
A Novel PDE10A Inhibitor for Tourette Syndrome and Other Movement Disorders



Abstract
:1. Introduction
2. Materials and Methods
2.1. PDE10A Inhibitory Activity
2.2. PDE10A Selectivity
2.3. In Vivo Activities in Rat
2.4. Determination of EM-221 in Plasma and Striatum
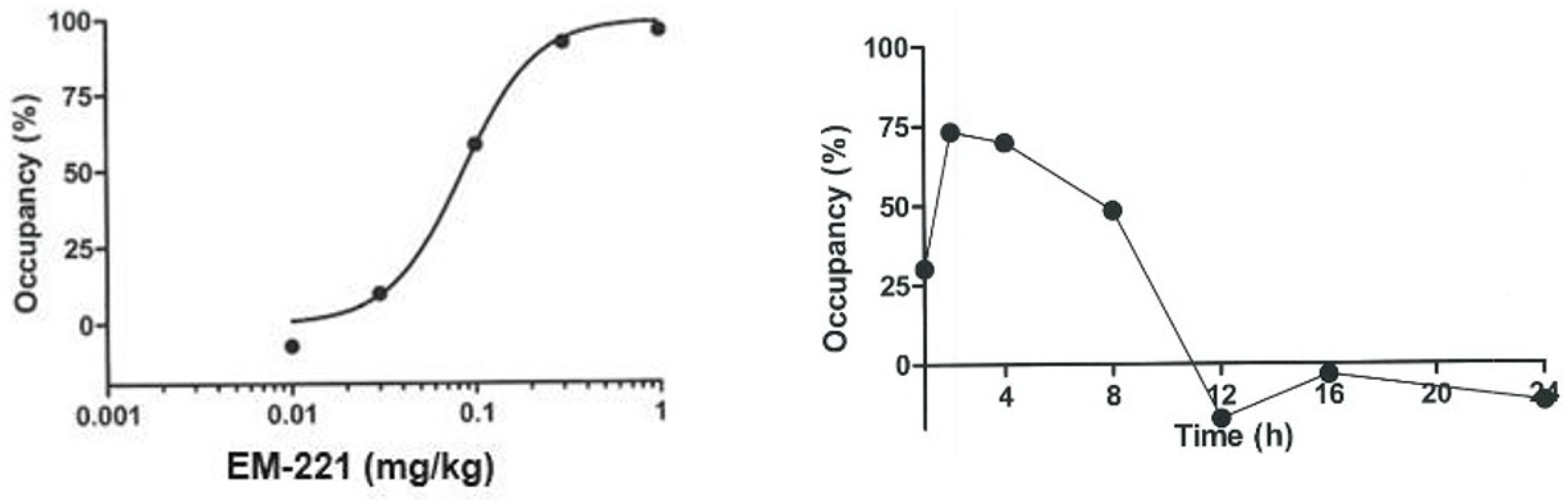
2.5. PDE10A Enzyme Occupancy
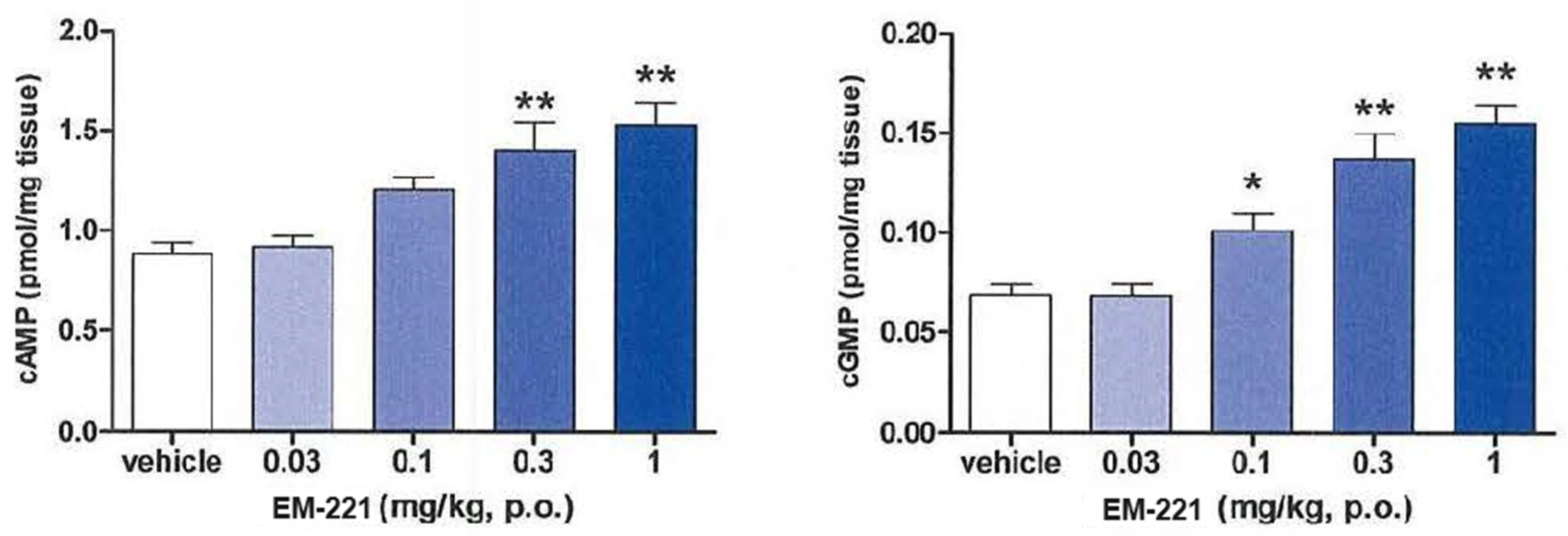
2.6. Effects of cAMP and cGMP in the Striatum
2.7. Effects on Enkephalin and Substance P mRNA Expression in the Striatum
2.8. Inhibition of MK-801-Induced Hyperlocomotion
2.9. Inhibition of Conditioned Avoidance Responding
2.10. Novel Object Recognition
2.11. Reversal of MK-801 Disruption of Prepulse Inhibition of Startle (PPI)
2.12. Clinical Studies
3. Results
3.1. Preclinical Pharmacology
3.1.1. In Vitro Assessments of PDE Potencies and Selectivity
3.1.2. Pharmacokinetics, Brain Penetrability, and Receptor Occupancy in Rodents
3.1.3. Pharmacodynamic Activities in Rats
3.1.4. Preclinical Safety Assessment
3.2. Clinical Results
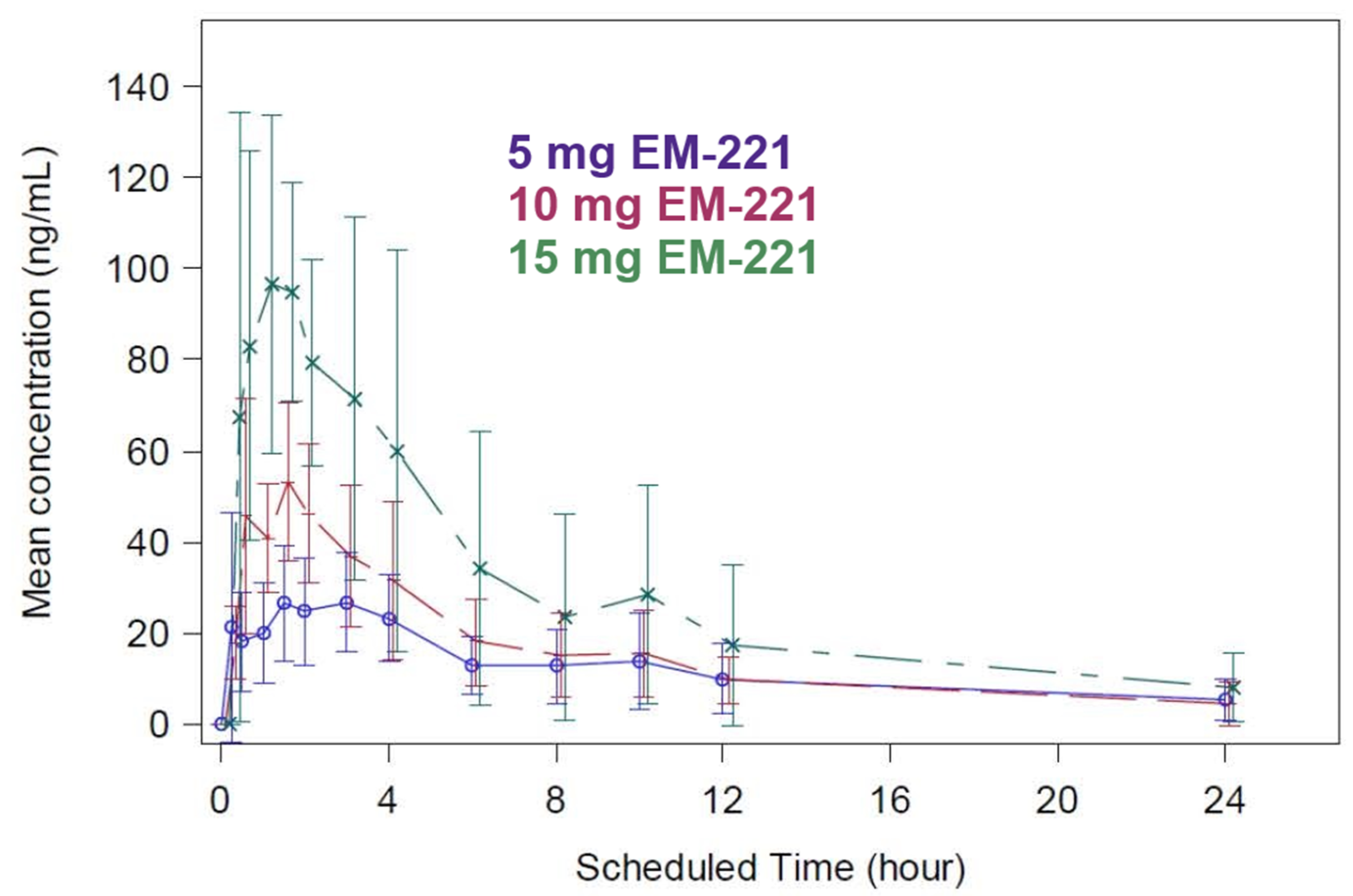
3.2.1. SAD Study Pharmacokinetics
3.2.2. MAD Study Pharmacokinetics
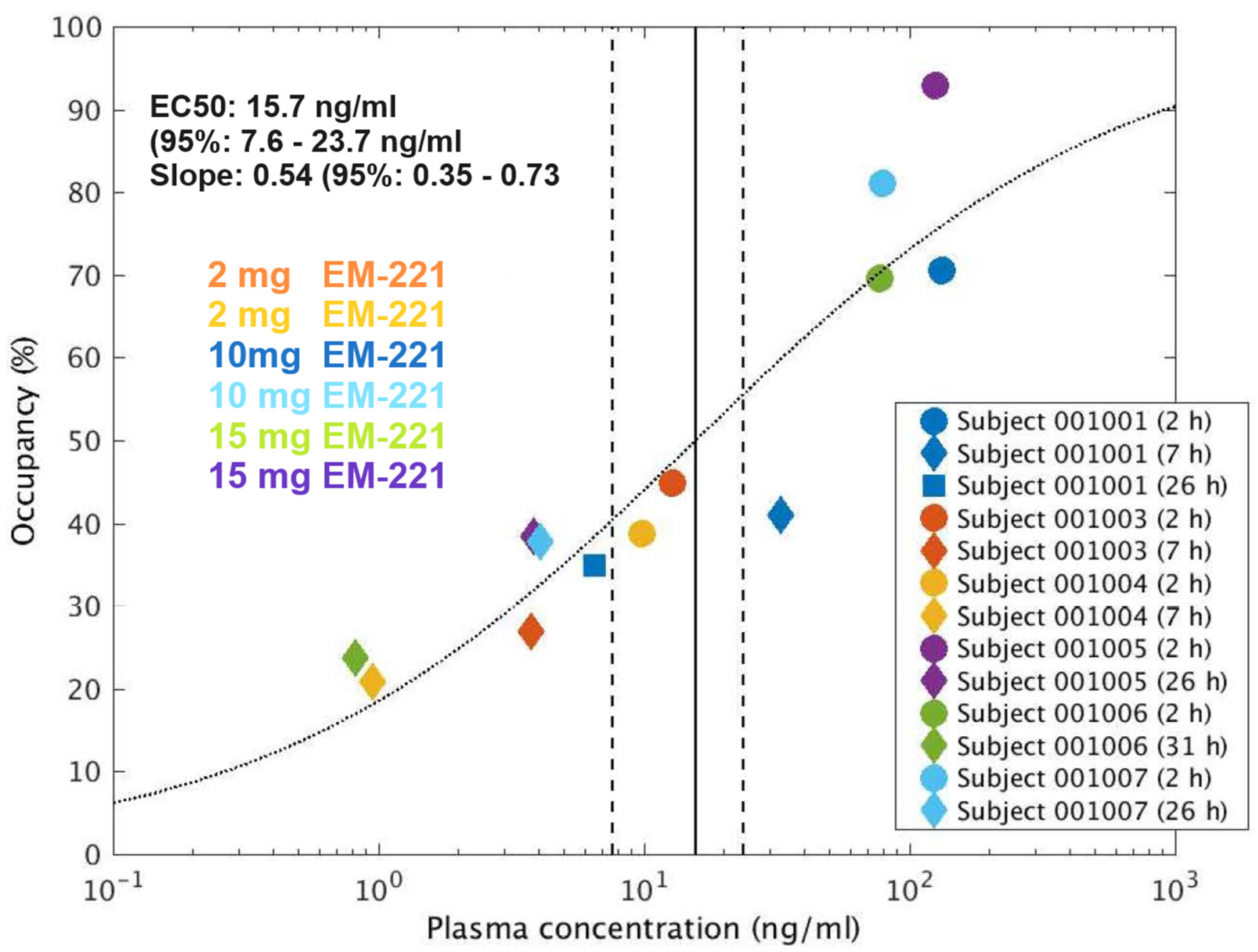
3.2.3. PET Study Results
3.2.4. SAD Study Safety Results (Table 3)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 0.25 mg N = 6 n (%) E | 0.75 mg N = 6 n (%) E | 2 mg N = 6 n (%) E | 5 mg N = 6 n (%) E | 10 mg N = 6 n (%) E | 15 mg N = 6 n (%) E | Placebo N = 12 n (%) E | Total (Subjects on EM-221) | |
|---|---|---|---|---|---|---|---|---|
| SAD study | 2 (33.3) 4 | 5 (83.3) 9 | 3 (50.0) 3 | 5 (83.3) 8 | 5 (83.3) 14 | 6 (100) 27 | 5 (41.7) 5 | 31 (83.3) 70 |
| MAD study | 4 (66.7) 12 | 3 (50%) 9 | 5 (83.3%) 35 | 5 (83.3%) 7 | 12 (94.4%) 56 |
3.2.5. Neurological Adverse Events at the Highest Dose of 15 mg in the SAD
3.2.6. MAD Study Safety Results (Table 3)
3.2.7. PET Study Safety Results
4. Discussion
4.1. Preclinical Pharmacology
4.2. Nonclinical Toxicity
4.3. Clinical
4.4. Tourette Syndrome
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Johnson, K.A.; Worbe, Y.; Foote, K.D.; Butson, C.R.; Gunduz, A.; Okun, M.S. Tourette syndrome: Clinical features, pathophysiology, and treatment. Lancet Neurol. 2023, 22, 147–158. [Google Scholar] [CrossRef]
- Felling, R.J.; Singer, H.S. Neurobiology of Tourette syndrome: Current status and need for further investigation. J. Neurosci. 2011, 31, 12387–12395. [Google Scholar] [CrossRef]
- Groth, C.; Skov, L.; Lange, T.; Debes, N.M. Predictors of the clinical course of Tourette syndrome: A longitudinal study. J. Child. Neurol. 2019, 34, 913–921. [Google Scholar] [CrossRef] [PubMed]
- Tinker, S.C.; Bitsko, R.H.; Danielson, M.L.; Newsome, K.; Kaminski, J.W. Estimating the number of people with Tourette syndrome and persistent tic disorder in the United States. Psychiatry Res. 2022, 314, 114684. [Google Scholar] [CrossRef] [PubMed]
- Knight, T.; Steeves, T.; Day, L.; Lowerison, M.; Jette, N.; Pringsheim, T. Prevalence of tic disorders: A systematic review and meta-analysis. Pediatr. Neurol. 2012, 47, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Bitsko, R.H.; Claussen, A.H.; Lichstein, J.; Black, L.I.; Jones, S.E.; Danielson, M.L.; Hoenig, J.M.; Jack, S.P.D.; Brody, D.J.; Gyawali, S. Mental health surveillance among children—United States, 2013–2019. MMWR Suppl. 2022, 71, 1. [Google Scholar] [CrossRef] [PubMed]
- Hirschtritt, M.E.; Lee, P.C.; Pauls, D.L.; Dion, Y.; Grados, M.A.; Illmann, C.; King, R.A.; Sandor, P.; McMahon, W.M.; Lyon, G.J.; et al. Lifetime Prevalence, Age of Risk, and Genetic Relationships of Comorbid Psychiatric Disorders in Tourette Syndrome. JAMA Psychiatry 2015, 72, 325–333. [Google Scholar] [CrossRef]
- Albin, R.L. Neurobiology of basal ganglia and Tourette syndrome: Striatal and dopamine function. Adv. Neurol. 2006, 99, 99–106. [Google Scholar]
- Ganos, C.; Roessner, V.; Münchau, A. The functional anatomy of Gilles de la Tourette syndrome. Neurosci. Biobehav. Rev. 2013, 37, 1050–1062. [Google Scholar] [CrossRef]
- Graybiel, A.M. The basal ganglia. Curr. Biol. 2000, 10, R509–R511. [Google Scholar] [CrossRef]
- DeLong, M.R.; Wichmann, T. Circuits and Circuit Disorders of the Basal Ganglia. Arch. Neurol. 2007, 64, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Albin, R.L.; Young, A.B.; Penney, J.B. The functional anatomy of basal ganglia disorders. Trends Neurosci. 1989, 12, 366–375. [Google Scholar] [CrossRef] [PubMed]
- Calabresi, P.; Picconi, B.; Tozzi, A.; Ghiglieri, V.; Di Filippo, M. Direct and indirect pathways of basal ganglia: A critical reappraisal. Nat. Neurosci. 2014, 17, 1022. [Google Scholar] [CrossRef] [PubMed]
- Wichmann, T. and M.R. DeLong, Functional and pathophysiological models of the basal ganglia. Curr. Opin. Neurobiol. 1996, 6, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Surmeier, D.J.; Ding, J.; Day, M.; Wang, Z.; Shen, W. D1 and D2 dopamine-receptor modulation of striatal glutamatergic signaling in striatal medium spiny neurons. Trends Neurosci. 2007, 30, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Nao, C.; Kenji, F.T.; René, H.; Stephen, R. Functional Connectome of the Striatal Medium Spiny Neuron. J. Neurosci. 2011, 31, 1183. [Google Scholar]
- Graybiel, A.M. The basal ganglia and chunking of action repertoires. Neurobiol. Learn. Mem. 1998, 70, 119–136. [Google Scholar] [CrossRef] [PubMed]
- Hirokane, K.; Nakamura, T.; Kubota, Y.; Hu, D.; Yagi, T.; Graybiel, A.M.; Kitsukawa, T. Emergence of rhythmic chunking in complex stepping of mice. Iscience 2023, 26, 106765. [Google Scholar] [CrossRef] [PubMed]
- Palminteri, S.; Pessiglione, M. Chapter Five—Reinforcement Learning and Tourette Syndrome. In International Review of Neurobiology; Martino, D., Cavanna, A.E., Eds.; Academic Press: Cambridge, MA, USA, 2013; pp. 131–153. [Google Scholar]
- Sandyk, R.; Allender, J. Brain Reward Systems and Tourette’s Syndrome. Int. J. Neurosci. 1989, 45, 255–257. [Google Scholar] [CrossRef]
- Isaacs, D.; Riordan, H. Sensory hypersensitivity in Tourette syndrome: A review. Brain Dev. 2020, 42, 627–638. [Google Scholar] [CrossRef]
- Kleimaker, A.; Kleimaker, M.; Bäumer, T.; Beste, C.; Münchau, A. Gilles de la Tourette Syndrome—A Disorder of Action-Perception Integration. Front. Neurol. 2020, 11, 597898. [Google Scholar] [CrossRef] [PubMed]
- Huys, D.; Hardenacke, K.; Poppe, P.; Bartsch, C.; Baskin, B.; Kuhn, J. Update on the role of antipsychotics in the treatment of Tourette syndrome. Neuropsychiatr. Dis. Treat. 2012, 8, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.D.; Barr, A.M.; Chung, Y.; Yuen, J.W.; Etminan, M.; Carleton, B.C.; White, R.F.; Honer, W.G.; Procyshyn, R.M. Antipsychotic-associated symptoms of tourette syndrome: A systematic review. CNS Drugs 2018, 32, 917–938. [Google Scholar] [CrossRef] [PubMed]
- Charych, E.I.; Brandon, N.J. Molecular and Cellular Understanding of PDE10A: A Dual-Substrate Phosphodiesterase with Therapeutic Potential to Modulate Basal Ganglia Function. In Cyclic-Nucleotide Phosphodiesterases in the Central Nervous System; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2014; pp. 247–268. [Google Scholar]
- Chappie, T.A.; Humphrey, M.; Menniti, F.S.; Schmidt, C.J. PDE10A Inhibitors: An Assessment of the Current CNS Drug Discovery Landscape. Curr. Opin. Investig. Drugs 2009, 12, 458–467. [Google Scholar]
- Menniti, F.S.; Chappie, T.A.; Schmidt, C.J. PDE10A inhibitors—Clinical failure or window into antipsychotic drug action? Front. Neuroscience 2021, 14, 600178. [Google Scholar] [CrossRef] [PubMed]
- Conti, M. and J. Beavo, Biochemistry and Physiology of Cyclic Nucleotide Phosphodiesterases: Essential Components in Cyclic Nucleotide Signaling. Annu. Rev. Biochem. 2007, 76, 481–511. [Google Scholar] [CrossRef] [PubMed]
- Seeger, T.F.; Bartlett, B.; Coskran, T.M.; Culp, J.S.; James, L.C.; Krull, D.L.; Lanfear, J.; Ryan, A.M.; Schmidt, C.J.; Strick, C.A.; et al. Immunohistochemical localization of PDE10A in the rat brain. Brain Res. 2003, 985, 113–126. [Google Scholar] [CrossRef]
- Coskran, T.M.; Morton, D.; Menniti, F.S.; Adamowicz, W.O.; Kleiman, R.J.; Ryan, A.M.; Strick, C.A.; Schmidt, C.J.; Stephenson, D.T. Immunohistochemical Localization of Phosphodiesterase 10A in Multiple Mammalian Species. J. Histochem. Cytochem. 2006, 54, 1205–1213. [Google Scholar] [CrossRef] [PubMed]
- Lakics, V.; Karran, E.H.; Boess, F.G. Quantitative comparison of phosphodiesterase mRNA distribution in human brain and peripheral tissues. Neuropharmacology 2010, 59, 367–374. [Google Scholar] [CrossRef]
- Schmidt, C.J.; Chapin, D.S.; Cianfrogna, J.; Corman, M.L.; Hajos, M.; Harms, J.F.; Hoffman, W.E.; Lebel, L.A.; McCarthy, S.A.; Nelson, F.R.; et al. Preclinical Characterization of Selective Phosphodiesterase 10A Inhibitors: A New Therapeutic Approach to the Treatment of Schizophrenia. J. Pharmacol. Exp. Ther. 2008, 325, 681–690. [Google Scholar] [CrossRef]
- Threlfell, S.; Sammut, S.; Menniti, F.S.; Schmidt, C.J.; West, A.R. Inhibition of Phosphodiesterase 10A Increases the Responsiveness of Striatal Projection Neurons to Cortical Stimulation. J. Pharmacol. Exp. Ther. 2009, 328, 785–795. [Google Scholar] [CrossRef] [PubMed]
- Polito, M.; Guiot, E.; Gangarossa, G.; Longueville, S.; Doulazmi, M.; Valjent, E.; Herve, D.; Girault, J.A.; Paupardin-Tritsch, D.; Castro, L.R.; et al. Selective Effects of PDE10A Inhibitors on Striatopallidal Neurons Require Phosphatase Inhibition by DARPP-32. eNeuro 2015, 2. [Google Scholar] [CrossRef] [PubMed]
- Strick, C.A.; James, L.C.; Fox, C.B.; Seeger, T.F.; Menniti, F.S.; Schmidt, C.J. Alterations in gene regulation following inhibition of the striatum-enriched phosphodiesterase, PDE10A. Neuropharmacology 2010, 58, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.M.; Uslaner, J.M.; Cox, C.D.; Huszar, S.L.; Cannon, C.E.; Vardigan, J.D.; Eddins, D.; Toolan, D.M.; Kandebo, M.; Yao, L. The novel phosphodiesterase 10A inhibitor THPP-1 has antipsychotic-like effects in rat and improves cognition in rat and rhesus monkey. Neuropharmacology 2013, 64, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Megens, A.A.; Hendrickx, H.M.; Mahieu, M.M.; Wellens, A.L.; de Boer, P.; Vanhoof, G. PDE10A inhibitors stimulate or suppress motor behavior dependent on the relative activation state of the direct and indirect striatal output pathways. Pharmacol. Res. Perspect. 2014, 2, e00057. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Harada, A.; Suzuki, H.; Miyamoto, M.; Kimura, H. TAK-063, a PDE10A inhibitor with balanced activation of direct and indirect pathways, provides potent antipsychotic-like effects in multiple paradigms. Neuropsychopharmacology 2016, 41, 2252–2262. [Google Scholar] [CrossRef] [PubMed]
- Nishi, A.; Kuroiwa, M.; Miller, D.B.; O’Callaghan, J.P.; Bateup, H.S.; Shuto, T.; Sotogaku, N.; Fukuda, T.; Heintz, N.; Greengard, P.; et al. Distinct Roles of PDE4 and PDE10A in the Regulation of cAMP/PKA Signaling in the Striatum. J. Neurosci. 2008, 28, 10460–10471. [Google Scholar] [CrossRef] [PubMed]
- Uthayathas, S.; Masilamoni, G.J.; Shaffer, C.L.; Schmidt, C.J.; Menniti, F.S.; Papa, S.M. Phosphodiesterase 10A inhibitor MP-10 effects in primates: Comparison with risperidone and mechanistic implications. Neuropharmacology 2014, 77, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Walling, D.P.; Banerjee, A.; Dawra, V.; Boyer, S.; Schmidt, C.J.; DeMartinis, N. Phosphodiesterase 10A Inhibitor Monotherapy Is Not an Effective Treatment of Acute Schizophrenia. J. Clin. Psychopharmacol. 2019, 39, 575–582. [Google Scholar] [CrossRef]
- DeMartinis, N., 3rd; Lopez, R.N.; Pickering, E.H.; Schmidt, C.J.; Gertsik, L.; Walling, D.P.; Ogden, A. A Proof-of-Concept Study Evaluating the Phosphodiesterase 10A Inhibitor PF-02545920 in the Adjunctive Treatment of Suboptimally Controlled Symptoms of Schizophrenia. J. Clin. Psychopharmacol. 2019, 39, 318–328. [Google Scholar] [CrossRef]
- Macek, T.A.; McCue, M.; Dong, X.; Hanson, E.; Goldsmith, P.; Affinito, J.; Mahableshwarkar, A.R. A phase 2, randomized, placebo-controlled study of the efficacy and safety of TAK-063 in subjects with an acute exacerbation of schizophrenia. Schizophr. Res. 2019, 204, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Layton, M.E.; Kern, J.C.; Hartingh, T.J.; Shipe, W.D.; Raheem, I.; Kandebo, M.; Hayes, R.P.; Huszar, S.; Eddins, D.; Ma, B. Discovery of MK-8189, a Highly Potent and Selective PDE10A Inhibitor for the Treatment of Schizophrenia. J. Med. Chem. 2023, 66, 1157–1171. [Google Scholar] [CrossRef] [PubMed]
- Delnomdedieu, M.; Tan, Y.; Ogden, A.; Berger, Z.; Reilmann, R. A randomized, double-blind, placebo-controlled phase ii efficacy and safety study of the PDE10A inhibitor PF-02545920 in Huntington Disease (AMARYLLIS). J. Neurol. Neurosurg. Psychiatry 2018, 89 (Suppl. S1), A99–A100. [Google Scholar]
- Arakawa, K.; Maehara, S.; Yuge, N.; Ishikawa, M.; Miyazaki, Y.; Naba, H.; Kato, Y.; Nakao, K. Pharmacological characterization of a novel potent, selective, and orally active phosphodiesterase 10A inhibitor, PDM-042 [(E)-4-(2-(2-(5,8-dimethyl-[1,2,4] triazolo [1,5-a] pyrazin-2-yl) vinyl)-6-(pyrrolidin-1-yl) pyrimidin-4-yl) morpholine] in rats: Potential for the treatment of schizophrenia. Pharmacol. Res. Perspect. 2016, 4, e00241. [Google Scholar] [PubMed]
- Wadenberg, M.-L.G.; Hicks, P.B. The conditioned avoidance response test re-evaluated: Is it a sensitive test for the detection of potentially atypical antipsychotics? Neurosci. Biobehav. Rev. 1999, 23, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Bond, A.; Lader, M. The use of analogue scales in rating subjective feelings. Br. J. Med. Psychol. 1974, 47, 211–218. [Google Scholar] [CrossRef]
- Chouinard, G.; Margolese, H.C. Manual for the extrapyramidal symptom rating scale (ESRS). Schizophr. Res. 2005, 76, 247–265. [Google Scholar] [CrossRef]
- Hoddes, E.; Zarcone, V.; Smythe, H.; Phillips, R.; Dement, W.C. Quantification of sleepiness: A new approach. Psychophysiology 1973, 10, 431–436. [Google Scholar] [CrossRef]
- Posner, K.; Brown, G.K.; Stanley, B.; Brent, D.A.; Yershova, K.V.; Oquendo, M.A.; Currier, G.W.; Melvin, G.A.; Greenhill, L.; Shen, S. The Columbia–Suicide Severity Rating Scale: Initial validity and internal consistency findings from three multisite studies with adolescents and adults. Am. J. Psychiatry 2011, 168, 1266–1277. [Google Scholar] [CrossRef]
- Plisson, C.; Weinzimmer, D.; Jakobsen, S.; Natesan, S.; Salinas, C.; Lin, S.-F.; Labaree, D.; Zheng, M.-Q.; Nabulsi, N.; Marques, T.R. Phosphodiesterase 10A PET radioligand development program: From pig to human. J. Nucl. Med. 2014, 55, 595–601. [Google Scholar] [CrossRef]
- Yokoi, F.; Gründer, G.; Biziere, K.; Stephane, M.; Dogan, A.S.; Dannals, R.F.; Ravert, H.; Suri, A.; Bramer, S.; Wong, D.F. Dopamine D2 and D3 receptor occupancy in normal humans treated with the antipsychotic drug aripiprazole (OPC 14597): A study using positron emission tomography and [11C] raclopride. Neuropsychopharmacology 2002, 27, 248–259. [Google Scholar] [CrossRef] [PubMed]
- Kapur, S.; Zipursky, R.; Roy, P.; Jones, C.; Remington, G.; Reed, K.; Houle, S. The relationship between D2 receptor occupancy and plasma levels on low dose oral haloperidol: A PET study. Psychopharmacology 1997, 131, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.K.; Joung, Y.S.; Lee, J.-S.; Song, D.H.; Lee, Y.S.; Kim, J.-W.; Kim, B.-N.; Cho, S.C. A multicenter, randomized, double-blind, placebo-controlled study of aripiprazole in children and adolescents with Tourette’s disorder. J. Clin. Psychiatry 2013, 74, 12352. [Google Scholar] [CrossRef] [PubMed]
- Nawrocki, A.R.; Rodriguez, C.G.; Toolan, D.M.; Price, O.; Henry, M.; Forrest, G.; Szeto, D.; Keohane, C.A.; Pan, Y.; Smith, K.M.; et al. Genetic deletion and pharmacological inhibition of phosphodiesterase 10A protects mice from diet-induced obesity and insulin resistance. Diabetes 2014, 63, 300–311. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Lindenberg, A.; Nielsen, J.; Such, P.; Lemming, O.M.; Zambori, J.; Buller, R.; der Goltz, C.V. A double-blind, randomized, placebo-controlled proof of concept study of the efficacy and safety of Lu AF11167 for persistent negative symptoms in people with schizophrenia. Eur. Neuropsychopharmacol. 2022, 61, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Delnomdedieu, M.; Forsberg, A.; Ogden, A.; Fazio, P.; Yu, C.R.; Stenkrona, P.; Duvvuri, S.; David, W.; Al-Tawil, N.; Vitolo, O.V.; et al. In vivo measurement of PDE10A enzyme occupancy by positron emission tomography (PET) following single oral dose administration of PF-02545920 in healthy male subjects. Neuropharmacology 2017, 117, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Macek, T.A.; Suzuki, K.; Asin, K.; Kimura, H. Translational Development Strategies for TAK-063, a Phosphodiesterase 10A Inhibitor. Int. J. Neuropsychopharmacol. 2020, 23, 524–532. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.; Yang, B.; Anair, J.D.; Martin, M.M.; Fleps, S.W.; Pamukcu, A.; Yeh, N.H.; Contractor, A.; Kennedy, A.; Parker, J.G. Antipsychotic drug efficacy correlates with the modulation of D1 rather than D2 receptor-expressing striatal projection neurons. Nat. Neurosci. 2023, 26, 1417–1428. [Google Scholar] [CrossRef] [PubMed]
- Aringhieri, S.; Carli, M.; Kolachalam, S.; Verdesca, V.; Cini, E.; Rossi, M.; McCormick, P.J.; Corsini, G.U.; Maggio, R.; Scarselli, M. Molecular targets of atypical antipsychotics: From mechanism of action to clinical differences. Pharmacol. Ther. 2018, 192, 20–41. [Google Scholar] [CrossRef]
- Klein, M.O.; Battagello, D.S.; Cardoso, A.R.; Hauser, D.N.; Bittencourt, J.C.; Correa, R.G. Dopamine: Functions, signaling, and association with neurological diseases. Cell. Mol. Neurobiol. 2019, 39, 31–59. [Google Scholar] [CrossRef]
- Giampà, C.; Laurenti, D.; Anizilotti, S.; Bernardi, G.; Menniti, F.S.; Fusco, F.R. Inhibition of the striatal specific phosphodiesterase PDE10A ameliorates striatal and cortical pathology in the R6/2 mouse model of Huntington’s disease. PLoS ONE, 2010; in press. [Google Scholar]
- Kleiman, R.J.; Kimmel, L.H.; Bove, S.E.; Lanz, T.A.; Harms, J.F.; Romegialli, A.; Miller, K.S.; Willis, A.; Etages, S.D.; Kuhn, M.; et al. Chronic suppression of phosphodiesterase 10A alters striatal expression of genes responsible for neurotransmitter synthesis, neurotransmission, and signaling pathways implicated in Huntington’s disease. J. Pharmacol. Exp. Ther. 2011, 336, 64–76. [Google Scholar] [CrossRef] [PubMed]
- van der Plas, E.; Schubert, R.; Reilmann, R.; Nopoulos, P.C. A feasibility study of quantitative motor assessments in children using the Q-motor suite. J. Huntington’s Dis. 2019, 8, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Russell, D.S.; Barret, O.; Jennings, D.L.; Friedman, J.H.; Tamagnan, G.D.; Thomae, D.; Alagille, D.; Morley, T.J.; Papin, C.; Papapetropoulos, S. The phosphodiesterase 10 positron emission tomography tracer, [18F] MNI-659, as a novel biomarker for early Huntington disease. JAMA Neurol. 2014, 71, 1520–1528. [Google Scholar] [CrossRef] [PubMed]
- Russell, D.S.; Jennings, D.L.; Barret, O.; Tamagnan, G.D.; Carroll, V.M.; Caillé, F.; Alagille, D.; Morley, T.J.; Papin, C.; Seibyl, J.P. Change in PDE10 across early Huntington disease assessed by [18F] MNI-659 and PET imaging. Neurology 2016, 86, 748–754. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, M.; D’Angelo, V.; Esposito, Z.; Nuccetelli, V.; Sorge, R.; Martorana, A.; Stefani, A.; Bernardi, G.; Sancesario, G. Lowered cAMP and cGMP signalling in the brain during levodopa-induced dyskinesias in hemiparkinsonian rats: New aspects in the pathogenetic mechanisms. Eur. J. Neurosci. 2008, 28, 941–950. [Google Scholar] [CrossRef] [PubMed]
- Sancesario, G.; Morrone, L.A.; D’Angelo, V.; Castelli, V.; Ferrazzoli, D.; Sica, F.; Martorana, A.; Sorge, R.; Cavaliere, F.; Bernardi, G. Levodopa-induced dyskinesias are associated with transient down- regulation of cAMP and cGMP in the caudate-putamen of hemiparkinsonian rats: Reduced synthesis or increased catabolism? Neurochem. Int. 2014, 79, 44–56. [Google Scholar] [CrossRef]
- Beck, G.; Maehara, S.; Chang, P.L.; Papa, S.M. A selective phosphodiesterase 10A inhibitor reduces L-Dopa-induced dyskinesias in Parkinsonian monkeys. Mov. Disord. 2018, 33, 805–814. [Google Scholar] [CrossRef] [PubMed]
- Potts, L.F.; Uthayathas, S.; Greven, A.C.; Dyavarshetty, B.; Mouradian, M.M.; Papa, S.M. A new quantitative rating scale for dyskinesia in nonhuman primates. Behav. Pharmacol. 2015, 26, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Uthayathas, S.; Shaffer, C.L.; Menniti, F.S.; Schmidt, C.J.; Papa, S.M. Assessment of adverse effects of neurotropic drugs in monkeys with the “drug effects on the nervous system” (DENS) scale. J. Neurosci. Methods 2013, 215, 97–102. [Google Scholar] [CrossRef]
- Chang, S.-E.; Angstadt, M.; Chow, H.M.; Etchell, A.C.; Garnett, E.O.; Choo, A.L.; Kessler, D.; Welsh, R.C.; Sripada, C. Anomalous network architecture of the resting brain in children who stutter. J. Fluen. Disord. 2018, 55, 46–67. [Google Scholar] [CrossRef]
- Qiao, J.; Wang, Z.; Zhao, G.; Huo, Y.; Herder, C.L.; Sikora, C.O.; Peterson, B.S. Functional neural circuits that underlie developmental stuttering. PLoS ONE 2017, 12, e0179255. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.-E.; Guenther, F.H. Involvement of the cortico-basal ganglia-thalamocortical loop in developmental stuttering. Front. Psychol. 2020, 10, 489833. [Google Scholar] [CrossRef] [PubMed]
- Maguire, G.A.; Riley, G.D.; Franklin, D.L.; Gottschalk, L.A. Risperidone for the treatment of stuttering. J. Clin. Psychopharmacol. 2000, 20, 479–482. [Google Scholar] [CrossRef] [PubMed]
- Shaygannejad, V.; Khatoonabadi, S.A.; Shafiei, B.; Ghasemi, M.; Fatehi, F.; Meamar, R.; Dehghani, L. Olanzapine versus haloperidol: Which can control stuttering better? Int. J. Prev. Med. 2013, 4 (Suppl. S2), S270. [Google Scholar] [PubMed]
- Iverach, L.; O’Brian, S.; Jones, M.; Block, S.; Lincoln, M.; Harrison, E.; Hewat, S.; Menzies, R.G.; Packman, A.; Onslow, M. Prevalence of anxiety disorders among adults seeking speech therapy for stuttering. J. Anxiety Disord. 2009, 23, 928–934. [Google Scholar] [CrossRef]
- Maguire, G.A.; Nguyen, D.L.; Simonson, K.C.; Kurz, T.L. The pharmacologic treatment of stuttering and its neuropharmacologic basis. Front. Neurosci. 2020, 14, 158. [Google Scholar] [CrossRef]


| Dose (mg/kg, p.o.) | Plasma (ng/mL) | Striatum (ng/g Tissue) | Striatum/Plasma |
|---|---|---|---|
| 0.025 | 2.3 ± 0.3 | 124 ± 16 | 55 |
| 0.05 | 5.5 ± 1.2 | 237 ± 403 | 43 |
| 0.1 | 11 ± 2 | 348 ± 38 | 33 |
| 0.2 | 19 ± 1 | 416 ± 43 | 22 |
| Subject | Dose (mg) | Scan Time (Hours Post-Dose) | Plasma Conc. (ng/mL) | ΔBPND (%) | ||||
|---|---|---|---|---|---|---|---|---|
| DCa | DPu | Acc | Mean | Reduction from 2 h to 26 or 31 h | ||||
| 1001 | 10 | 2 | 132 | 84.8 | 64.0 | 62.8 | 70.5 | |
| 7 | 32.6 | 58.5 | 41.9 | 22.4 | 40.9 | |||
| 26 | 6.51 | 45.6 | 26.9 | 32.3 | 34.9 | 50.5% | ||
| 1003 | 2 | 2 | 12.8 | 49.0 | 42.0 | 43.2 | 44.7 | |
| 7 | 3.74 | 30.1 | 24.9 | 25.8 | 27.0 | |||
| 1004 | 2 | 2 | 9.88 | 37.2 | 30.8 | 48.4 | 38.8 | |
| 7 | 0.951 | 21.1 | 12.8 | 28.8 | 20.9 | |||
| 1005 | 15 | 2 | 125 | 106.8 | 80.1 | 91.6 | 92.8 | |
| 26 | 3.86 | 36.4 | 32.0 | 46.9 | 38.5 | 58.5% | ||
| 1006 | 15 | 2 | 77.5 | 75.3 | 66.2 | 67.4 | 69.6 | |
| 31 | 0.819 | 31.2 | 21.9 | 18.2 | 23.8 | 65.8% | ||
| 1007 | 10 | 2 | 78.9 | 88.0 | 76.5 | 78.5 | 81.0 | |
| 26 | 4.08 | 42.3 | 29.2 | 41.9 | 37.8 | 53.3% | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marshall, R.D.; Menniti, F.S.; Tepper, M.A. A Novel PDE10A Inhibitor for Tourette Syndrome and Other Movement Disorders. Cells 2024, 13, 1230. https://doi.org/10.3390/cells13141230
Marshall RD, Menniti FS, Tepper MA. A Novel PDE10A Inhibitor for Tourette Syndrome and Other Movement Disorders. Cells. 2024; 13(14):1230. https://doi.org/10.3390/cells13141230
Chicago/Turabian StyleMarshall, Randall D., Frank S. Menniti, and Mark A. Tepper. 2024. "A Novel PDE10A Inhibitor for Tourette Syndrome and Other Movement Disorders" Cells 13, no. 14: 1230. https://doi.org/10.3390/cells13141230
APA StyleMarshall, R. D., Menniti, F. S., & Tepper, M. A. (2024). A Novel PDE10A Inhibitor for Tourette Syndrome and Other Movement Disorders. Cells, 13(14), 1230. https://doi.org/10.3390/cells13141230