J. A Novel Curcumin-like Dienone Induces Apoptosis in Triple-Negative Breast Cancer Cells. Cell. Oncol. 2016, 39, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Robles-Escajeda, E.; Lerma, D.; Nyakeriga, A.M.; Ross, J.A.; Kirken, R.A. Searching in Mother Nature for Anti-Cancer Activity: Anti-Proliferative and Pro-Apoptotic Effect Elicited by Green Barley on Leukemia/Lymphoma Cells. PLoS ONE 2013, 8, 73508. [Google Scholar] [CrossRef]
- Abdelhameed, R.F.A.; Habib, E.S.; Ibrahim, A.K.; Yamada, K.; Abdel-Kader, M.S.; Ahmed, S.A.; Ibrahim, A.K.; Badr, J.M.; Nafie, M.S. Chemical Constituent Profiling of Phyllostachys Heterocycla Var. Pubescens with Selective Cytotoxic Polar Fraction through EGFR Inhibition in HepG2 Cells. Molecules 2021, 26, 940. [Google Scholar] [CrossRef]
- Contreras, L.; Medina, S.; Schiaffino Bustamante, A.Y.; Borrego, E.A.; Valenzuela, C.A.; Das, U.; Karki, S.S.; Dimmock, J.R.; Aguilera, R.J. Three Novel Piperidones Exhibit Tumor-Selective Cytotoxicity on Leukemia Cells via Protein Degradation and Stress-Mediated Mechanisms. Pharmacol. Rep. 2022, 3, 159–174. [Google Scholar] [CrossRef]
- Martínez-Castillo, M.; Villegas-Sepúlveda, N.; Meraz-Rios, M.A.; Hernández-Zavala, A.; Berumen, J.; Coleman, M.A.; Orozco, L.; Cordova, E.J. Curcumin Differentially Affects Cell Cycle and Cell Death in Acute and Chronic Myeloid Leukemia Cells. Oncol. Lett. 2018, 15, 6777–6783. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, D.A.; Contreras, L.; Villanueva, P.J.; Borrego, E.A.; Morán-Santibañez, K.; Hess, J.D.; Dejesus, R.; Larragoity, M.; Betancourt, A.P.; Mohl, J.E.; et al. Identification of a Potent Cytotoxic Pyrazole with Anti-Breast Cancer Activity That Alters Multiple Pathways. Cells 2022, 11, 254. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Parrish, N.; Hormozdiari, F.; Eskin, E. Assembly of Non-Unique Insertion Content Using next-Generation Sequencing. In The Impact of Accurate Quantification on Proteomic and Genetic Analysis and Research; Apple Academic Press: Palm Bay, FL, USA, 2014; pp. 21–40. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Lyons, N.; Koedam, M.; van de Peppel, J.; van Leeuwen, J.P.T.M.; van der Eerden, B.C.J. Identification of Small Molecules as Novel Anti-Adipogenic Compounds Based on Connectivity Map. Front. Endocrinol. 2022, 13, 1017832. [Google Scholar] [CrossRef]
- Musa, A.; Ghoraie, L.S.; Zhang, S.-D.; Glazko, G.; Yli-Harja, O.; Dehmer, M.; Haibe-Kains, B.; Emmert-Streib, F. A Review of Connectivity Map and Computational Approaches in Pharmacogenomics. Brief. Bioinform. 2018, 19, 506–523. [Google Scholar] [CrossRef] [PubMed]
- Swain, R.M.; Contreras, L.; Varela-Ramirez, A.; Hossain, M.; Das, U.; Valenzuela, C.A.; Penichet, M.L.; Dimmock, J.R.; Aguilera, R.J. Two Novel Piperidones Induce Apoptosis and Antiproliferative Effects on Human Prostate and Lymphoma Cancer Cell Lines. Investig. New Drugs 2022, 40, 905–921. [Google Scholar] [CrossRef]
- David, T.I.; Adelakun, N.S.; Omotuyi, O.I.; Metibemu, D.S.; Ekun, O.E.; Eniafe, G.O.; Inyang, O.K.; Adewumi, B.; Enejoh, O.A.; Owolabi, R.T.; et al. Molecular Docking Analysis of Phyto-Constituents from Cannabis Sativa with PfDHFR. Bioinformation 2018, 14, 574–579. [Google Scholar] [CrossRef]
- Van Engeland, M.; Nieland, L.J.W.; Ramaekers, F.C.S.; Schutte, B.; Reutelingsperger, C.P.M. Annexin V-Affinity Assay: A Review on an Apoptosis Detection System Based on Phosphatidylserine Exposure. Cytometry 1998, 31, 1–9. [Google Scholar] [CrossRef]
- Nagata, S.; Tanaka, M. Programmed Cell Death and the Immune system. Nat. Rev. Immunol. 2017, 17, 333–340. [Google Scholar] [CrossRef]
- Hoose, S.A.; Duran, C.; Malik, I.; Eslamfam, S.; Shasserre, S.C.; Downing, S.S.; Hoover, E.M.; Dowd, K.E.; Smith, R., 3rd; Polymenis, M. Systematic Analysis of Cell Cycle Effects of Common Drugs Leads to the Discovery of a Suppressive Interaction between Gemfibrozil and Fluoxetine. PLoS ONE 2012, 7, e36503. [Google Scholar] [CrossRef] [PubMed]
- Kluska, M.; Woźniak, K. Natural Polyphenols as Modulators of Etoposide Anti-Cancer Activity. Int. J. Mol. Sci. 2021, 22, 6602. [Google Scholar] [CrossRef]
- Iorio, F.; Bosotti, R.; Scacheri, E.; Belcastro, V.; Mithbaokar, P.; Ferriero, R.; Murino, L.; Tagliaferri, R.; Brunetti-Pierri, N.; Isacchi, A.; et al. Discovery of Drug Mode of Action and Drug Repositioning from Transcriptional Responses. Proc. Natl. Acad. Sci. USA 2010, 107, 14621–14626. [Google Scholar] [CrossRef] [PubMed]
- Pabon, N.A.; Xia, Y.; Estabrooks, S.K.; Ye, Z.; Herbrand, A.K.; Süß, E.; Biondi, R.M.; Assimon, V.A.; Gestwicki, J.E.; Brodsky, J.L.; et al. Predicting Protein Targets for Drug-like Compounds Using Transcriptomics. PLoS Comput. Biol. 2018, 14, e1006651. [Google Scholar] [CrossRef]
- Lim, N.; Pavlidis, P. Evaluation of Connectivity Map Shows Limited Reproducibility in Drug Repositioning. Sci. Rep. 2021, 11, 17624. [Google Scholar] [CrossRef] [PubMed]
- How Do I Interpret Connectivity Scores, and What Is a “Good” Score? Available online: https://clue.io/connectopedia/connectivity_scores (accessed on 5 October 2022).
- Schrödinger. What Is Considered a Good GlideScore? Available online: https://www.schrodinger.com/kb/639 (accessed on 5 October 2022).
- What Is the Difference between the Docking Score and GlideScore from the Results of a Docking Run?|Schrödinger. Available online: https://www.schrodinger.com/kb/348 (accessed on 5 October 2022).
- Schrödinger Can I Relate MM-GBSA Energies to Binding Affinity? Available online: https://www.schrodinger.com/kb/1647 (accessed on 5 October 2022).
- Stanton, R.A.; Gernert, K.M.; Nettles, J.H.; Aneja, R. ChemInform Abstract: Drugs That Target Dynamic Microtubules: A New Molecular Perspective. ChemInform 2011, 42, 443–481. [Google Scholar] [CrossRef]
- Kapoor, S.; Srivastava, S.; Panda, D. Indibulin Dampens Microtubule Dynamics and Produces Synergistic Antiproliferative Effect with Vinblastine in MCF-7 Cells: Implications in Cancer Chemotherapy. Sci. Rep. 2018, 8, 2–13. [Google Scholar] [CrossRef]
- Bukowski, K.; Kciuk, M.; Kontek, R. Molecular Sciences Mechanisms of Multidrug Resistance in Cancer Chemotherapy. Int. J. Mol. Sci. 2020, 21, 3233. [Google Scholar] [CrossRef]
- Villanueva, P.J.; Gutierrez, D.A.; Contreras, L.; Parra, K.; Segura-Cabrera, A.; Varela-Ramirez, A.; Aguilera, R.J. The Antimalarial Drug Pyronaridine Inhibits Topoisomerase II in Breast Cancer Cells and Hinders Tumor Progression In Vivo HHS Public Access. Clin. Cancer Drugs 2021, 8, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.R.; Wang, Z.F.; Shi, L.; Wang, Z.C.; Zhu, H.L. Design, Synthesis, and Biological Evaluation of Pyrazole Derivatives Containing Acetamide Bond as Potential BRAFV600E Inhibitors. Bioorg. Med. Chem. Lett. 2018, 28, 2382–2390. [Google Scholar] [CrossRef] [PubMed]
- Balbi, A.; Anzaldi, M.; MacCi, C.; Aiello, C.; Mazzei, M.; Gangemi, R.; Castagnola, P.; Miele, M.; Rosano, C.; Viale, M. Synthesis and Biological Evaluation of Novel Pyrazole Derivatives with Anticancer Activity. Eur. J. Med. Chem. 2011, 46, 5293–5309. [Google Scholar] [CrossRef] [PubMed]
- Mellini, P.; Marrocco, B.; Borovika, D.; Polletta, L.; Carnevale, I.; Saladini, S.; Stazi, G.; Zwergel, C.; Trapencieris, P.; Ferretti, E.; et al. Pyrazole-Based Inhibitors of Enhancer of Zeste Homologue 2 Induce Apoptosis and Autophagy in Cancer Cells. Philos. Trans. R. Soc. B Biol. Sci. 2018, 373, 20170150. [Google Scholar] [CrossRef]
- Birge, R.B.; Boeltz, S.; Kumar, S.; Carlson, J.; Wanderley, J.; Calianese, D.; Barcinski, M.; Brekken, R.A.; Huang, X.; Hutchins, J.T.; et al. Phosphatidylserine Is a Global Immunosuppressive Signal in Efferocytosis, Infectious Disease, and Cancer. Cell Death Differ. 2016, 23, 962–978. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Sun, J.; Zhou, S.; Wan, C.; Qin, S. Prediction of Drug-Target Interactions for Drug Repositioning Only Based on Genomic Expression Similarity. PLoS Comput. Biol. 2013, 9, 1003315. [Google Scholar] [CrossRef]
- Subramanian, A.; Narayan, R.; Corsello, S.M.; Peck, D.D.; Natoli, T.E.; Lu, X.; Gould, J.; Davis, J.F.; Tubelli, A.A.; Asiedu, J.K.; et al. A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell 2017, 171, 1437–1452.e17. [Google Scholar] [CrossRef] [PubMed]
- Kline-Smith, S.L.; Walczak, C.E. Mitotic Spindle Assembly and Chromosome Segregation: Refocusing on Microtubule Dynamics. Mol. Cell 2004, 15, 317–327. [Google Scholar] [CrossRef]
- Bunning, A.R.; Gupta, M.L.J. The Importance of Microtubule-Dependent Tension in Accurate Chromosome Segregation. Front. Cell Dev. Biol. 2023, 11, 1096333. [Google Scholar] [CrossRef]
- Salerni, B.L.; Bates, D.J.; Albershardt, T.C.; Lowrey, C.H.; Eastman, A. Vinblastine Induces Acute, Cell Cycle Phase—Independent Apoptosis in Some Leukemias and Lymphomas and Can Induce Acute Apoptosis in Others When Mcl-1 Is Suppressed. Mol. Cancer Ther. 2010, 9, 791–802. [Google Scholar] [CrossRef] [PubMed]
- Thomas, E.; Gopalakrishnan, V.; Hegde, M.; Kumar, S.; Karki, S.S.; Raghavan, S.C.; Choudhary, B. A Novel Resveratrol Based Tubulin Inhibitor Induces Mitotic Arrest and Activates Apoptosis in Cancer Cells. Sci. Rep. 2016, 6, 34653. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Tissue Origin | CC50 (µM) * | SCI ** |
|---|---|---|---|
| Solid Tumor/Adherent Cell Lines | 72 h | ||
| MCF-10A | Breast Non-Cancerous | 4.40 | N/A |
| MDA-MB-231 | Breast Cancer | 0.93 | 4.73 |
| MCF-7 | “ | 0.23 | 19.13 |
| HCC 70 | “ | 0.74 | 5.94 |
| T47D | “ | 0.63 | 6.98 |
| A375 | Melanoma | 0.34 | 12.94 |
| HT-29 | Colon Cancer | 0.47 | 9.36 |
| Colo 205 | “ | 0.30 | 14.66 |
| A549 | Lung Cancer | 0.17 | 25.88 |
| HepG2 | Liver Cancer | 0.36 | 12.22 |
| PANC-1 | Pancreatic Cancer | 0.28 | 1571 |
| PC3 | Prostate Cancer | 0.92 | 4.78 |
| Blood Non-Adherent Cell Lines | 48 h | ||
| CEM | Lymphomas/Leukemias | 0.42 | 10.47 |
| HL-60 | “ | 0.82 | 5.36 |
| Ramos | “ | 0.75 | 5.86 |
| NALM6 | “ | 2.07 | 2.12 |
| Jurkat | “ | 0.32 | 13.75 |
| KCL22 | “ | 0.72 | 6.11 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borrego, E.A.; Guerena, C.D.; Schiaffino Bustamante, A.Y.; Gutierrez, D.A.; Valenzuela, C.A.; Betancourt, A.P.; Varela-Ramirez, A.; Aguilera, R.J. A Novel Pyrazole Exhibits Potent Anticancer Cytotoxicity via Apoptosis, Cell Cycle Arrest, and the Inhibition of Tubulin Polymerization in Triple-Negative Breast Cancer Cells. Cells 2024, 13, 1225. https://doi.org/10.3390/cells13141225
Borrego EA, Guerena CD, Schiaffino Bustamante AY, Gutierrez DA, Valenzuela CA, Betancourt AP, Varela-Ramirez A, Aguilera RJ. A Novel Pyrazole Exhibits Potent Anticancer Cytotoxicity via Apoptosis, Cell Cycle Arrest, and the Inhibition of Tubulin Polymerization in Triple-Negative Breast Cancer Cells. Cells. 2024; 13(14):1225. https://doi.org/10.3390/cells13141225
Chicago/Turabian StyleBorrego, Edgar A., Cristina D. Guerena, Austre Y. Schiaffino Bustamante, Denisse A. Gutierrez, Carlos A. Valenzuela, Ana P. Betancourt, Armando Varela-Ramirez, and Renato J. Aguilera. 2024. "A Novel Pyrazole Exhibits Potent Anticancer Cytotoxicity via Apoptosis, Cell Cycle Arrest, and the Inhibition of Tubulin Polymerization in Triple-Negative Breast Cancer Cells" Cells 13, no. 14: 1225. https://doi.org/10.3390/cells13141225