
Blocking Thromboxane-Prostanoid Receptor Signaling Attenuates Lipopolysaccharide- and Stearic Acid-Induced Inflammatory Response in Human PBMCs

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Human Subjects
2.3. PBMC Isolation and Culture
2.4. Real-Time Quantitative Polymerase Chain Reaction
2.5. Enzyme-Linked Immunosorbent Assay (ELISA)
2.6. Thromboxane B2 Assay
2.7. LDH Cytotoxicity Assay
2.8. Statistical Analysis
3. Results
3.1. TPR Expression Is Increased in PBMCs in Individuals with Obesity
3.2. Blockade of TPR Signaling Reduces Lipopolysaccharide (LPS)-Induced Pro-Inflammatory Response in Human PBMCs
3.3. Inhibition of TPR Attenuates LPS-Induced TXB2 Secretion
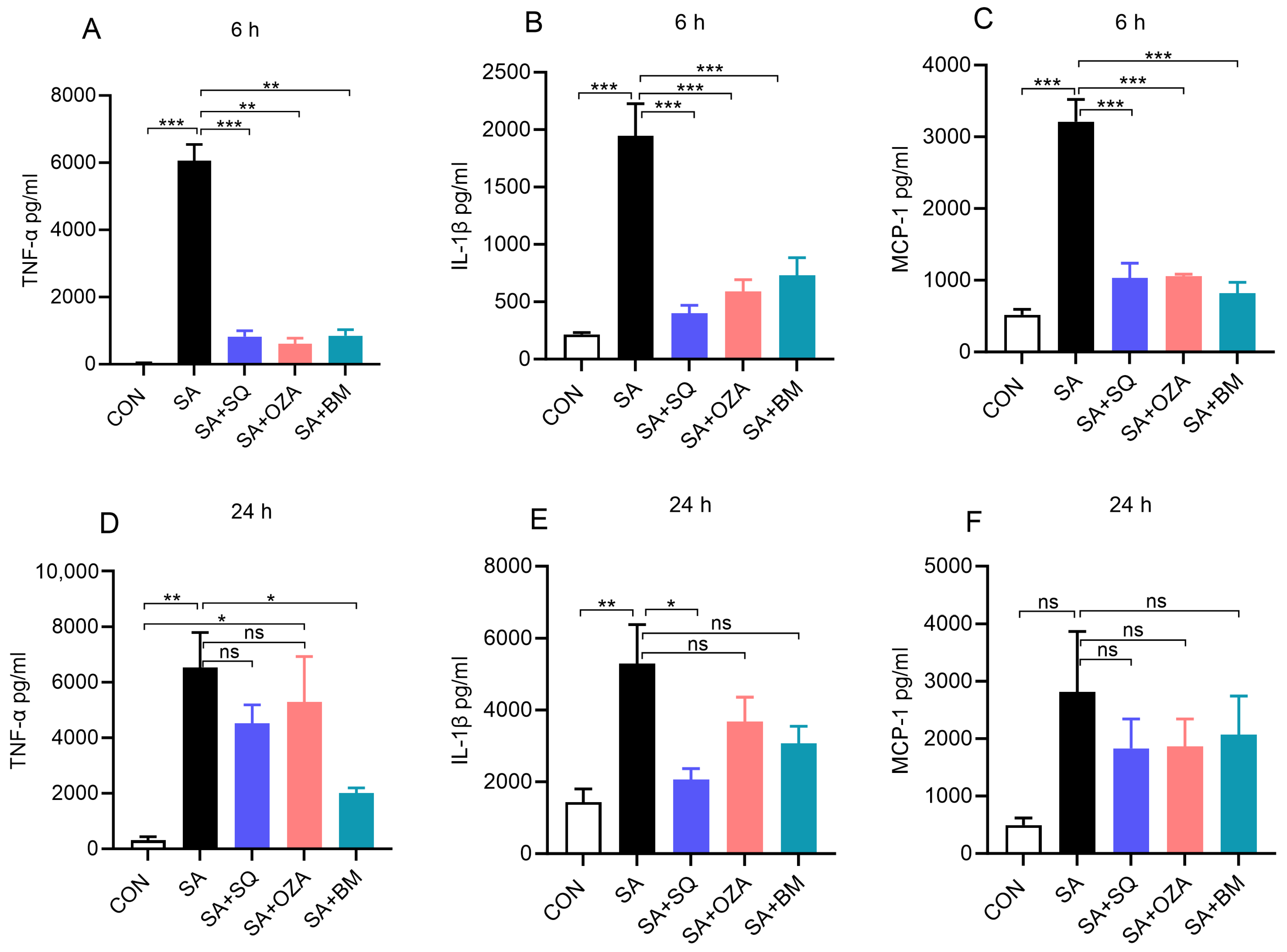
3.4. Blockade of TPR Signaling Reduces Stearic Acid-Induced Pro-Inflammatory Response in Human PBMCs
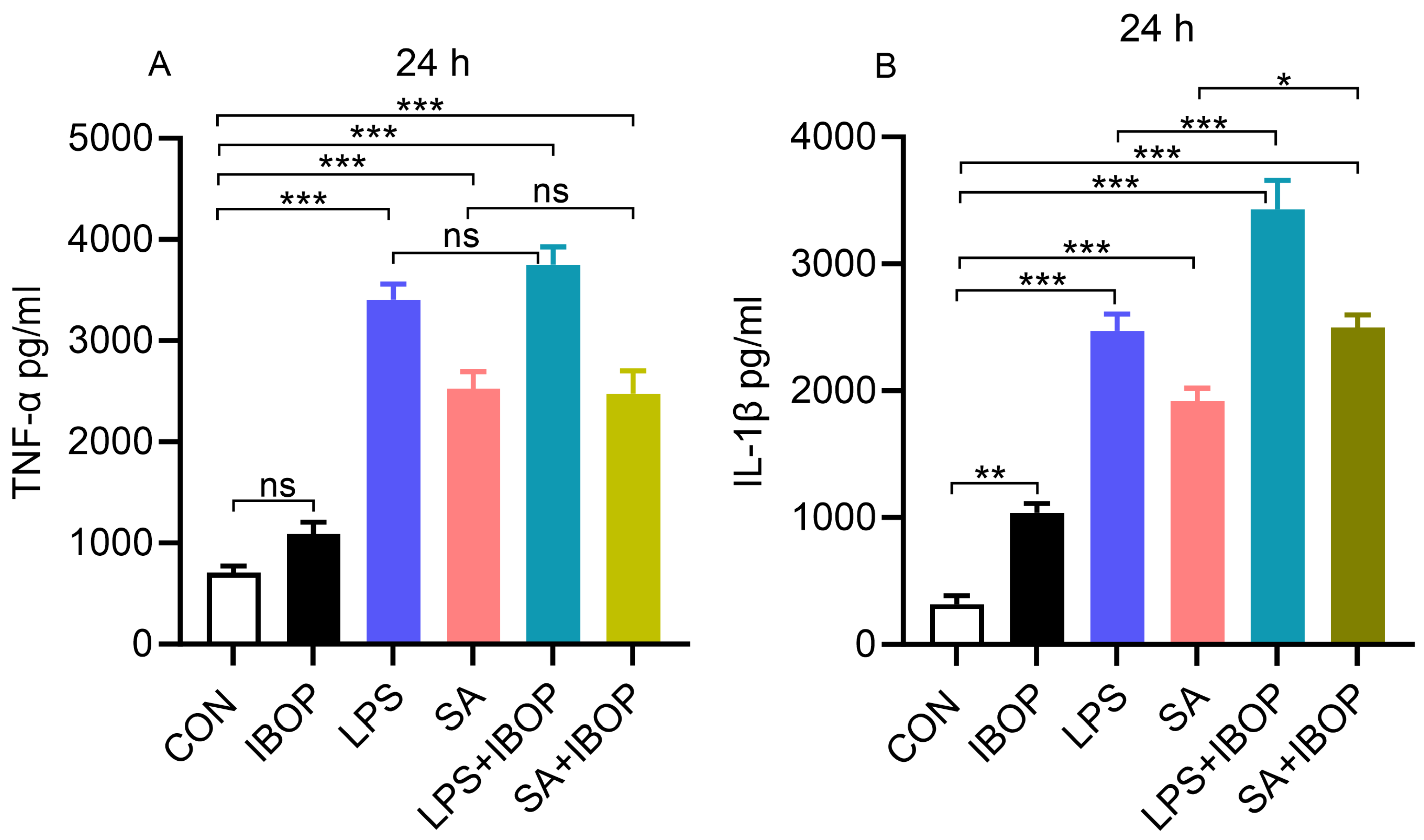
3.5. Activation of TPR Potentiates LPS- and SA-Induced Pro-Inflammatory Response in PBMCs
3.6. BM567, a Dual-Inhibitor, Attenuates TPR Agonist-Mediated Inflammatory Response in PBMCs
3.7. Inhibition of ROCK Activity Blocks IBOP-Induced Pro-Inflammatory Response in PBMCs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9(th) edition. Diabetes Res. Clin. Pract. 2019, 157, 107843. [Google Scholar] [CrossRef] [PubMed]
- Al-Goblan, A.S.; Al-Alfi, M.A.; Khan, M.Z. Mechanism linking diabetes mellitus and obesity. Diabetes Metab. Syndr. Obes. Targets Ther. 2014, 7, 587–591. [Google Scholar] [CrossRef] [PubMed]
- Ellulu, M.S.; Patimah, I.; Khaza’ai, H.; Rahmat, A.; Abed, Y. Obesity and inflammation: The linking mechanism and the complications. Arch. Med. Sci. 2017, 13, 851–863. [Google Scholar] [CrossRef] [PubMed]
- Rucker, D.; Dhamoon, A.S. Physiology, Thromboxane A2. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Cherdon, C.; Rolin, S.; Hanson, J.; Ooms, A.; de Leval, L.; Drion, P.; Michiels, C.; Pirotte, B.; Masereel, B.; Sakalihassan, N.; et al. BM-573 inhibits the development of early atherosclerotic lesions in Apo E deficient mice by blocking TP receptors and thromboxane synthase. Prostaglandins Other Lipid Mediat. 2011, 94, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Petri, M.H.; Tellier, C.; Michiels, C.; Ellertsen, I.; Dogne, J.M.; Back, M. Effects of the dual TP receptor antagonist and thromboxane synthase inhibitor EV-077 on human endothelial and vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 2013, 441, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.W.; Lien, J.C.; Kuo, S.C.; Huang, T.F. Inhibitory Effects of an Orally Active Thromboxane A2 Receptor Antagonist, nstpbp5185, on Atherosclerosis in ApoE-Deficient Mice. Thromb. Haemost. 2018, 118, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Korneszczuk, K.; Karaa, A.; Lin, T.; Clemens, M.G.; Zhang, J.X. Thromboxane A2 from Kupffer cells contributes to the hyperresponsiveness of hepatic portal circulation to endothelin-1 in endotoxemic rats. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 288, G277–G283. [Google Scholar] [CrossRef] [PubMed]
- Collins, B.J.; Blum, M.G.; Parker, R.E.; Chang, A.C.; Blair, K.S.; Zorn, G.L., 3rd; Christman, B.W.; Pierson, R.N., 3rd. Thromboxane mediates pulmonary hypertension and lung inflammation during hyperacute lung rejection. J. Appl. Physiol. 2001, 90, 2257–2268. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, H.; Ito, Y.; Hosono, K.; Tsuru, S.; Inoue, T.; Nakamoto, S.; Kurashige, C.; Hirashima, M.; Narumiya, S.; Okamoto, H.; et al. Roles of Thromboxane Receptor Signaling in Enhancement of Lipopolysaccharide-Induced Lymphangiogenesis and Lymphatic Drainage Function in Diaphragm. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 1390–1407. [Google Scholar] [CrossRef]
- Hartney, J.M.; Gustafson, C.E.; Bowler, R.P.; Pelanda, R.; Torres, R.M. Thromboxane receptor signaling is required for fibronectin-induced matrix metalloproteinase 9 production by human and murine macrophages and is attenuated by the Arhgef1 molecule. J. Biol. Chem. 2011, 286, 44521–44531. [Google Scholar] [CrossRef]
- Altavilla, D.; Squadrito, F.; Canale, P.; Ioculano, M.; Squadrito, G.; Campo, G.M.; Serrano, M.; Sardella, A.; Urna, G.; Spignoli, G.; et al. G 619, a dual thromboxane synthase inhibitor and thromboxane A2 receptor antagonist, inhibits tumor necrosis factor-α biosynthesis. Eur. J. Pharmacol. 1995, 286, 31–39. [Google Scholar] [CrossRef]
- Yan, A.; Cai, G.; Xia, W.; Fu, Y. Thromboxane A2 receptor antagonist SQ29548 suppresses the LPS-induced release of inflammatory cytokines in BV2 microglia cells via suppressing MAPK and NF-κB signaling pathways. Mol. Med. Rep. 2017, 16, 2491–2496. [Google Scholar] [CrossRef]
- Yang, W.; Yan, A.; Zhang, T.; Shao, J.; Liu, T.; Yang, X.; Xia, W.; Fu, Y. Thromboxane A2 Receptor Stimulation Enhances Microglial Interleukin-1β and NO Biosynthesis Mediated by the Activation of ERK Pathway. Front. Aging Neurosci. 2016, 8, 8. [Google Scholar] [CrossRef]
- Hersoug, L.G.; Moller, P.; Loft, S. Role of microbiota-derived lipopolysaccharide in adipose tissue inflammation, adipocyte size and pyroptosis during obesity. Nutr. Res. Rev. 2018, 31, 153–163. [Google Scholar] [CrossRef]
- Ngkelo, A.; Meja, K.; Yeadon, M.; Adcock, I.; Kirkham, P.A. LPS induced inflammatory responses in human peripheral blood mononuclear cells is mediated through NOX4 and Giα dependent PI-3kinase signalling. J. Inflamm. 2012, 9, 1. [Google Scholar] [CrossRef]
- Rocha, D.M.; Caldas, A.P.; Oliveira, L.L.; Bressan, J.; Hermsdorff, H.H. Saturated fatty acids trigger TLR4-mediated inflammatory response. Atherosclerosis 2016, 244, 211–215. [Google Scholar] [CrossRef]
- Henderson, G.C. Plasma Free Fatty Acid Concentration as a Modifiable Risk Factor for Metabolic Disease. Nutrients 2021, 13, 2590. [Google Scholar] [CrossRef]
- Zhou, H.; Urso, C.J.; Jadeja, V. Saturated Fatty Acids in Obesity-Associated Inflammation. J. Inflamm. Res. 2020, 13, 1–14. [Google Scholar] [CrossRef]
- Ueki, K.; Kondo, T.; Kahn, C.R. Suppressor of cytokine signaling 1 (SOCS-1) and SOCS-3 cause insulin resistance through inhibition of tyrosine phosphorylation of insulin receptor substrate proteins by discrete mechanisms. Mol. Cell Biol. 2004, 24, 5434–5446. [Google Scholar] [CrossRef]
- Shi, H.; Kokoeva, M.V.; Inouye, K.; Tzameli, I.; Yin, H.; Flier, J.S. TLR4 links innate immunity and fatty acid-induced insulin resistance. J. Clin. Investig. 2006, 116, 3015–3025. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Barnes, G.T.; Yang, Q.; Tan, G.; Yang, D.; Chou, C.J.; Sole, J.; Nichols, A.; Ross, J.S.; Tartaglia, L.A.; et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Investig. 2003, 112, 1821–1830. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.C.; Smith, G.I.; Palacios, H.H.; Farabi, S.S.; Yoshino, M.; Yoshino, J.; Cho, K.; Davila-Roman, V.G.; Shankaran, M.; Barve, R.A.; et al. Cardiometabolic characteristics of people with metabolically healthy and unhealthy obesity. Cell Metab. 2024, 36, 745–761.e5. [Google Scholar] [CrossRef] [PubMed]
- Stepien, M.; Stepien, A.; Wlazel, R.N.; Paradowski, M.; Banach, M.; Rysz, J. Obesity indices and inflammatory markers in obese non-diabetic normo- and hypertensive patients: A comparative pilot study. Lipids Health Dis. 2014, 13, 29. [Google Scholar] [CrossRef]
- Hishinuma, T.; Tsukamoto, H.; Suzuki, K.; Mizugaki, M. Relationship between thromboxane/prostacyclin ratio and diabetic vascular complications. Prostaglandins Leukot. Essent. Fat. Acids 2001, 65, 191–196. [Google Scholar] [CrossRef]
- Lasserre, B.; Navarro-Delmasure, C.; Pham Huu Chanh, A.; Catala, J.; Hollande, E. Modifications in the TXA(2) and PGI(2) plasma levels and some other biochemical parameters during the initiation and development of non-insulin-dependent diabetes mellitus (NIDDM) syndrome in the rabbit. Prostaglandins Leukot. Essent. Fat. Acids 2000, 62, 285–291. [Google Scholar] [CrossRef]
- Li, R.H.L.; Nguyen, N.; Tablin, F. Canine platelets express functional Toll-like receptor-4: Lipopolysaccharide-triggered platelet activation is dependent on adenosine diphosphate and thromboxane A2 in dogs. BMC Vet. Res. 2019, 15, 245. [Google Scholar] [CrossRef]
- Xie, X.; Sun, W.; Wang, J.; Li, X.; Liu, X.; Liu, N. Activation of thromboxane A2 receptors mediates endothelial dysfunction in diabetic mice. Clin. Exp. Hypertens. 2017, 39, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Hu, J.; Gao, X.; Liang, H.; Yu, H.; Liu, S.; Liu, Z. Hyperglycemia via activation of thromboxane A2 receptor impairs the integrity and function of blood-brain barrier in microvascular endothelial cells. Oncotarget 2017, 8, 30030–30038. [Google Scholar] [CrossRef]
- He, J.; Zhou, Y.; Xing, J.; Wang, Q.; Zhu, H.; Zhu, Y.; Zou, M.H. Liver kinase B1 is required for thromboxane receptor-dependent nuclear factor-κB activation and inflammatory responses. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1297–1305. [Google Scholar] [CrossRef]
- Stinson, M.W.; Liu, S.; Laurenson, A.J.; Rotty, J.D. Macrophage migration is differentially regulated by fibronectin and laminin through altered adhesion and myosin II localization. Mol. Biol. Cell 2024, 35, ar22. [Google Scholar] [CrossRef] [PubMed]
- Autissier, P.; Soulas, C.; Burdo, T.H.; Williams, K.C. Evaluation of a 12-color flow cytometry panel to study lymphocyte, monocyte, and dendritic cell subsets in humans. Cytom. Part A 2010, 77, 410–419. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.W.; Rocha, P.N.; Nataraj, C.; Robinson, L.A.; Spurney, R.F.; Koller, B.H.; Coffman, T.M. Proinflammatory actions of thromboxane receptors to enhance cellular immune responses. J. Immunol. 2003, 171, 6389–6395. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Oosting, M.; Deelen, P.; Ricano-Ponce, I.; Smeekens, S.; Jaeger, M.; Matzaraki, V.; Swertz, M.A.; Xavier, R.J.; Franke, L.; et al. Inter-individual variability and genetic influences on cytokine responses to bacteria and fungi. Nat. Med. 2016, 22, 952–960. [Google Scholar] [CrossRef] [PubMed]
- Capra, V.; Back, M.; Angiolillo, D.J.; Cattaneo, M.; Sakariassen, K.S. Impact of vascular thromboxane prostanoid receptor activation on hemostasis, thrombosis, oxidative stress, and inflammation. J. Thromb. Haemost. 2014, 12, 126–137. [Google Scholar] [CrossRef]
- Dogne, J.M.; de Leval, X.; Benoit, P.; Rolin, S.; Pirotte, B.; Masereel, B. Therapeutic potential of thromboxane inhibitors in asthma. Expert Opin. Investig. Drugs 2002, 11, 275–281. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Measurement | Normal (n = 15) | Obesity (n = 14) | p Value |
|---|---|---|---|
| Age (years) | 40.20 ± 4.27 | 47.43 ± 2.22 | 0.1537 |
| Weight (lbs) | 168.7 ± 5.80 | 241.1 ± 7.15 | <0.0001 |
| Body mass index (kg/m2) | 24.24 ± 0.61 | 34.79 ± 0.96 | <0.0001 |
| Insulin (µIU/mL) | 4.53 ± 1.02 | 12.85 ± 3.64 | 0.0275 |
| Glucose (mg/dL) | 90.00 ± 1.60 | 92.00 ± 2.67 | 0.5206 |
| HOMA-IR | 1.01 ± 0.23 | 2.88 ± 0.71 | 0.0144 |
| Total cholesterol (mg/dL) | 197.4 ± 6.66 | 208.3 ± 14.54 | 0.4922 |
| Triglycerides (mg/dL) | 114.8 ± 18.23 | 168.8 ± 42.18 | 0.2394 |
| LDL cholesterol (mg/dL) | 121.3 ± 6.92 | 140.5 ± 13.21 | 0.1941 |
| HDL cholesterol (mg/dL) | 53.13 ± 3.65 | 41.07 ± 2.67 | 0.0140 |
| Creatinine (mg/dL) | 1.05 ± 0.03 | 1.02 ± 0.05 | 0.7158 |
| ALT (U/L) | 17.80 ± 2.88 | 38.07 ± 13.29 | 0.1353 |
| AST (U/L) | 31.00 ± 2.12 | 34.64 ± 4.51 | 0.4616 |
| HA1C (%) | 5.43 ± 0.51 | 5.77 ± 1.85 | 0.0063 |
| GFR (mL/min) | 82.87 ± 4.08 | 82.36 ± 5.03 | 0.9375 |
| CRP (mg/L) | 2.69 ± 1.03 | 2.94 ± 0.68 | 0.8442 |
| Fat mass (%) | 19.53 ± 1.14 | 35.51 ± 1.32 | <0.0001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rajamanickam, V.; Desouza, C.V.; Castillo, R.T.; Saraswathi, V. Blocking Thromboxane-Prostanoid Receptor Signaling Attenuates Lipopolysaccharide- and Stearic Acid-Induced Inflammatory Response in Human PBMCs. Cells 2024, 13, 1320. https://doi.org/10.3390/cells13161320
Rajamanickam V, Desouza CV, Castillo RT, Saraswathi V. Blocking Thromboxane-Prostanoid Receptor Signaling Attenuates Lipopolysaccharide- and Stearic Acid-Induced Inflammatory Response in Human PBMCs. Cells. 2024; 13(16):1320. https://doi.org/10.3390/cells13161320
Chicago/Turabian StyleRajamanickam, Vinothkumar, Cyrus V. Desouza, Romilia T. Castillo, and Viswanathan Saraswathi. 2024. "Blocking Thromboxane-Prostanoid Receptor Signaling Attenuates Lipopolysaccharide- and Stearic Acid-Induced Inflammatory Response in Human PBMCs" Cells 13, no. 16: 1320. https://doi.org/10.3390/cells13161320