
Investigating Splice Defects in USH2A Using Targeted Long-Read Sequencing
 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Cohort
2.2. Variant Analysis
2.2.1. Variant Filtering
2.2.2. In Silico Predictions
2.3. Cell Collection, RNA Extraction, and RT-PCR
2.4. ONT Sequencing
3. Results
3.1. Patient 1 (GC20114)
3.2. Patient 2 (GC19437)
3.3. Patient 3 (GC2569)
3.4. Patient 4 (GC20993)
3.5. Patient 5 (GC22929)
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Von Graefe, A. Vereizelte Beobachtungen und Bemerkungen. Exceptionnelles verhalter des Gesichts feldes bei Pigmentenarter der Netzhalt. Arch. Klin. Ophtalmol. 1858, 4, 250–253. [Google Scholar]
- Rivolta, C.; Sweklo, E.A.; Berson, E.L.; Dryja, T.P. Missense mutation in the USH2A gene: Association with recessive retinitis pigmentosa without hearing loss. Am. J. Hum. Genet. 2000, 66, 1975–1978. [Google Scholar] [CrossRef] [PubMed]
- Hufnagel, R.B.; Liang, W.; Duncan, J.L.; Brewer, C.C.; Audo, I.; Ayala, A.R.; Branham, K.; Cheetham, J.K.; Daiger, S.P.; Durham, T.A.; et al. Tissue-specific genotype-phenotype correlations among USH2A-related disorders in the RUSH2A study. Hum. Mutat. 2022, 43, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Lenassi, E.; Vincent, A.; Li, Z.; Saihan, Z.; Coffey, A.J.; Steele-Stallard, H.B.; Moore, A.T.; Steel, K.P.; Luxon, L.M.; Héon, E.; et al. A detailed clinical and molecular survey of subjects with nonsyndromic USH2A retinopathy reveals an allelic hierarchy of disease-causing variants. Eur. J. Hum. Genet. 2015, 23, 1318–1327. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Vermeirsch, S.; Pontikos, N.; Martin-Gutierrez, M.P.; Daich Varela, M.; Malka, S.; Schiff, E.; Knight, H.; Wright, G.; Jurkute, N.; et al. Spectrum of Genetic Variants in the Most Common Genes Causing Inherited Retinal Disease in a Large Molecularly Characterized United Kingdom Cohort. Ophthalmol. Retina 2024, 12, 699–709. [Google Scholar] [CrossRef]
- Pontikos, N.; Arno, G.; Jurkute, N.; Schiff, E.; Ba-Abbad, R.; Malka, S.; Gimenez, A.; Georgiou, M.; Wright, G.; Armengol, M.; et al. Genetic Basis of Inherited Retinal Disease in a Molecularly Characterized Cohort of More Than 3000 Families from the United Kingdom. Ophthalmology 2020, 127, 1384–1394. [Google Scholar] [CrossRef]
- Sibley, C.R.; Blazquez, L.; Ule, J. Lessons from non-canonical splicing. Nat. Rev. Genet. 2016, 17, 407–421. [Google Scholar] [CrossRef]
- Ellard, S.; Baple, E.L.; Callaway, A.; Berry, I.; Forrester, N.; Turnbull, C.; Owens, M.; Eccles, D.M.; Abbs, S.; Scott, R.; et al. ACGS Best Practice Guidelines for Variant Classification in Rare Disease 2020; ACGS: Auckland, New Zealand, 2020. [Google Scholar]
- Varela, M.D.; Bellingham, J.; Motta, F.; Jurkute, N.; Ellingford, J.M.; Quinodoz, M.; Oprych, K.; Niblock, M.; Janeschitz-Kriegl, L.; Kaminska, K.; et al. Multidisciplinary team directed analysis of whole genome sequencing reveals pathogenic non-coding variants in molecularly undiagnosed inherited retinal dystrophies. Hum. Mol. Genet. 2022, 32, 595–607. [Google Scholar] [CrossRef]
- 100000 Genomes Project Pilot Investigators; Smedley, D.; Smith, K.R.; Martin, A.; Thomas, E.A.; McDonagh, E.M.; Cipriani, V.; Ellingford, J.M.; Arno, G.; Tucci, A.; et al. 100,000 Genomes Pilot on Rare-Disease Diagnosis in Health Care—Preliminary Report. N. Engl. J. Med. 2021, 385, 1868–1880. [Google Scholar]
- Toualbi, L.; Toms, M.; Moosajee, M. USH2A-retinopathy: From genetics to therapeutics. Exp. Eye Res. 2020, 201, 108330. [Google Scholar] [CrossRef]
- Dulla, K.; Slijkerman, R.; van Diepen, H.C.; Albert, S.; Dona, M.; Beumer, W.; Turunen, J.J.; Chan, H.L.; Schulkens, I.A.; Vorthoren, L.; et al. Antisense oligonucleotide-based treatment of retinitis pigmentosa caused by USH2A exon 13 mutations. Mol. Ther. 2021, 29, 2441–2455. [Google Scholar] [CrossRef] [PubMed]
- McAllister, M.; Dearing, A. Patient reported outcomes and patient empowerment in clinical genetics services. Clin. Genet. 2015, 88, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Carss, K.J.; Arno, G.; Erwood, M.; Stephens, J.; Sanchis-Juan, A.; Hull, S.; Megy, K.; Grozeva, D.; Dewhurst, E.; Malka, S.; et al. Comprehensive Rare Variant Analysis via Whole-Genome Sequencing to Determine the Molecular Pathology of Inherited Retinal Disease. Am. J. Human. Genet. 2017, 100, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.R.; Williams, E.; Foulger, R.E.; Leigh, S.; Daugherty, L.C.; Niblock, O.; Leong, I.U.S.; Smith, K.R.; Gerasimenko, O.; Haraldsdottir, E.; et al. PanelApp crowdsources expert knowledge to establish consensus diagnostic gene panels. Nat. Genet. 2019, 51, 1560–1565. [Google Scholar] [CrossRef] [PubMed]
- Jaganathan, K.; Panagiotopoulou, S.K.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019, 176, 535–548.e24. [Google Scholar] [CrossRef] [PubMed]
- Brunak, S.; Engelbrecht, J.; Knudsen, S. Prediction of human mRNA donor and acceptor sites from the DNA sequence. J. Mol. Biol. 1991, 220, 49–65. [Google Scholar] [CrossRef] [PubMed]
- Lenassi, E.; Saihan, Z.; Bitner-Glindzicz, M.; Webster, A.R. The effect of the Common c.2299delG mutation in USH2A on RNA splicing. Exp. Eye Res. 2014, 122, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Jiman, O.A.; UK Inherited Retinal Disease Consortium; Taylor, R.L.; Lenassi, E.; Smith, J.C.; Douzgou, S.; Ellingford, J.M.; Barton, S.; Hardcastle, C.; Fletcher, T.; et al. Diagnostic yield of panel-based genetic testing in syndromic inherited retinal disease. Eur. J. Hum. Genet. 2020, 28, 576–586. [Google Scholar] [CrossRef] [PubMed]
- Downes, S.M.; Nguyen, T.; Tai, V.; Broadgate, S.; Shah, M.; Al-Khuzaei, S.; MacLaren, R.E.; Shanks, M.; Clouston, P.; Halford, S. Genetic and Clinical Findings in an Ethnically Diverse Cohort with Retinitis Pigmentosa Associated with Pathogenic Variants in CERKL. Genes 2020, 11, 1497. [Google Scholar] [CrossRef]
- Qian, X.; Wang, J.; Wang, M.; Igelman, A.D.; Jones, K.D.; Li, Y.; Wang, K.; Goetz, K.E.; Birch, D.G.; Yang, P.; et al. Identification of Deep-Intronic Splice Mutations in a Large Cohort of Patients With Inherited Retinal Diseases. Front. Genet. 2021, 12, 647400. [Google Scholar] [CrossRef]
- Slijkerman, R.W.; Vaché, C.; Dona, M.; García-García, G.; Claustres, M.; Hetterschijt, L.; A Peters, T.; Hartel, B.P.; Pennings, R.J.; Millan, J.M.; et al. Antisense Oligonucleotide-based Splice Correction for USH2A-associated Retinal Degeneration Caused by a Frequent Deep-intronic Mutation. Mol. Ther. Nucleic Acids 2016, 5, e381. [Google Scholar] [CrossRef] [PubMed]
- Reurink, J.; Oostrik, J.; Aben, M.; Ramos, M.G.; van Berkel, E.; Ołdak, M.; van Wijk, E.; Kremer, H.; Roosing, S.; Cremers, F.P.M. Minigene-Based Splice Assays Reveal the Effect of Non-Canonical Splice Site Variants in USH2A. Int. J. Mol. Sci. 2022, 23, 13343. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient (ID) | Candidate Variant (GRCh38, NM_206933.4) | Previously Reported | Gnomad Frequency (v4.1.0) | ClinVar Prediction | Predicted Protein Consequence | Observed Protein Consequence |
|---|---|---|---|---|---|---|
| 1 (GC20114) | chr1:g.215793253G>A c.9959-2971C>T | No | Absent | Absent | p.(Met3321LeufsTer8) | p.Met3321LeufsTer8 |
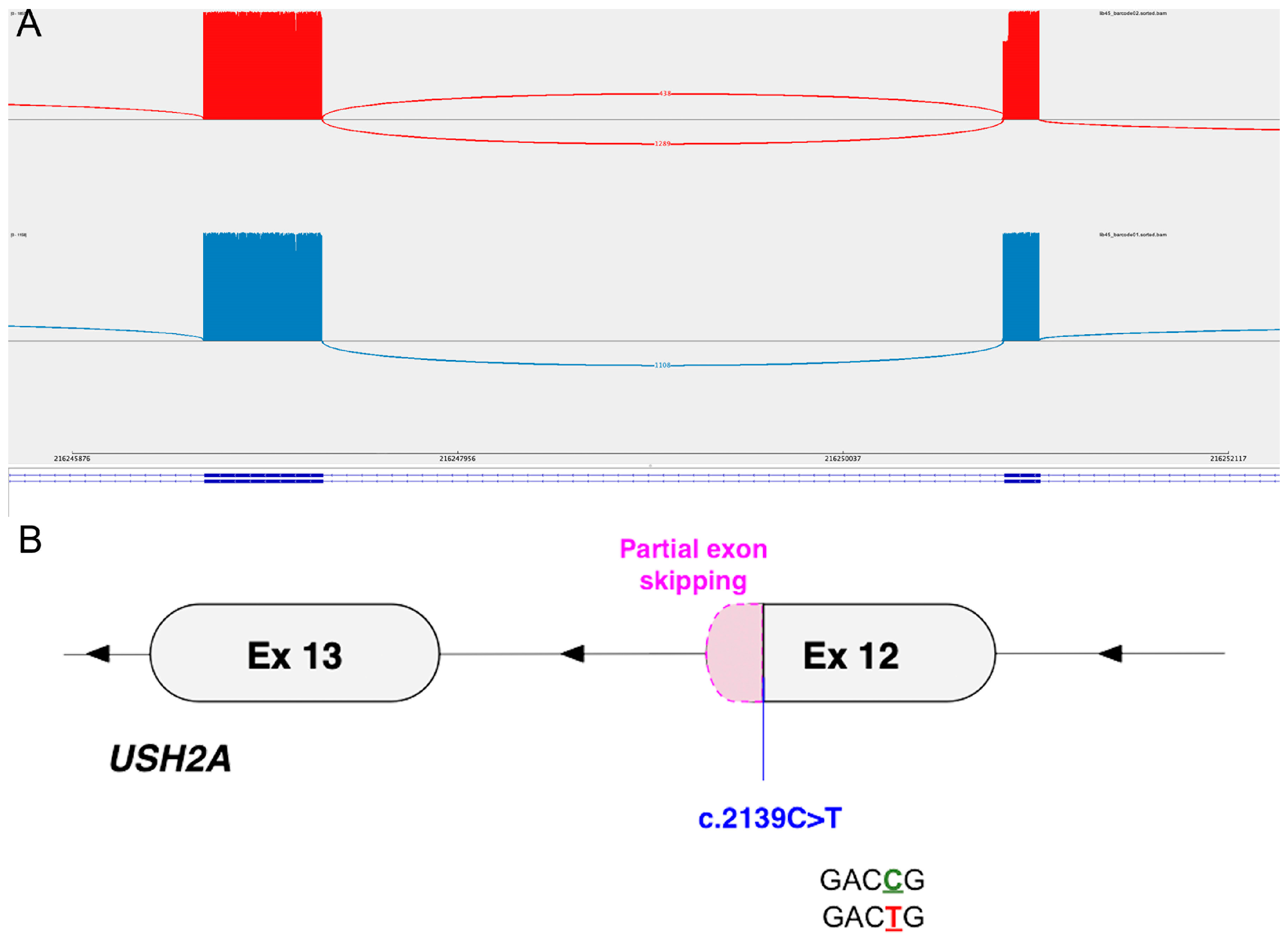
| 2 (GC19437) | chr1:g.216250931G>A c.2139C>T | Yes [19] | 0.00001549 | Conflicting (VUS ×3, Pathogenic ×1) | p.(Gly713=) | p.Glu714_Gly723del |
| 3 (GC2569) | chr1:g.216198560_216198588dup c.3812-3_3837dup | Yes (carrier state in an affected individual with an alternative molecular diagnosis) [20] | 0.0007429 | Conflicting (LP ×1, VUS ×2, LB ×1) | p.(Met1280Ter) | p.Gly1271_Ser1360del |
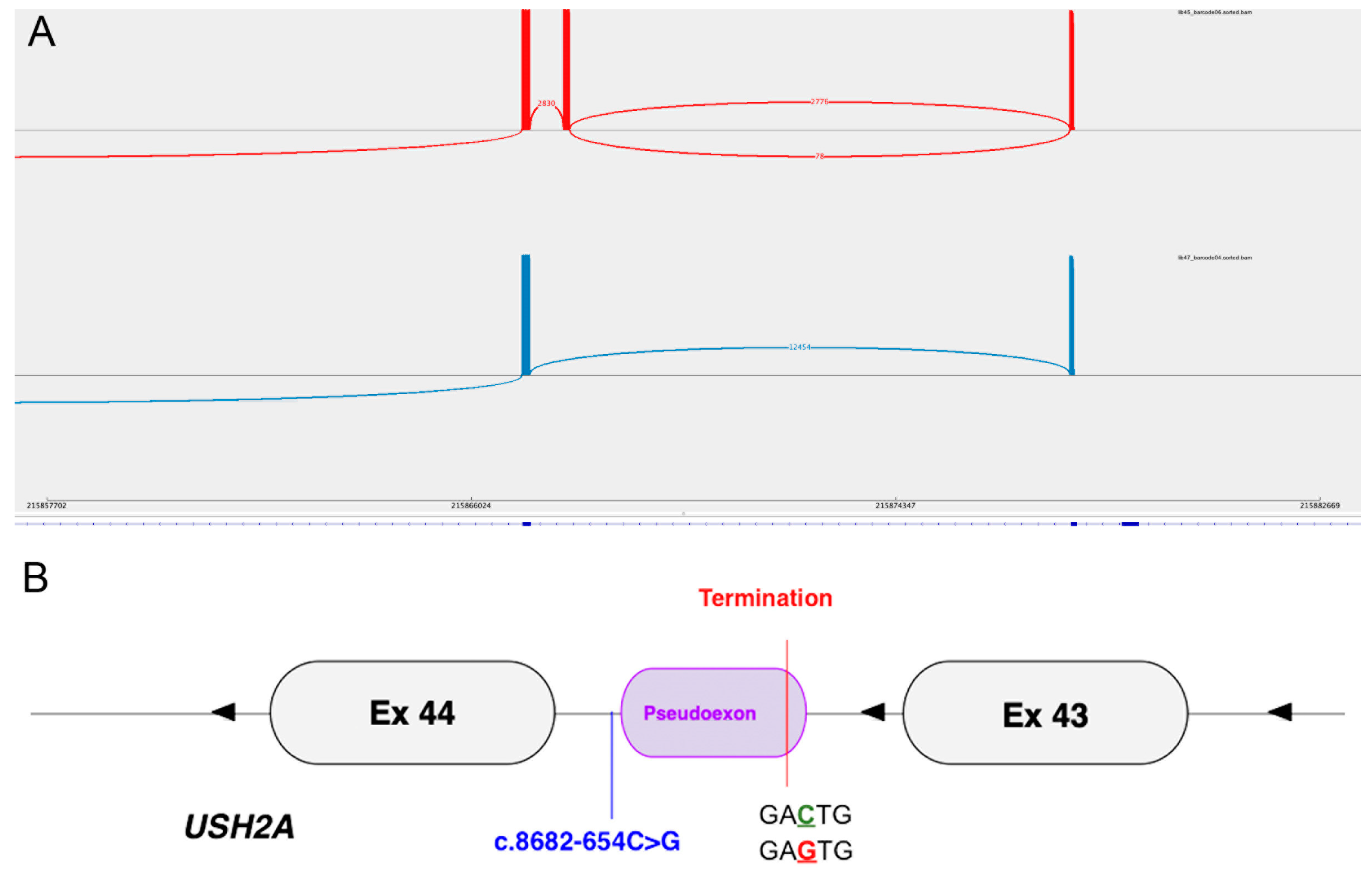
| 4 (GC20993) | chr1:g.215867824G>C c.8682-654C>G | Yes [21] | 0.000006570 | Absent | p.(Phe2895Ter) | p.Phe2895Ter |
| 5 (GC22929) | chr1:g.215845435C>T c.9055+389G>A | No | 0.00001971 | Absent | p.(Glu3019AspfsTer65) | p.Glu3019AspfsTer65 |
| Patient (ID) | Clinical Diagnosis | USH2A Variants (NM_206933.4) | |
|---|---|---|---|
| Variant 1 | Variant 2 | ||
| 1 (GC20114) | USH2 | c.9959-2971C>T | c.6289_6302del14 p.(Ile2097Ter) |
| 2 (GC19437) | USH2 | c.2139C>T | c.7595-2144A>G |
| 3 (GC2569) | RP | c.3812-3_3837dup | c.4222C>T p.(Gln1408Ter) |
| 4 (GC20993) | RP | c.8682-654C>G | c.9882C>G p.(Cys3294Trp) |
| 5 (GC22929) | USH2 | c.9055 + 389G>A | c.2299del p.(Glu767SerfsTer21). |
| Patient and Variant | (Δ) Type | (Δ) Score | Position | Consequence |
|---|---|---|---|---|
| 1 (GC20114) c.9959-2971C>T | Acceptor gain | 0.68 | +114 | Pseudoexon inclusion |
| Donor gain | 0.67 | +2 | 113 bp (intron 50) | |
| 2 (GC19437) c.2139C>T | Donor loss | 0.89 | −28 | Partial exon skipping |
| Donor gain | 0.86 | +2 | 30 bp (exon 12) | |
| 3 (GC2569) c.3812-3_3837dup | Acceptor loss | 0.57 | −3 | Weakening of canonical site |
| Acceptor gain | 0.29 | 0 | Use of first copy of the acceptor motif and stop gain within exon 18 | |
| 4 (GC20993) c.8682-654C>G | Acceptor gain | 0.36 | +131 | Pseudoexon inclusion |
| Donor gain | 0.50 | +3 | 130 bp (intron 43) | |
| 5 (GC22929) c.9055+389G>A | Acceptor gain | 0.26 | +80 | Pseudoexon inclusion |
| Donor Gain | 0.29 | +4 | 78 bp (intron 45) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chandrasekhar, S.; Lin, S.; Jurkute, N.; Oprych, K.; Estramiana Elorrieta, L.; Schiff, E.; Malka, S.; Wright, G.; Michaelides, M.; Mahroo, O.A.; et al. Investigating Splice Defects in USH2A Using Targeted Long-Read Sequencing. Cells 2024, 13, 1261. https://doi.org/10.3390/cells13151261
Chandrasekhar S, Lin S, Jurkute N, Oprych K, Estramiana Elorrieta L, Schiff E, Malka S, Wright G, Michaelides M, Mahroo OA, et al. Investigating Splice Defects in USH2A Using Targeted Long-Read Sequencing. Cells. 2024; 13(15):1261. https://doi.org/10.3390/cells13151261
Chicago/Turabian StyleChandrasekhar, Shwetha, Siying Lin, Neringa Jurkute, Kathryn Oprych, Leire Estramiana Elorrieta, Elena Schiff, Samantha Malka, Genevieve Wright, Michel Michaelides, Omar A. Mahroo, and et al. 2024. "Investigating Splice Defects in USH2A Using Targeted Long-Read Sequencing" Cells 13, no. 15: 1261. https://doi.org/10.3390/cells13151261