
An Epilepsy-Associated CILK1 Variant Compromises KATNIP Regulation and Impairs Primary Cilia and Hedgehog Signaling

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents and Antibodies
2.2. MEF Cell Isolation and Culture
2.3. Immunoblotting
2.4. Immunofluorescence
2.5. Cilia Length Measurement
2.6. RNA-Seq Analysis and Enrichment Analysis
2.7. Statistical Analysis
3. Results
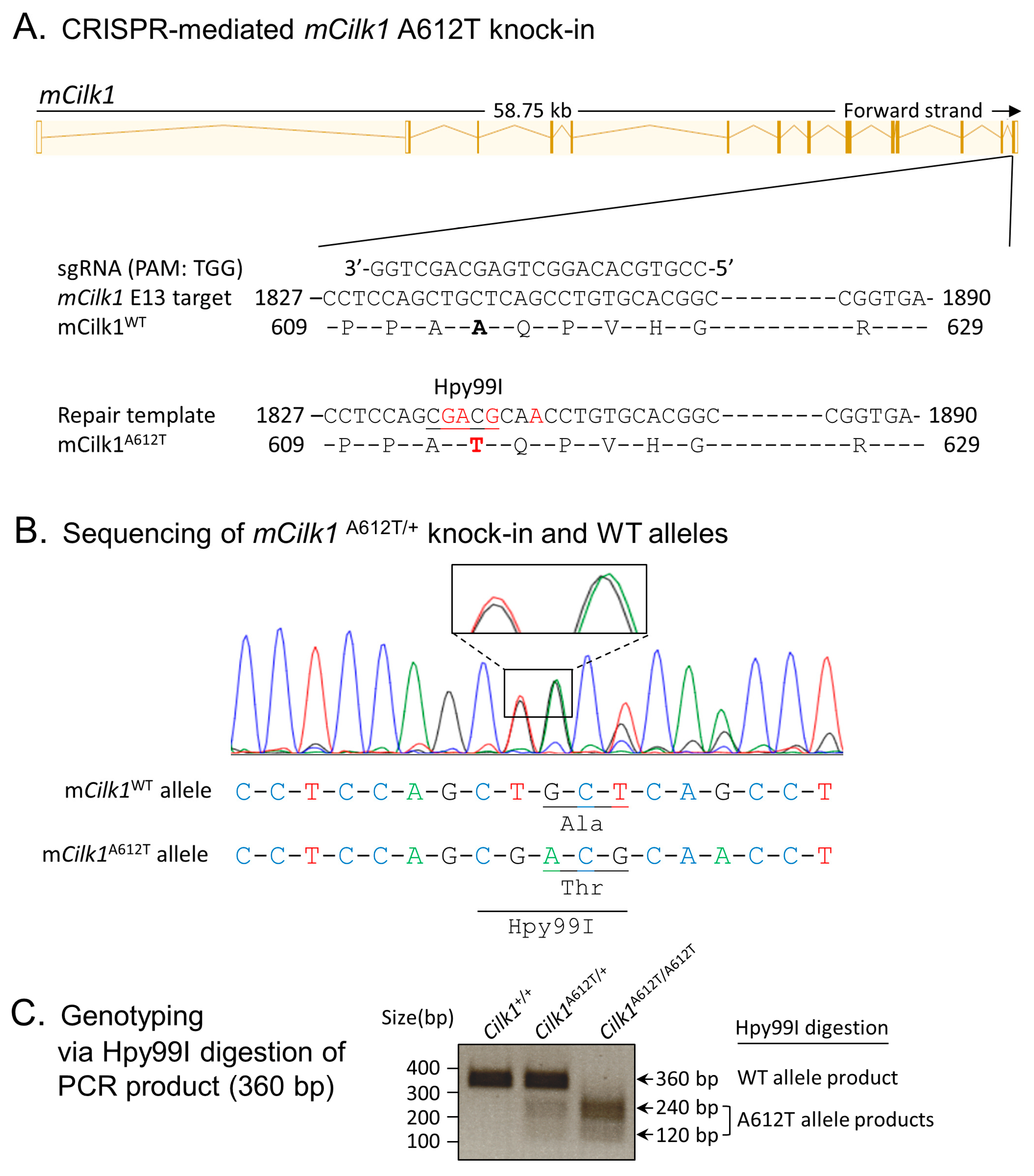
3.1. Cells Expressing the Mouse Equivalent of Human Variant CILK1 A615T
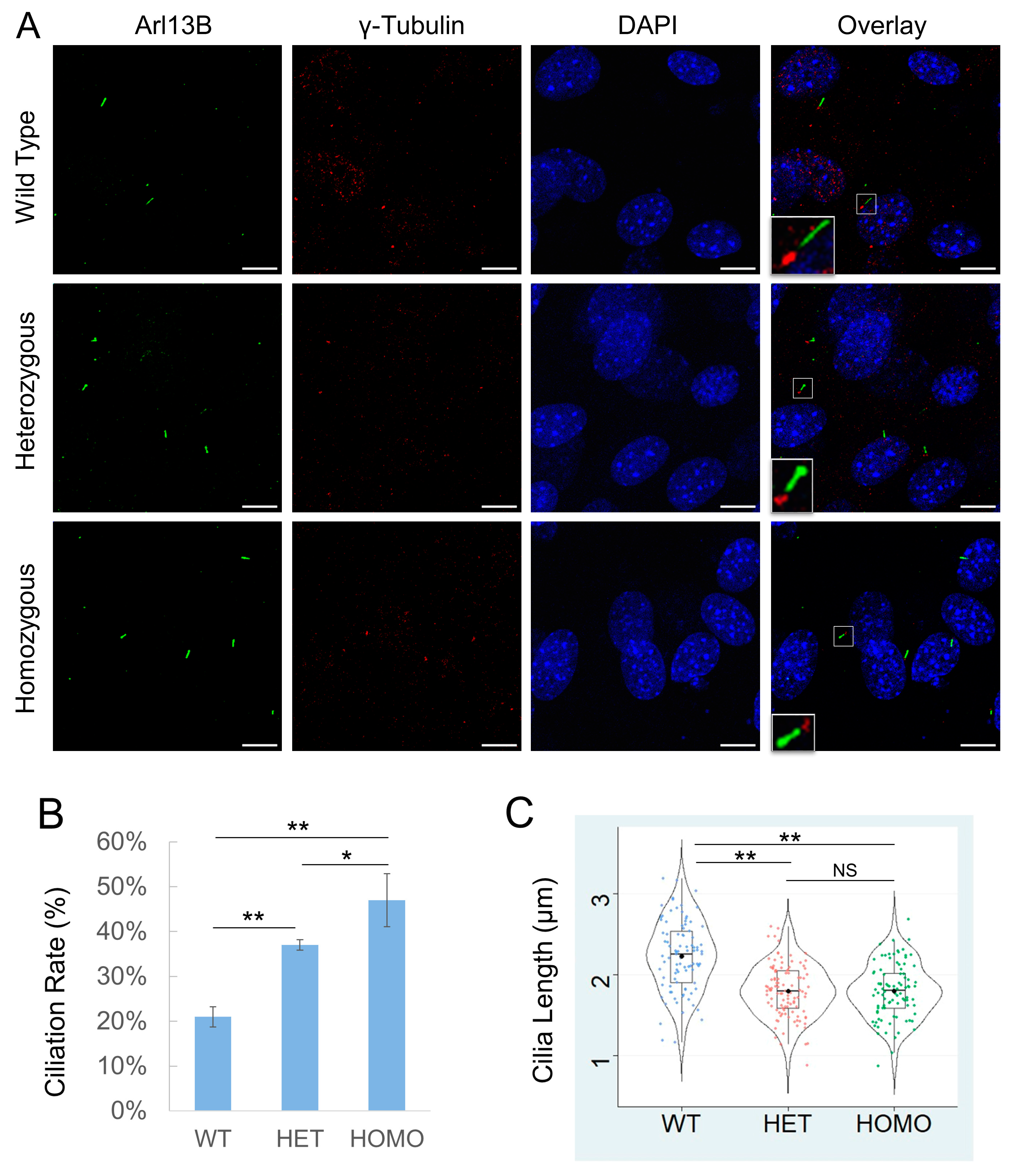
3.2. Cilk1 A612T Mutant Allele Induces Higher Ciliation Frequency but Shorter Cilia
3.3. Cilk1 A612T Mutant Allele Upregulates Hedgehog Signaling
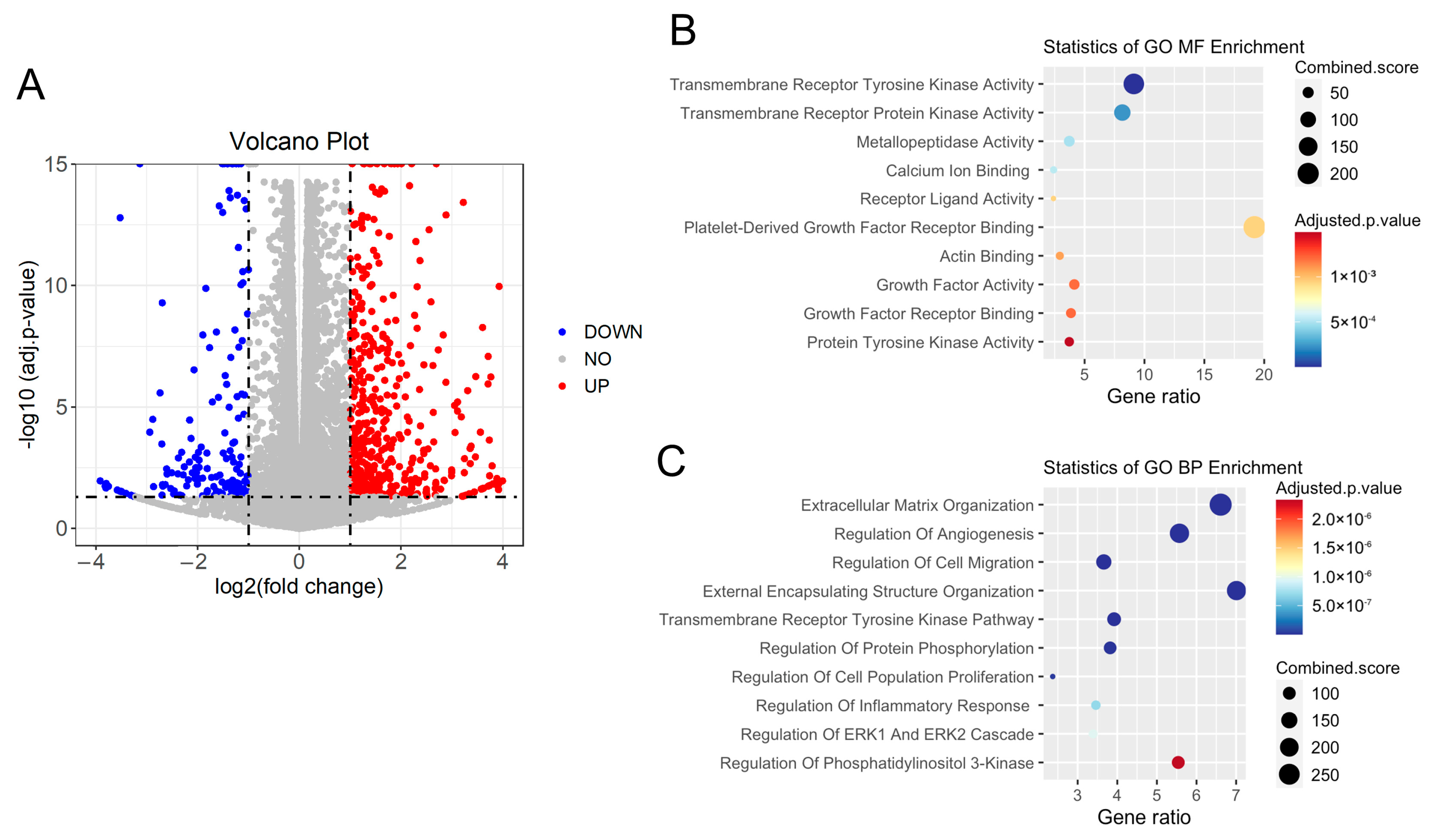
3.4. Cilk1 A612T Variant Produced Alterations in Cilia-Related Receptor Tyrosine Kinase (RTK) Pathways
3.5. The A615T Variant Compromises KATNIP Regulation of CILK1
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Singla, V.; Reiter, J.F. The primary cilium as the cell’s antenna: Signaling at a sensory organelle. Science 2006, 313, 629–633. [Google Scholar] [CrossRef]
- Mill, P.; Christensen, S.T.; Pedersen, L.B. Primary cilia as dynamic and diverse signalling hubs in development and disease. Nat. Rev. Genet. 2023, 24, 421–441. [Google Scholar] [CrossRef]
- Reiter, J.F.; Leroux, M.R. Genes and molecular pathways underpinning ciliopathies. Nat. Rev. Mol. Cell Biol. 2017, 18, 533–547. [Google Scholar] [CrossRef]
- Waters, A.M.; Beales, P.L. Ciliopathies: An expanding disease spectrum. Pediatr. Nephrol. 2011, 26, 1039–1056. [Google Scholar] [CrossRef]
- Baker, K.; Beales, P.L. Making sense of cilia in disease: The human ciliopathies. Am. J. Med. Genet. Part C Semin. Med. Genet. 2009, 151C, 281–295. [Google Scholar] [CrossRef]
- Kirschen, G.W.; Xiong, Q. Primary cilia as a novel horizon between neuron and environment. Neural Regen. Res. 2017, 12, 1225–1230. [Google Scholar]
- Fu, Z.; Gailey, C.D.; Wang, E.J.; Brautigan, D.L. Ciliogenesis associated kinase 1: Targets and functions in various organ systems. FEBS Lett. 2019, 593, 2990–3002. [Google Scholar] [CrossRef]
- Turner, J.S.; McCabe, E.A.; Kuang, K.W.; Gailey, C.D.; Brautigan, D.L.; Limerick, A.; Wang, E.X.; Fu, Z. The Scaffold Protein KATNIP Enhances CILK1 Control of Primary Cilia. Mol. Cell. Biol. 2023, 43, 472–480. [Google Scholar] [CrossRef]
- Tong, Y.; Park, S.H.; Wu, D.; Xu, W.; Guillot, S.J.; Jin, L.; Li, X.; Wang, Y.; Lin, C.S.; Fu, Z. An essential role of intestinal cell kinase in lung development is linked to the perinatal lethality of human ECO syndrome. FEBS Lett. 2017, 591, 1247–1257. [Google Scholar] [CrossRef]
- Chaya, T.; Omori, Y.; Kuwahara, R.; Furukawa, T. ICK is essential for cell type-specific ciliogenesis and the regulation of ciliary transport. EMBO J. 2014, 33, 1227–1242. [Google Scholar] [CrossRef]
- Burghoorn, J.; Dekkers, M.P.; Rademakers, S.; de Jong, T.; Willemsen, R.; Jansen, G. Mutation of the MAP kinase DYF-5 affects docking and undocking of kinesin-2 motors and reduces their speed in the cilia of Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2007, 104, 7157–7162. [Google Scholar] [CrossRef]
- Broekhuis, J.R.; Verhey, K.J.; Jansen, G. Regulation of cilium length and intraflagellar transport by the RCK-kinases ICK and MOK in renal epithelial cells. PLoS ONE 2014, 9, e108470. [Google Scholar] [CrossRef]
- Nakamura, K.; Noguchi, T.; Takahara, M.; Omori, Y.; Furukawa, T.; Katoh, Y.; Nakayama, K. Anterograde trafficking of ciliary MAP kinase-like ICK/CILK1 by the intraflagellar transport machinery is required for intraciliary retrograde protein trafficking. J. Biol. Chem. 2020, 295, 13363–13376. [Google Scholar] [CrossRef]
- Satoda, Y.; Noguchi, T.; Fujii, T.; Taniguchi, A.; Katoh, Y.; Nakayama, K. BROMI/TBC1D32 together with CCRK/CDK20 and FAM149B1/JBTS36 contributes to intraflagellar transport turnaround involving ICK/CILK1. Mol. Biol. Cell 2022, 33, ar79. [Google Scholar] [CrossRef]
- Rah, G.; Cha, H.; Kim, J.; Song, J.; Kim, H.; Oh, Y.K.; Ahn, C.; Kang, M.; Kim, J.; Yoo, K.H.; et al. KLC3 Regulates Ciliary Trafficking and Cyst Progression in CILK1 Deficiency-Related Polycystic Kidney Disease. J. Am. Soc. Nephrol. 2022, 33, 1726–1741. [Google Scholar] [CrossRef]
- Oh, Y.S.; Wang, E.J.; Gailey, C.D.; Brautigan, D.L.; Allen, B.L.; Fu, Z. Ciliopathy-Associated Protein Kinase ICK Requires Its Non-Catalytic Carboxyl-Terminal Domain for Regulation of Ciliogenesis. Cells 2019, 8, 677. [Google Scholar] [CrossRef]
- Gailey, C.D.; Wang, E.J.; Jin, L.; Ahmadi, S.; Brautigan, D.L.; Li, X.; Xu, W.; Scott, M.M.; Fu, Z. Phosphosite T674A mutation in kinesin family member 3A fails to reproduce tissue and ciliary defects characteristic of CILK1 loss of function. Dev. Dyn. 2021, 250, 263–273. [Google Scholar] [CrossRef]
- Bailey, J.N.; de Nijs, L.; Bai, D.; Suzuki, T.; Miyamoto, H.; Tanaka, M.; Patterson, C.; Lin, Y.C.; Medina, M.T.; Alonso, M.E.; et al. Variant Intestinal-Cell Kinase in Juvenile Myoclonic Epilepsy. N. Engl. J. Med. 2018, 378, 1018–1028. [Google Scholar] [CrossRef]
- Wang, E.J.; Gailey, C.D.; Brautigan, D.L.; Fu, Z. Functional Alterations in Ciliogenesis-Associated Kinase 1 (CILK1) that Result from Mutations Linked to Juvenile Myoclonic Epilepsy. Cells 2020, 9, 694. [Google Scholar] [CrossRef]
- Durkin, M.E.; Qian, X.; Popescu, N.C.; Lowy, D.R. Isolation of Mouse Embryo Fibroblasts. Bio Protoc. 2013, 3, e908. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef]
- Moon, H.; Song, J.; Shin, J.O.; Lee, H.; Kim, H.K.; Eggenschwiller, J.T.; Bok, J.; Ko, H.W. Intestinal cell kinase, a protein associated with endocrine-cerebro-osteodysplasia syndrome, is a key regulator of cilia length and Hedgehog signaling. Proc. Natl. Acad. Sci. USA 2014, 111, 8541–8546. [Google Scholar] [CrossRef]
- Collins, I.; Wann, A.K.T. Regulation of the Extracellular Matrix by Ciliary Machinery. Cells 2020, 9, 278. [Google Scholar] [CrossRef]
- Pala, R.; Jamal, M.; Alshammari, Q.; Nauli, S.M. The Roles of Primary Cilia in Cardiovascular Diseases. Cells 2018, 7, 233. [Google Scholar] [CrossRef]
- Stoufflet, J.; Caille, I. The Primary Cilium and Neuronal Migration. Cells 2022, 11, 3384. [Google Scholar] [CrossRef]
- Nakazato, R.; Matsuda, Y.; Ijaz, F.; Ikegami, K. Circadian oscillation in primary cilium length by clock genes regulates fibroblast cell migration. EMBO Rep. 2023, 24, e56870. [Google Scholar] [CrossRef]
- Pruski, M.; Lang, B. Primary Cilia-An Underexplored Topic in Major Mental Illness. Front. Psychiatry 2019, 10, 104. [Google Scholar] [CrossRef]
- Liu, J.P.; Zeitlin, S.O. The long and the short of aberrant ciliogenesis in Huntington disease. J. Clin. Investig. 2011, 121, 4237–4241. [Google Scholar] [CrossRef]
- Munoz-Estrada, J.; Lora-Castellanos, A.; Meza, I.; Alarcon Elizalde, S.; Benitez-King, G. Primary cilia formation is diminished in schizophrenia and bipolar disorder: A possible marker for these psychiatric diseases. Schizophr. Res. 2018, 195, 412–420. [Google Scholar] [CrossRef]
- Parker, A.K.; Le, M.M.; Smith, T.S.; Hoang-Minh, L.B.; Atkinson, E.W.; Ugartemendia, G.; Semple-Rowland, S.; Coleman, J.E.; Sarkisian, M.R. Neonatal seizures induced by pentylenetetrazol or kainic acid disrupt primary cilia growth on developing mouse cortical neurons. Exp. Neurol. 2016, 282, 119–127. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Limerick, A.; McCabe, E.A.; Turner, J.S.; Kuang, K.W.; Brautigan, D.L.; Hao, Y.; Chu, C.Y.; Fu, S.H.; Ahmadi, S.; Xu, W.; et al. An Epilepsy-Associated CILK1 Variant Compromises KATNIP Regulation and Impairs Primary Cilia and Hedgehog Signaling. Cells 2024, 13, 1258. https://doi.org/10.3390/cells13151258
Limerick A, McCabe EA, Turner JS, Kuang KW, Brautigan DL, Hao Y, Chu CY, Fu SH, Ahmadi S, Xu W, et al. An Epilepsy-Associated CILK1 Variant Compromises KATNIP Regulation and Impairs Primary Cilia and Hedgehog Signaling. Cells. 2024; 13(15):1258. https://doi.org/10.3390/cells13151258
Chicago/Turabian StyleLimerick, Ana, Ellie A. McCabe, Jacob S. Turner, Kevin W. Kuang, David L. Brautigan, Yi Hao, Cheuk Ying Chu, Sean H. Fu, Sean Ahmadi, Wenhao Xu, and et al. 2024. "An Epilepsy-Associated CILK1 Variant Compromises KATNIP Regulation and Impairs Primary Cilia and Hedgehog Signaling" Cells 13, no. 15: 1258. https://doi.org/10.3390/cells13151258
APA StyleLimerick, A., McCabe, E. A., Turner, J. S., Kuang, K. W., Brautigan, D. L., Hao, Y., Chu, C. Y., Fu, S. H., Ahmadi, S., Xu, W., & Fu, Z. (2024). An Epilepsy-Associated CILK1 Variant Compromises KATNIP Regulation and Impairs Primary Cilia and Hedgehog Signaling. Cells, 13(15), 1258. https://doi.org/10.3390/cells13151258