


The Need to Identify Novel Markers for Early Renal Injury in Cardiorenal Syndrome



Abstract
:1. Introduction
1.1. Renal Diagnostic Criteria
1.2. Cardiac Diagnostic Criteria
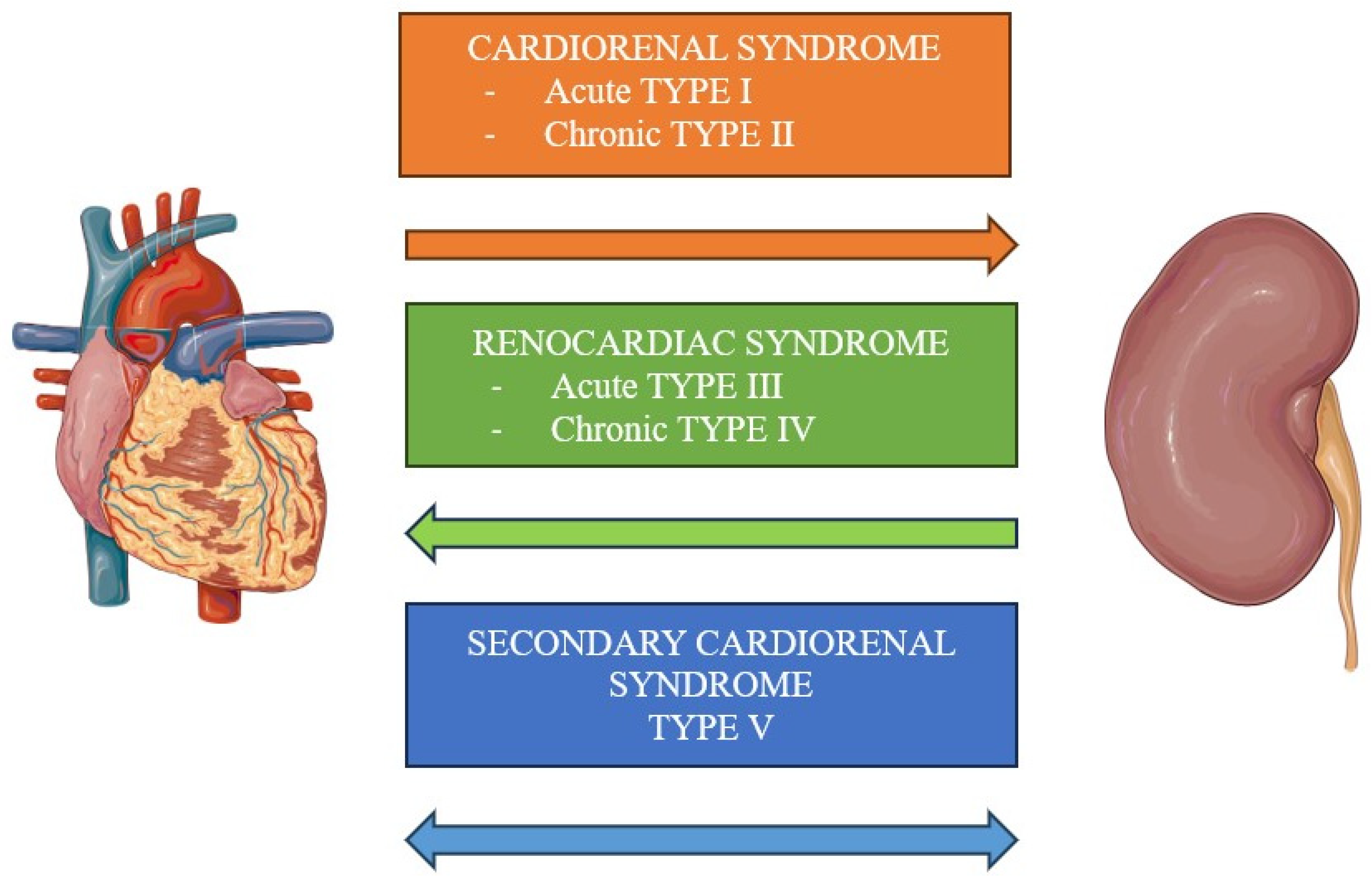
2. Classification
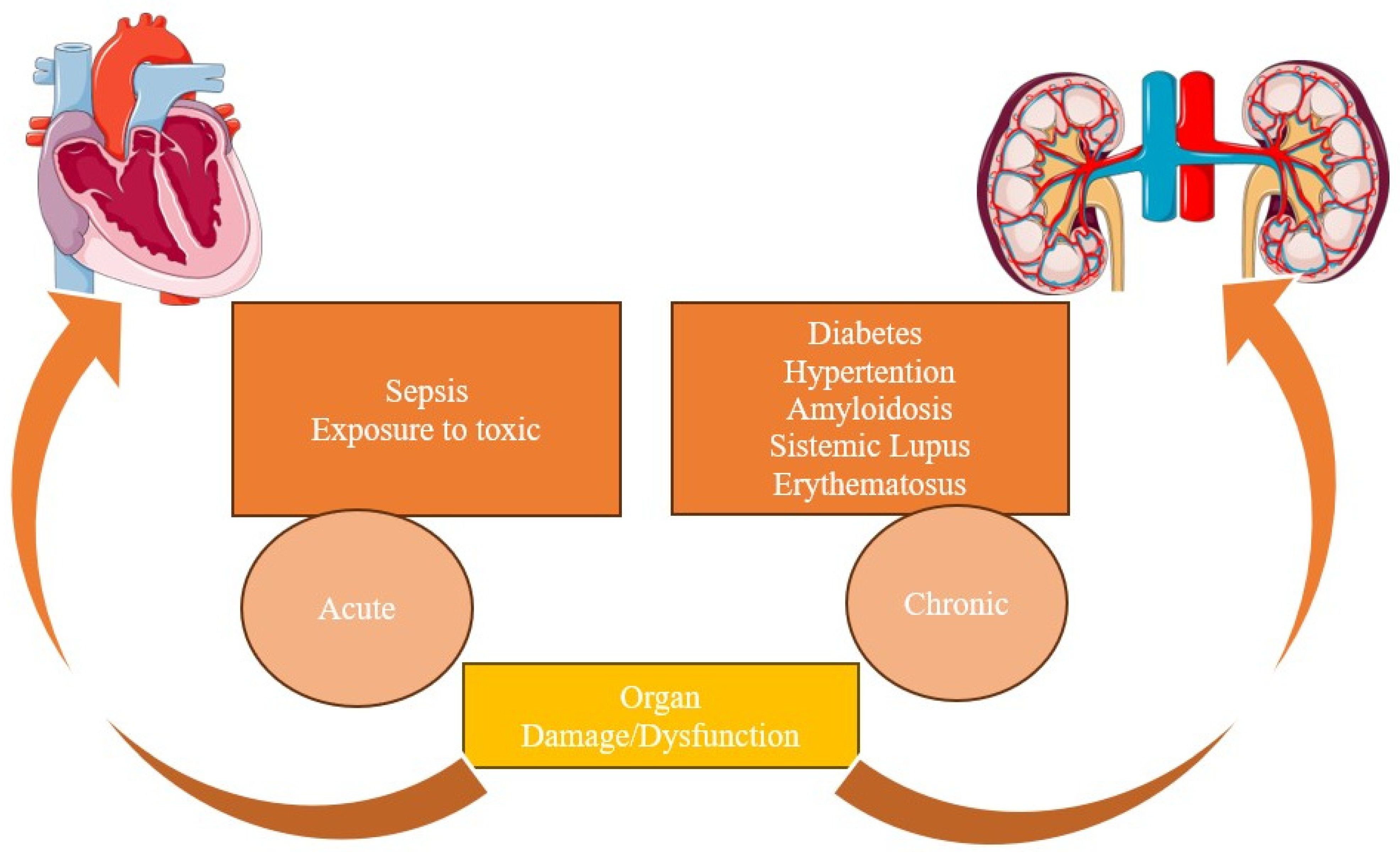
2.1. Type I
2.2. Type II
2.3. Type III
2.4. Type IV
2.5. Type V
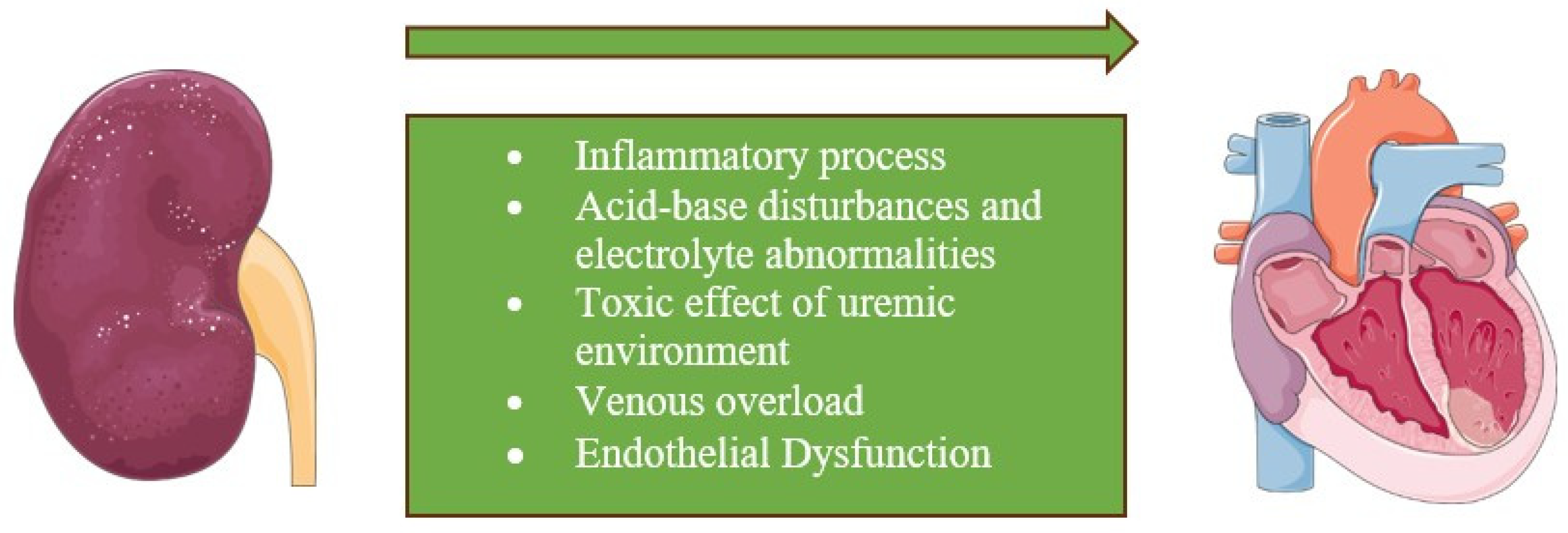
3. Pathophysiology
3.1. Hemodynamic Alterations
3.1.1. Venous Congestion
3.1.2. Intra-Abdominal Pressure
3.2. Non-Hemodynamic Alterations
3.2.1. Neurohormonal Pathways
3.2.2. Oxidative Stress
3.2.3. Inflammation
4. Biomarkers of Renal Injury
4.1. Established Biomarkers
4.1.1. Creatinine
4.1.2. Glomerular Filtration Rate (GFR)
4.1.3. Brain Natriuretic Peptide
4.2. Novel Biomarkers (Table 2)
4.2.1. Neutrophil Gelatinase-Associated Lipocalin (NGAL)
4.2.2. Cystatin C (CysC)
4.2.3. Kidney Injury Molecule-1 (KIM-1)
4.2.4. N-Acetyl-β-d-Glucosaminidase (NAG)
4.2.5. Interleukin 18 (IL-18)
4.2.6. Galectin-3

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Novel Biomarker | Description | References | Results |
|---|---|---|---|
| NGAL | Marker secreted in urine and blood. An early marker of renal damage. | Song et al. [84] | Diagnosis of CRS type I: ROC curve AUC 0.875 [0.813–0.937] p < 0.001 |
| Alvelos et al. [85] | Development of CRS type I in patients with acute heart failure: AUC 0.93 [0.88–0.98] p < 0.001 | ||
| Chen et al. [86] | AKI progression in patients with CRS type I: OR, 4.7; 95% CI, 1.7–13.4 p < 0.001 | ||
| CysC | Assessment of kidney function. In CKD, it is better than serum creatinine. Predictor of adverse outcomes | Pinsino et al. [87] | CysC-based estimated glomerular filtration rate predicts a composite endpoint of in-hospital mortality, renal replacement therapy, or severe right ventricular failure in patients with LVAD: OR per 5 mL/(min·1.73 m2) decrease 1.16 (1.02–1.31) |
| Ruan et al. [88] | Levels of Cys C are independently associated with in-hospital and 12-months mortality in patients with CRS type I OR, 1.48; 95% CI, 1.75–4.16, p = 0.027 and OR, 2.72; 95% CI, 1.92–4.28, p = 0.017, respectively | ||
| Rafouli-Stergiou et al. [68] | In-hospital changes in CysC predicted cardiac death or rehospitalization for heart failure decompensation at 60 days in patients with CRS: ROC curve, AUC 0.681 [0.549–0.812], p = 0.014 | ||
| NAG | Marker of acute kidney injury. | Liangos et al. [74] | The second, third, and fourth quartile groups of NAG associated with an increased risk of dialysis requirement or hospital death in patients with AKI: OR 3.0 (95% CI 1.3–7.2); OR 3.7 (95% CI 1.6–8.8) and OR 9.1 (95% CI 3.7–22.7), respectively |
| KIM-1 | Marker of acute kidney injury. It is measured in urine. | Liangos et al. [74] | The second, third, and fourth quartile groups of KIM associated with an increased risk of dialysis requirement or hospital death in patients with AKI: OR 1.4 (95% CI 0.6–3.0), OR 1.4 (95% CI 0.6–3.0), and OR 3.2 (95% CI 1.4 to 7.4), respectively |
| Kaddourah et al. [71] | In children with dilated cardiomyopathy, a combined model using cut-off values of KIM-1 ≥ 235, IL-18 ≥ 17.5, and (BNP) > 15 pg/mL resulted in a distinction between patients with mildly depressed LV (55 > LVEF ≥ 45) and those with LVEF < 45%: ROC curve AUC 0.70 | ||
| IL-18 | Early marker of acute kidney injury. | Parikh et al. [77,78] | Levels of IL-18 predicted the development of AKI in ICU patients: IL-18 > 100 pg/mL OR 6.5 (95% CI 2.1–20.4) p < 0.001 Levels of IL-18 predicted mortality in ICU patients: IL-18 >200 pg/mL OR 2.32 (95% CI 1.2–4.4) p < 0.001 |
| Chen et al. [86] | AKI progression in patients with CRS type I: OR 3.6 (95% CI 1.4–9.5) | ||
| Kaddourah et al. [71] | In children with dilated cardiomyopathy, a combined model using cut-off values of KIM-1 ≥ 235, IL-18 ≥ 17.5, and (BNP) > 15 pg/mL distinguished patients with mildly depressed LV (55 > LVEF ≥ 45) and those with LVEF < 45%: ROC curve AUC 0.70 | ||
| Gal-3 | Marker of cardio-renal fibrosis and dysfunction. | Iacoviello et al. [83] | Gal-3 associated with kidney injury in patients with chronic heart failure: OR 1.08 (95% CI 1.02–1.14), p = 0.012 |
5. Conclusions and Perspectives
Funding
Conflicts of Interest
References
- Cases and Observations, Illustrative of Renal Disease, Accompanied with the Secretion of Albuminous Urine. Br. Foreign Med. Rev. 1840, 10, 301–329.
- Ronco, C.; Haapio, M.; House, A.A.; Anavekar, N.; Bellomo, R. Cardiorenal syndrome. J. Am. Coll. Cardiol. 2008, 52, 1527–1539. [Google Scholar] [CrossRef]
- House, A.A.; Anand, I.; Bellomo, R.; Cruz, D.; Bobek, I.; Anker, S.D.; Aspromonte, N.; Bagshaw, S.; Berl, T.; Acute Dialysis Quality Initiative Consensus Group; et al. Definition and classification of Cardio-Renal Syndromes: Workgroup statements from the 7th ADQI Consensus Conference. Nephrol. Dial. Transplant. 2010, 25, 1416–1420. [Google Scholar] [CrossRef] [PubMed]
- Berl, T.; Henrich, W. Kidney-heart interactions: Epidemiology, pathogenesis, and treatment. Clin. J. Am. Soc. Nephrol. CJASN 2006, 1, 8–18. [Google Scholar] [CrossRef]
- Kumar, U.; Wettersten, N.; Garimella, P.S. Cardiorenal Syndrome: Pathophysiology. Cardiol Clin. 2019, 37, 251–265. [Google Scholar] [CrossRef] [PubMed]
- Mavrakanas, T.A.; Khattak, A.; Singh, K.; Charytan, D.M. Epidemiology and Natural History of the Cardiorenal Syndromes in a Cohort with Echocardiography. Clin. J. Am. Soc. Nephrol. 2017, 12, 1624–1633. [Google Scholar] [CrossRef]
- Ronco, C.; Bellasi, A.; Di Lullo, L. Cardiorenal Syndrome: An Overview. Adv. Chronic Kidney Dis. 2018, 25, 382–390. [Google Scholar] [CrossRef]
- Levey, A.S. Defining AKD: The Spectrum of AKI, AKD, and CKD. Nephron 2022, 146, 302–305. [Google Scholar] [CrossRef]
- Ponikowski, P.; Voors, A.A.; Anker, S.D.; Bueno, H.; Cleland, J.G.F.; Coats, A.J.S.; Document Reviewers. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. J. Heart Fail. 2016, 18, 891–975. [Google Scholar] [CrossRef] [PubMed]
- Soni, S.S.; Ronco, C.; Pophale, R.; Bhansali, A.S.; Nagarik, A.P.; Barnela, S.R.; Raman, A. Cardio-Renal Syndrome Type 5: Epidemiology, Pathophysiology, and Treatment. Semin. Nephrol. 2012, 32, 49–56. [Google Scholar] [CrossRef]
- Clementi, A.; Virzì, G.M.; Goh, C.Y.; Cruz, D.N.; Granata, A.; Vescovo, G.; Ronco, C. Cardiorenal Syndrome Type 4: A Review. Cardiorenal Med. 2013, 3, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Ronco, C.; Cicoira, M.; McCullough, P.A. Cardiorenal syndrome type 1: Pathophysiological crosstalk leading to combined heart and kidney dysfunction in the setting of acutely decompensated heart failure. J. Am. Coll. Cardiol. 2012, 60, 1031–1042. [Google Scholar] [CrossRef] [PubMed]
- Haase, M.; Müller, C.; Damman, K.; Murray, P.T.; Kellum, J.A.; Ronco, C.; McCullough, P.A. Pathogenesis of cardiorenal syndrome type 1 in acute decompensated heart failure: Workgroup statements from the eleventh consensus conference of the Acute Dialysis Quality Initiative (ADQI). Contrib. Nephrol. 2013, 182, 99–116. [Google Scholar] [CrossRef] [PubMed]
- Cruz, D.N.; Bagshaw, S.M. Heart-kidney interaction: Epidemiology of cardiorenal syndromes. Int. J. Nephrol. 2011, 2011, 351291. [Google Scholar] [CrossRef] [PubMed]
- Hebert, K.; Dias, A.; Delgado, M.C.; Franco, E.; Tamariz, L.; Steen, D.; Trahan, P.; Major, B.; Arcement, L.M. Epidemiology and survival of the five stages of chronic kidney disease in a systolic heart failure population. Eur. J. Heart Fail. 2010, 12, 861–865. [Google Scholar] [CrossRef]
- Heywood, J.T.; Fonarow, G.C.; Costanzo, M.R.; Mathur, V.S.; Wigneswaran, J.R.; Wynne, J. ADHERE Scientific Advisory Committee and Investigators. High prevalence of renal dysfunction and its impact on outcome in 118, 465 patients hospitalized with acute decompensated heart failure: A report from the ADHERE database. J. Card. Fail. 2007, 13, 422–430. [Google Scholar] [CrossRef] [PubMed]
- Cruz, D.N.; Schmidt-Ott, K.M.; Vescovo, G.; House, A.A.; Kellum, J.A.; Ronco, C.; McCullough, P.A. Pathophysiology of cardiorenal syndrome type 2 in stable chronic heart failure: Workgroup statements from the eleventh consensus conference of the Acute Dialysis Quality Initiative (ADQI). Contrib. Nephrol. 2013, 182, 117–136. [Google Scholar] [CrossRef] [PubMed]
- Hanberg, J.S.; Rao, V.S.; Ahmad, T.; Chunara, Z.; Mahoney, D.; Jackson, K.; Jacoby, D.; Chen, M.; Wilson, F.P.; Tang, W.H.W.; et al. Inflammation and cardio-renal interactions in heart failure: A potential role for interleukin-6. Eur. J. Heart Fail. 2018, 20, 933–934. [Google Scholar] [CrossRef] [PubMed]
- Bagshaw, S.M.; Cruz, D.N.; Aspromonte, N.; Daliento, L.; Ronco, F.; Sheinfeld, G.; Acute Dialysis Quality Initiative (ADQI) Consensus Group. Epidemiology of cardio-renal syndromes: Workgroup statements from the 7th ADQI Consensus Conference. Nephrol. Dial. Transplant. 2010, 25, 1406–1416. [Google Scholar] [CrossRef]
- Chuasuwan, A.; Kellum, J.A. Cardio-renal syndrome type 3: Epidemiology, pathophysiology, and treatment. Semin Nephrol. 2012, 32, 31–39. [Google Scholar] [CrossRef]
- Shastri, S.; Sarnak, M.J. Cardiovascular disease and CKD: Core curriculum 2010. Am. J. Kidney Dis. 2010, 56, 399–417. [Google Scholar] [CrossRef] [PubMed]
- Ishani, A.; Grandits, G.A.; Grimm, R.H.; Svendsen, K.H.; Collins, A.J.; Prineas, R.J.; Neaton, J.D. Association of single measurements of dipstick proteinuria, estimated glomerular filtration rate, and hematocrit with 25-year incidence of end-stage renal disease in the multiple risk factor intervention trial. J. Am. Soc. Nephrol. 2006, 17, 1444–1452. [Google Scholar] [CrossRef] [PubMed]
- Raina, R.; Nair, N.; Chakraborty, R.; Nemer, L.; Dasgupta, R.; Varian, K. An Update on the Pathophysiology and Treatment of Cardiorenal Syndrome. Cardiol. Res. 2020, 11, 76–88. [Google Scholar] [CrossRef] [PubMed]
- Rowntree, L.G.; Fitz, R.; Geraghty, J.T. The effects of experimental chronic passive congestion on renal function. Arch. Intern. Med. 1913, XI, 121–147. [Google Scholar] [CrossRef]
- Win, F.R. The influence of venous pressure on the isolated mammalian kidney. J. Physiol. 1931, 72, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Blake, W.D.; Wégria, R.; Keating, R.P.; Ward, H.P. Effect of increased renal venous pressure on renal function. Am. J. Physiol. 1949, 157, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Nohria, A.; Hasselblad, V.; Stebbins, A.; Pauly, D.F.; Fonarow, G.C.; Shah, M.; Yancy, C.W.; Califf, R.M.; Stevenson, L.W.; Hill, J.A. Cardiorenal interactions: Insights from the ESCAPE trial. J. Am. Coll. Cardiol. 2008, 51, 1268–1274. [Google Scholar] [CrossRef] [PubMed]
- Mullens, W.; Verbrugge, F.H.; Nijst, P.; Tang, W.H.W. Renal sodium avidity in heart failure: From pathophysiology to treatment strategies. Eur. Heart J. 2017, 38, 1872–1882. [Google Scholar] [CrossRef] [PubMed]
- Mullens, W.; Abrahams, Z.; Skouri, H.N.; Francis, G.S.; Taylor, D.O.; Starling, R.C.; Paganini, E.; Tang, W.H.W. Elevated intra-abdominal pressure in acute decompensated heart failure: A potential contributor to worsening renal function? J. Am. Coll. Cardiol. 2008, 51, 300–306. [Google Scholar] [CrossRef]
- Malbrain, M.L.N.G.; Deeren, D.; De Potter, T.J.R. Intra-abdominal hypertension in the critically ill: It is time to pay attention. Curr. Opin. Crit. Care 2005, 11, 156–171. [Google Scholar] [CrossRef]
- Jackson, G.; Gibbs, C.R.; Davies, M.K.; Lip, G.Y.H. ABC of heart failure. Pathophysiol. BMJ. 2000, 320, 167–170. [Google Scholar] [CrossRef]
- Cadnapaphornchai, M.A.; Gurevich, A.K.; Weinberger, H.D.; Schrier, R.W. Pathophysiology of sodium and water retention in heart failure. Cardiology 2001, 96, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Liang, K.V.; Williams, A.W.; Greene, E.L.; Redfield, M.M. Acute decompensated heart failure and the cardiorenal syndrome. Crit Care Med. 2008, 36 (Suppl. S1), S75–S88. [Google Scholar] [CrossRef] [PubMed]
- Peoples, J.N.; Saraf, A.; Ghazal, N.; Pham, T.T.; Kwong, J.Q. Mitochondrial dysfunction and oxidative stress in heart disease. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Milkovic, L.; Gasparovic, A.C.; Cindric, M.; Mouthuy, P.-A.; Zarkovic, N. Short Overview of ROS as Cell Function Regulators and Their Implications in Therapy Concepts. Cells 2019, 8, 793. [Google Scholar] [CrossRef]
- Thannickal, V.J.; Fanburg, B.L. Reactive oxygen species in cell signaling. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2000, 279, L1005–L1028. [Google Scholar] [CrossRef] [PubMed]
- Gallo, G.; Lanza, O.; Savoia, C. New Insight in Cardiorenal Syndrome: From Biomarkers to Therapy. Int. J. Mol. Sci. 2023, 24, 5089. [Google Scholar] [CrossRef] [PubMed]
- McWilliam, S.J.; Wright, R.D.; Welsh, G.I.; Tuffin, J.; Budge, K.L.; Swan, L.; Wilm, T.; Martinas, I.-R.; Littlewood, J.; Oni, L. The complex interplay between kidney injury and inflammation. Clin. Kidney J. 2020, 14, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Ganda, A.; Onat, D.; Demmer, R.T.; Wan, E.; Vittorio, T.J.; Sabbah, H.N.; Colombo, P.C. Venous Congestion and Endothelial Cell Activation in Acute Decompensated Heart Failure. Curr. Heart Fail. Rep. 2010, 7, 66–74. [Google Scholar] [CrossRef]
- Virzì, G.M.; Zhang, J.; Nalesso, F.; Ronco, C.; McCullough, P.A. The Role of Dendritic and Endothelial Cells in Cardiorenal Syndrome. Cardiorenal Med. 2018, 8, 92–104. [Google Scholar] [CrossRef]
- Zager, R.A.; Johnson, A.C.M.; Becker, K.; Erpicum, P.; Rowart, P.; Defraigne, J.-O.; Krzesinski, J.-M.; Jouret, F.; Black, L.M.; Lever, J.M.; et al. Acute unilateral ischemic renal injury induces progressive renal inflammation, lipid accumulation, histone modification, and “end-stage” kidney disease. Am. J. Physiol. Physiol. 2011, 301, F1334–F1345. [Google Scholar] [CrossRef] [PubMed]
- Basile, D.P. The endothelial cell in ischemic acute kidney injury: Implications for acute and chronic function. Kidney Int. 2007, 72, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Anders, H.-J. Immune system modulation of kidney regeneration—Mechanisms and implications. Nat. Rev. Nephrol. 2014, 10, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Yanagita, M. Immune cells and inflammation in AKI to CKD progression. Am. J. Physiol. Physiol. 2018, 315, F1501–F1512. [Google Scholar] [CrossRef] [PubMed]
- Faul, C.; Amaral, A.P.; Oskouei, B.; Hu, M.-C.; Sloan, A.; Isakova, T.; Gutiérrez, O.M.; Aguillon-Prada, R.; Lincoln, J.; Hare, J.M.; et al. FGF23 induces left ventricular hypertrophy. J. Clin. Investig. 2011, 121, 4393–4408. [Google Scholar] [CrossRef] [PubMed]
- Pelliccia, F.; Cecchi, F.; Olivotto, I.; Camici, P.G. Microvascular Dysfunction in Hypertrophic Cardiomyopathy. J. Clin. Med. 2022, 11, 6560. [Google Scholar] [CrossRef] [PubMed]
- Liberale, L.; Badimon, L.; Montecucco, F.; Lüscher, T.F.; Libby, P.; Camici, G.G. Inflammation, Aging, and Cardiovascular Disease. J. Am. Coll. Cardiol. 2022, 79, 837–847. [Google Scholar] [CrossRef] [PubMed]
- Akhter, M.S.; Goodwin, J.E. Endothelial Dysfunction in Cardiorenal Conditions: Implications of Endothelial Glucocorticoid Receptor-Wnt Signaling. Int. J. Mol. Sci. 2023, 24, 14261. [Google Scholar] [CrossRef] [PubMed]
- Cruz, D.N.; Goh, C.Y.; Haase-Fielitz, A.; Ronco, C.; Haase, M. Early biomarkers of renal injury. Congest. Heart Fail. 2010, 16 (Suppl. 1), S25–S31. [Google Scholar] [CrossRef]
- Soni, S.S.; Ronco, C.; Katz, N.; Cruz, D.N. Early diagnosis of acute kidney injury: The promise of novel biomarkers. Blood Purif. 2009, 28, 165–174. [Google Scholar] [CrossRef]
- Kellum, J.A.; Mehta, R.L.; Levin, A.; Molitoris, B.A.; Warnock, D.G.; Shah, S.V.; Joannidis, M.; Ron, C. Acute Kidney Injury Network (AKIN). Development of a clinical research agenda for acute kidney injury using an international, interdisciplinary, three-step modified Delphi process. Clin. J. Am. Soc. Nephrol. 2008, 3, 887–894. [Google Scholar] [CrossRef] [PubMed]
- Molitor, B.A. Measuring glomerular filtration rate in acute kidney injury: Yes, but not yet. Crit. Care 2012, 16, 158. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Ni, G.; Wu, Q.; Zhou, Y.; Yao, W.; Zhang, H.; Li, X. Prognostic Value of N-Terminal Pro-B-Type Natriuretic Peptide and Glomerular Filtration Rate in Patients With Acute Heart Failure. Front. Cardiovasc. Med. 2020, 7, 123. [Google Scholar] [CrossRef] [PubMed]
- Takahama, H.; Nishikimi, T.; Takashio, S.; Hayashi, T.; Nagai-Okatani, C.; Asada, T.; Fujiwara, A.; Nakagawa, Y.; Amano, M.; Hamatani, Y.; et al. Change in the NT-proBNP/Mature BNP Molar Ratio Precedes Worsening Renal Function in Patients with Acute Heart Failure: A Novel Predictor Candidate for Cardiorenal Syndrome. J. Am. Heart Assoc. 2019, 8, e011468. [Google Scholar] [CrossRef] [PubMed]
- Welsh, P.; Campbell, R.T.; Mooney, L.; Kimenai, D.M.; Hayward, C.; Campbell, A.; Porteous, D.; Mills, N.L.; Lang, N.N.; Petrie, M.C.; et al. Reference Ranges for NT-proBNP (N-Terminal Pro-B-Type Natriuretic Peptide) and Risk Factors for Higher NT-proBNP Concentrations in a Large General Population Cohort. Circ. Heart Fail. 2022, 15, e009427. [Google Scholar] [CrossRef] [PubMed]
- Kjeldsen, L.; Cowland, J.B.; Borregaard, N. Human neutrophil gelatinase-associated lipocalin and homologous proteins in rat and mouse. Biochim. Biophys. Acta 2000, 1482, 272–283. [Google Scholar] [CrossRef] [PubMed]
- Stoesz, S.P.; Friedl, A.; Haag, J.D.; Lindstrom, M.J.; Clark, G.M.; Gould, M.N. Heterogeneous expression of the lipocalin NGAL in primary breast cancers. Int. J. Cancer 1998, 79, 565–572. [Google Scholar] [CrossRef]
- Cowland, J.B.; Borregaard, N. Molecular characterization and pattern of tissue expression of the gene for neutrophil gelatinase-associated lipocalin from humans. Genomics 1997, 45, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Friedl, A.; Stoesz, S.P.; Buckley, P.; Gould, M.N. Neutrophil gelatinase-associated lipocalin in normal and neoplastic human tissues. Cell type-specific pattern of expression. Histochem. J. 1999, 31, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, B.S.; Borregaard, N.; Bundgaard, J.R.; Timshel, S.; Sehested, M.; Kjeldsen, L. Induction of NGAL synthesis in epithelial cells of human colorectal neoplasia and inflammatory bowel diseases. Gut 1996, 38, 414–420. [Google Scholar] [CrossRef]
- Mishra, J.; Ma, Q.; Prada, A.; Mitsnefes, M.; Zahedi, K.; Yang, J.; Barasch, J.; Devarajan, P. Identification of neutrophil gelatinase-associated lipocalin as a novel early urinary biomarker for ischemic renal injury. J. Am. Soc. Nephrol. 2003, 14, 2534–2543. [Google Scholar] [CrossRef] [PubMed]
- Devarajan, P. Emerging urinary biomarkers in the diagnosis of acute kidney injury. Expert Opin. Med. Diagn. 2008, 2, 387–398. [Google Scholar] [CrossRef]
- Mårtensson, J.; Bellomo, R. The rise and fall of NGAL in acute kidney injury. Blood Purif. 2014, 37, 304–310. [Google Scholar] [CrossRef] [PubMed]
- Abrahamson, M.; Alvarez-Fernandez, M.; Nathanson, C.-M. Cystatins. Biochem. Soc. Symp. 2003, 70, 179–199. [Google Scholar] [CrossRef]
- Shlipak, M.G.; Matsushita, K.; Ärnlöv, J.; Inker, L.A.; Katz, R.; Polkinghorne, K.R.; Rothenbacher, D.; Sarnak, M.J.; Astor, B.C.; Coresh, J.; et al. Sevoort; CKD Prognosis Consortium. Cystatin C versus creatinine in determining risk based on kidney function. N. Engl. J. Med. 2013, 369, 932–943. [Google Scholar] [CrossRef] [PubMed]
- Filler, G.; Bökenkamp, A.; Hofmann, W.; Bricon, T.L.; Martínez-Brú, C.; Grubb, A. Cystatin C as a marker of GFRhistory, indications, and future research. Clin. Biochem. 2005, 38, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Shardlow, A.; McIntyre, N.J.; Fraser, S.D.S.; Roderick, P.; Raftery, J.; Fluck, R.J.; McIntyre, C.W.; Taal, M.W. The clinical utility and cost impact of cystatin C measurement in the diagnosis and management of chronic kidney disease: A primary care cohort study. PLoS Med. 2017, 14, e1002400. [Google Scholar] [CrossRef] [PubMed]
- Rafouli-Stergiou, P.; Parissis, J.; Farmakis, D.; Bistola, V.; Nikolaou, M.; Vasiliadis, K.; Ikonomidis, I.; Kremastinos, D.; Lekakis, J.; Filippatos, G. Prognostic value of in-hospital change in cystatin C in patients with acutely decompensated heart failure and renal dysfunction. Int. J. Cardiol. 2015, 182, 74–76. [Google Scholar] [CrossRef]
- Zhang, Z.; Humphreys, B.D.; Bonventre, J.V. Shedding of the urinary biomarker kidney injury molecule-1 (KIM-1) is regulated by MAP kinases and juxtamembrane region. J. Am. Soc. Nephrol. 2007, 18, 2704–2714. [Google Scholar] [CrossRef]
- Song, J.; Yu, J.; Prayogo, G.W.; Cao, W.; Wu, Y.; Jia, Z.; Zhang, A. Understanding kidney injury molecule 1: A novel immune factor in kidney pathophysiology. Am. J. Transl. Res. 2019, 11, 1219–1229. [Google Scholar]
- Kaddourah, A.; Goldstein, S.L.; Basu, R.; Nehus, E.J.; Terrell, T.C.; Brunner, L.; Bennett, M.R.; Haffner, C.; Jefferies, J.L. Novel urinary tubular injury markers reveal an evidence of underlying kidney injury in children with reduced left ventricular systolic function: A pilot study. Pediatr. Nephrol. 2016, 31, 1637–1645. [Google Scholar] [CrossRef]
- Vaidya, V.S.; Ramirez, V.; Ichimura, T.; Bobadilla, N.A.; Bonventre, J.V. Urinary kidney injury molecule-1: A sensitive quantitative biomarker for early detection of kidney tubular injury. Am. J. Physiol. Physiol. 2006, 290, F517–F529. [Google Scholar] [CrossRef]
- Hashimoto, R.; Adachi, H.; Nishida, H.; Tsuruta, M.; Nomura, G. Serum N-acetyl-beta-D-glucosaminidase activity in predicting the development of hypertension. Hypertension 1995, 25, 1311–1314. [Google Scholar] [CrossRef] [PubMed]
- Liangos, O.; Perianayagam, M.C.; Vaidya, V.S.; Han, W.K.; Wald, R.; Tighiouart, H.; MacKinnon, R.W.; Li, L.; Balakrishnan, V.S.; Pereira, B.J.G.; et al. Urinary N-acetyl-beta-(D)-glucosaminidase activity and kidney injury molecule-1 level are associated with adverse outcomes in acute renal failure. J. Am. Soc. Nephrol. 2007, 18, 904–912. [Google Scholar] [CrossRef]
- Ronco, C.; McCullough, P.; Anker, S.D.; Anand, I.; Aspromonte, N.; Bagshaw, S.M.; Acute Dialysis Quality Initiative (ADQI) Consensus Group. Cardio-renal syndromes: Report from the consensus conference of the acute dialysis quality initiative. Eur. Heart J. 2010, 31, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Brisco, M.A.; Testani, J.M. Novel renal biomarkers to assess cardiorenal syndrome. Curr. Heart Fail. Rep. 2014, 11, 485–499. [Google Scholar] [CrossRef] [PubMed]
- Parikh, C.R.; Abraham, E.; Ancukiewicz, M.; Edelstein, C.L. Urine IL-18 is an early diagnostic marker for acute kidney injury and predicts mortality in the intensive care unit. J. Am. Soc. Nephrol. 2005, 16, 3046–3052. [Google Scholar] [CrossRef] [PubMed]
- Parikh, C.R.; Jani, A.; Melnikov, V.Y.; Faubel, S.; Edelstein, C.L. Urinary interleukin-18 is a marker of human acute tubular necrosis. Am. J. Kidney Dis. 2004, 43, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Gembillo, G.; Visconti, L.; Giusti, M.A.; Siligato, R.; Gallo, A.; Santoro, D.; Mattina, A. Cardiorenal Syndrome: New Pathways and Novel Biomarkers. Biomolecules 2021, 11, 1581. [Google Scholar] [CrossRef]
- Sharma, U.C.; Pokharel, S.; Van Brakel, T.J.; Van Berlo, J.H.; Cleutjens, J.P.M.; Schroen, B.; André, S.; Crijns, H.J.G.M.; Gabius, H.-J.; Maessen, J.; et al. Galectin-3 marks activated macrophages in failure-prone hypertrophied hearts and contributes to cardiac dysfunction. Circulation 2004, 110, 3121–3128. [Google Scholar] [CrossRef]
- Rubinstein, N.; Ilarregui, J.M.; Toscano, M.A.; Rabinovich, G.A. The role of galectins in the initiation, amplification and resolution of the inflammatory response. Tissue Antigens 2004, 64, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Goffredo, G.; Barone, R.; Di Terlizzi, V.; Correale, M.; Brunetti, N.D.; Iacoviello, M. Biomarkers in Cardiorenal Syndrome. J. Clin. Med. 2021, 10, 3433. [Google Scholar] [CrossRef] [PubMed]
- Iacoviello, M.; Di Serio, F.; Rizzo, C.; Leone, M.; Grande, D.; Guida, P.; Gioia, M.I.; Parisi, G.; Leopizzi, T.; Caldarola, P.; et al. Association between high Gal-3 serum levels and worsening of renal function in chronic heart failure outpatients. Biomark. Med. 2019, 13, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Cai, D.; Zhang, B. Clinical values of serum NGAL combined with NT-proBNP in the early prognosis of type 1 cardiorenal syndrome. Am. J. Transl. Res. 2021, 13, 3363–3368. [Google Scholar] [PubMed]
- Alvelos, M.; Pimentel, R.; Pinho, E.; Gomes, A.; Lourenço, P.; Teles, M.J.; Almeida, P.; Guimarães, J.T.; Bettencourt, P. Neutrophil gelatinase-associated lipocalin in the diagnosis of type 1 cardio-renal syndrome in the general ward. Clin. J. Am. Soc. Nephrol. 2011, 6, 476–481. [Google Scholar] [CrossRef]
- Chen, C.; Yang, X.; Lei, Y.; Zha, Y.; Liu, H.; Ma, C.; Tian, J.; Chen, P.; Yang, T.; Hou, F.F. Urinary Biomarkers at the Time of AKI Diagnosis as Predictors of Progression of AKI among Patients with Acute Cardiorenal Syndrome. Clin. J. Am. Soc. Nephrol. 2016, 11, 1536–1544. [Google Scholar] [CrossRef] [PubMed]
- Pinsino, A.; Mondellini, G.M.; Royzman, E.A.; Hoffman, K.L.; D’Angelo, D.; Mabasa, M.; Gaudig, A.; Zuver, A.M.; Masoumi, A.; Garan, A.R.; et al. Cystatin C-Versus Creatinine-Based Assessment of Renal Function and Prediction of Early Outcomes among Patients with a Left Ventricular Assist Device. Circ. Heart Fail. 2020, 13, e006326. [Google Scholar] [CrossRef]
- Ruan, Z.-B.; Zhu, L.; Yin, Y.-G.; Chen, G.-C. Cystatin C, N-terminal probrain natriuretic peptides and outcomes in acute heart failure with acute kidney injury in a 12-month follow-up: Insights into the cardiorenal syndrome. J. Res. Med. Sci. 2014, 19, 404–409. [Google Scholar]

| AKI | AKD | CKD | |
|---|---|---|---|
| Temporal Pattern | ≤7 days | ≤3 months | ≥3 months |
| Functional Criteria | Increase in serum creatinine ≥ 50% within 7 days, or increase ≥0.3 mg/dL within 2 days, or oliguria for ≥4 h. | The same as AKI, or GFR < 60 mL/min/1.73 m2, or decrease in GFR by ≥35% with respect to baseline, or increase in serum creatinine by ≥50% with respect to baseline. | GFR < 60 mL/min/1.73 m2 |
| Structural Criteria | Albuminuria, hematuria, acid–base and electrolyte disturbances or sediment abnormalities. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lisa, A.; Carbone, F.; Liberale, L.; Montecucco, F. The Need to Identify Novel Markers for Early Renal Injury in Cardiorenal Syndrome. Cells 2024, 13, 1283. https://doi.org/10.3390/cells13151283
Lisa A, Carbone F, Liberale L, Montecucco F. The Need to Identify Novel Markers for Early Renal Injury in Cardiorenal Syndrome. Cells. 2024; 13(15):1283. https://doi.org/10.3390/cells13151283
Chicago/Turabian StyleLisa, Anna, Federico Carbone, Luca Liberale, and Fabrizio Montecucco. 2024. "The Need to Identify Novel Markers for Early Renal Injury in Cardiorenal Syndrome" Cells 13, no. 15: 1283. https://doi.org/10.3390/cells13151283
APA StyleLisa, A., Carbone, F., Liberale, L., & Montecucco, F. (2024). The Need to Identify Novel Markers for Early Renal Injury in Cardiorenal Syndrome. Cells, 13(15), 1283. https://doi.org/10.3390/cells13151283