
Activation of VGLL4 Suppresses Cardiomyocyte Maturational Hypertrophic Growth
 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Animals
2.2. Adenoviruses
2.3. Neonatal Rat Ventricular Myocytes (NRVMs)
2.4. AAV Transduction and Echocardiography Measurement
2.5. Seahorse XF Mito Stress Test
2.6. Statistics
3. Results
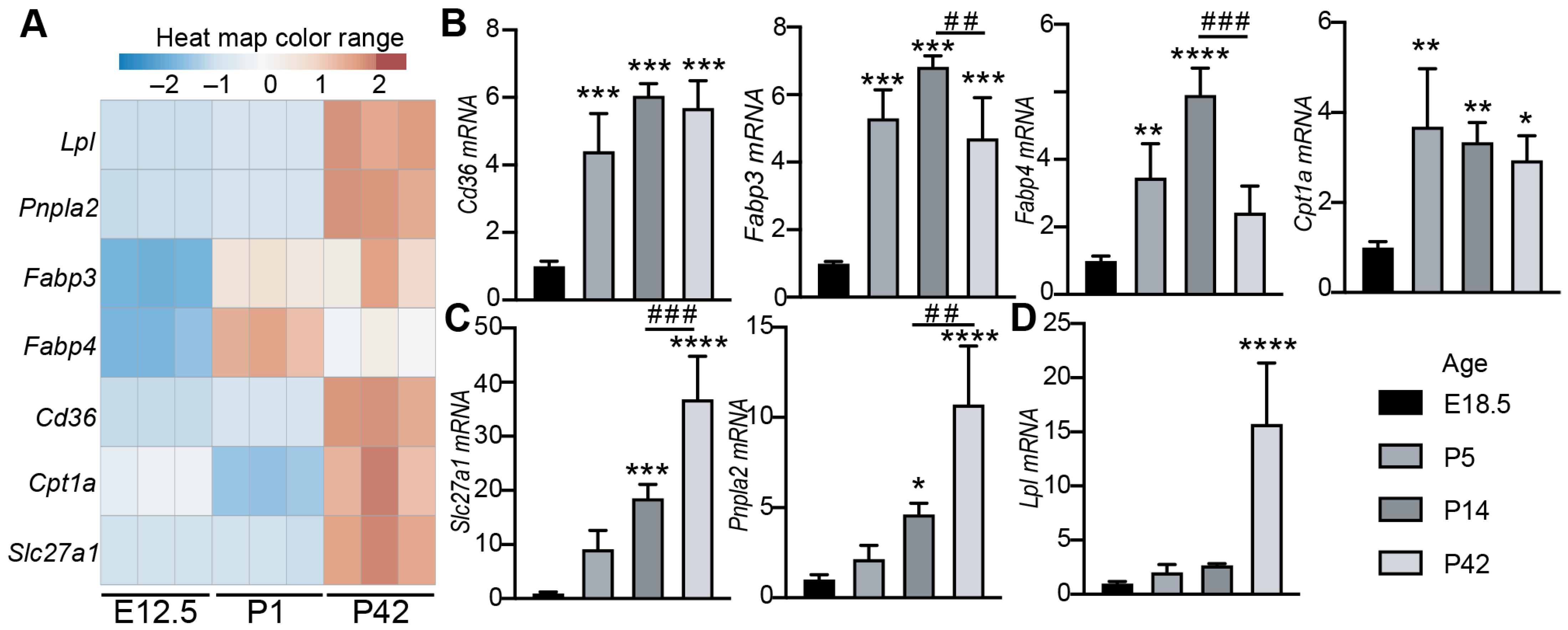
3.1. The Expression of Cardiac Lipid Metabolic Genes Increases with Age
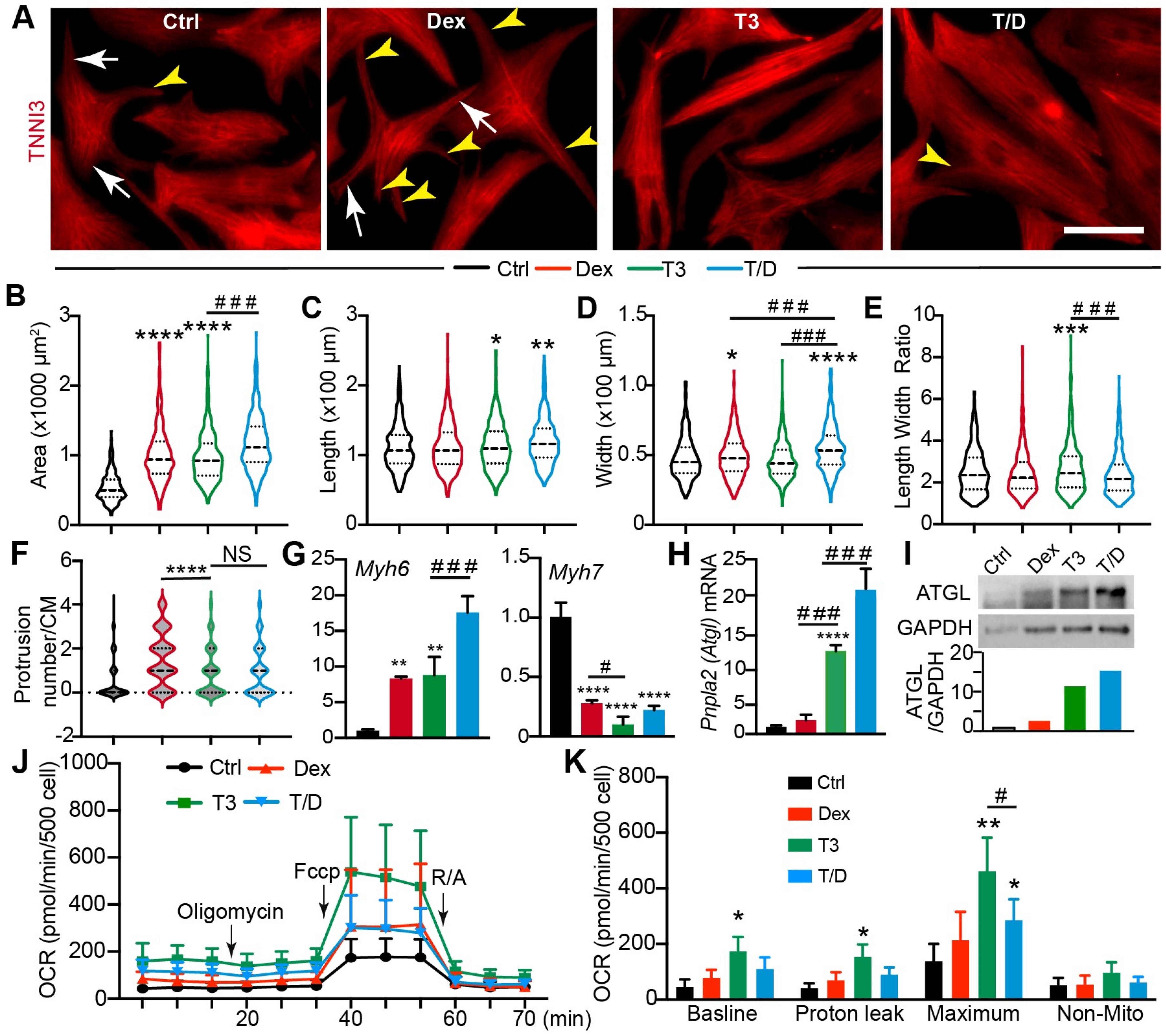
3.2. T3/Dex Treatment Promotes CM Maturational Hypertrophy
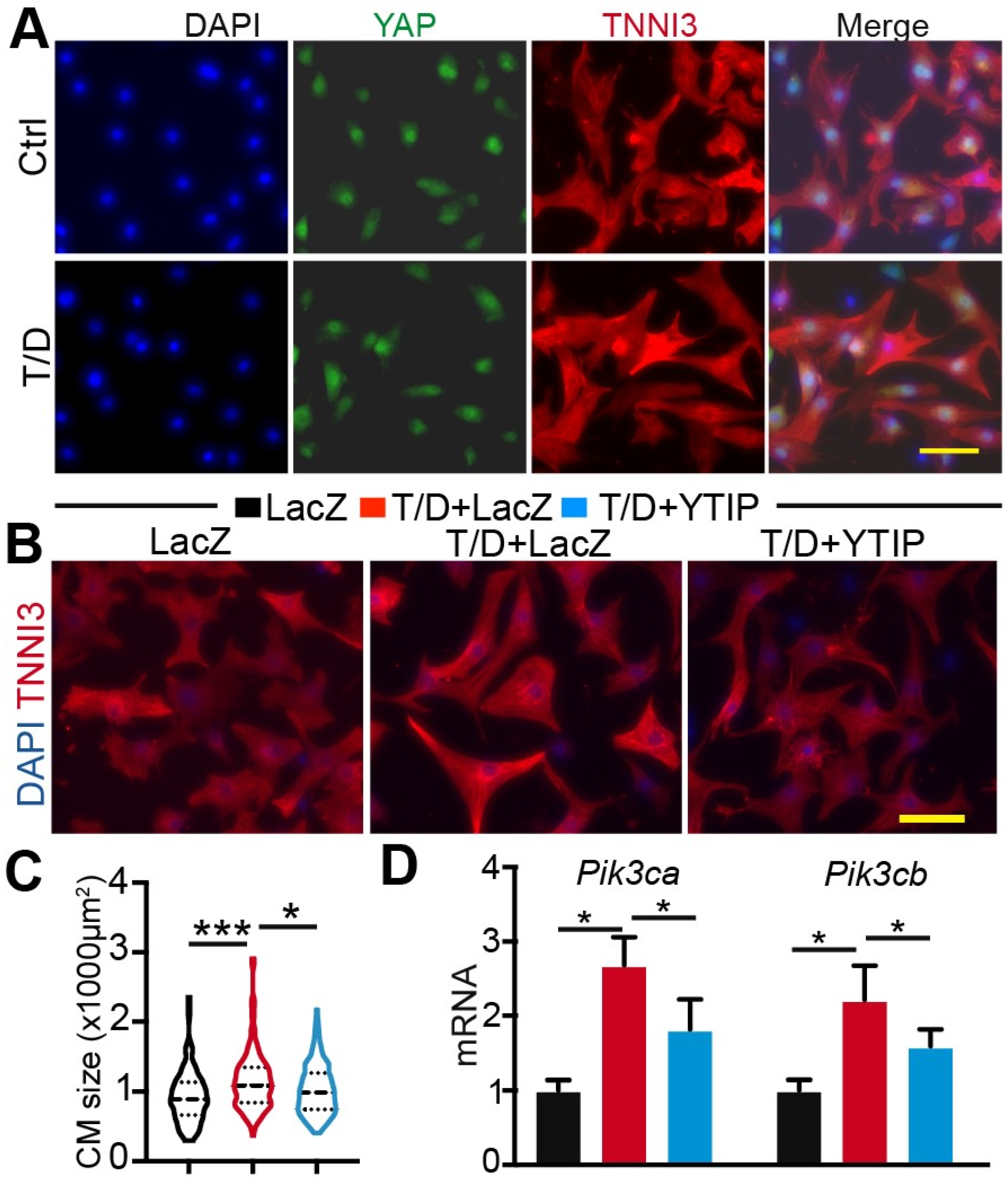
3.3. Reducing YAP/TEAD Complex Activity Attenuates CM Maturational Hypertrophy
3.4. Activation of VGLL4 Suppresses CM Maturational Hypertrophy In Vitro
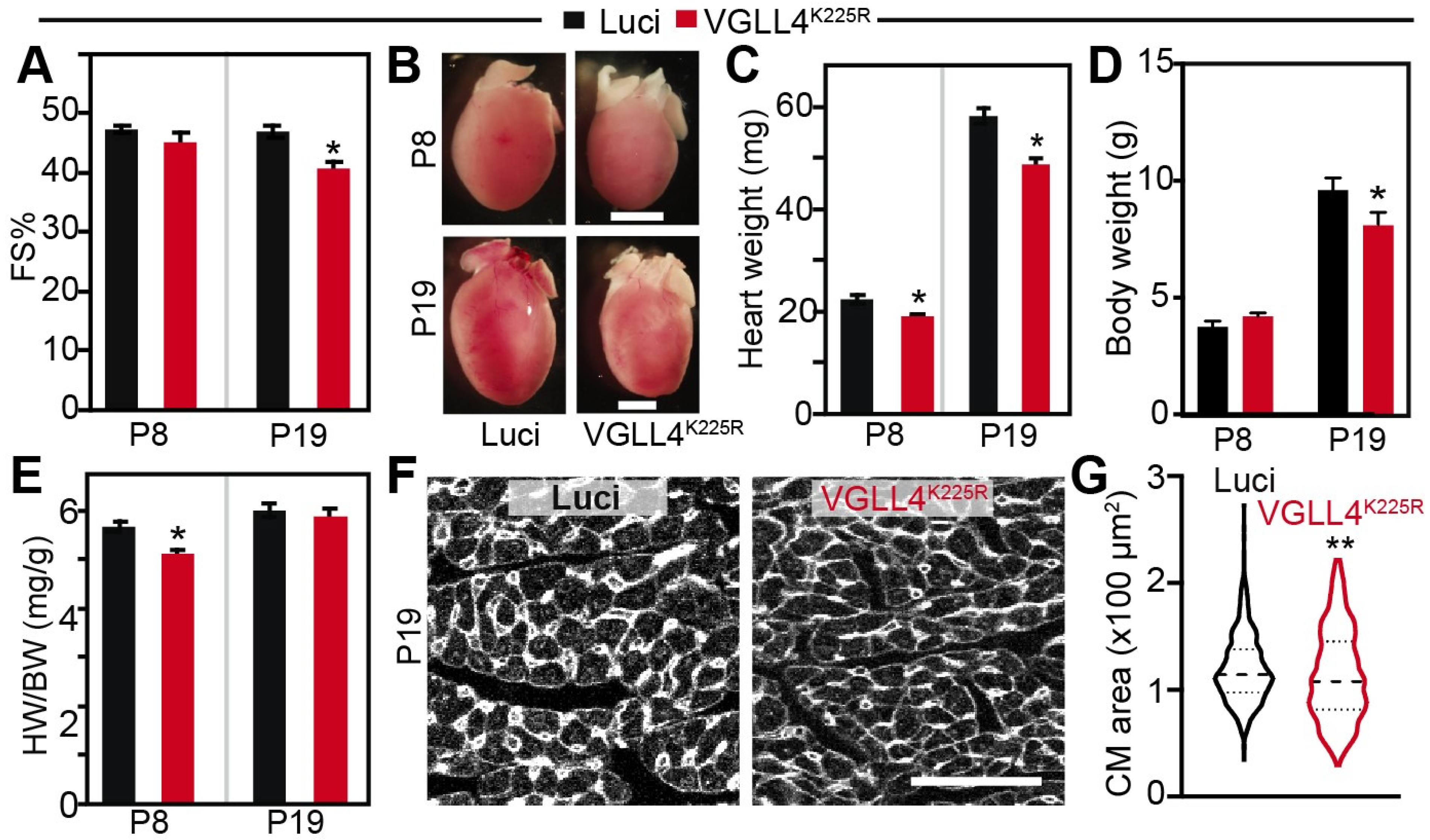
3.5. VGLL4 Suppresses CM Maturational Hypertrophic Growth In Vivo
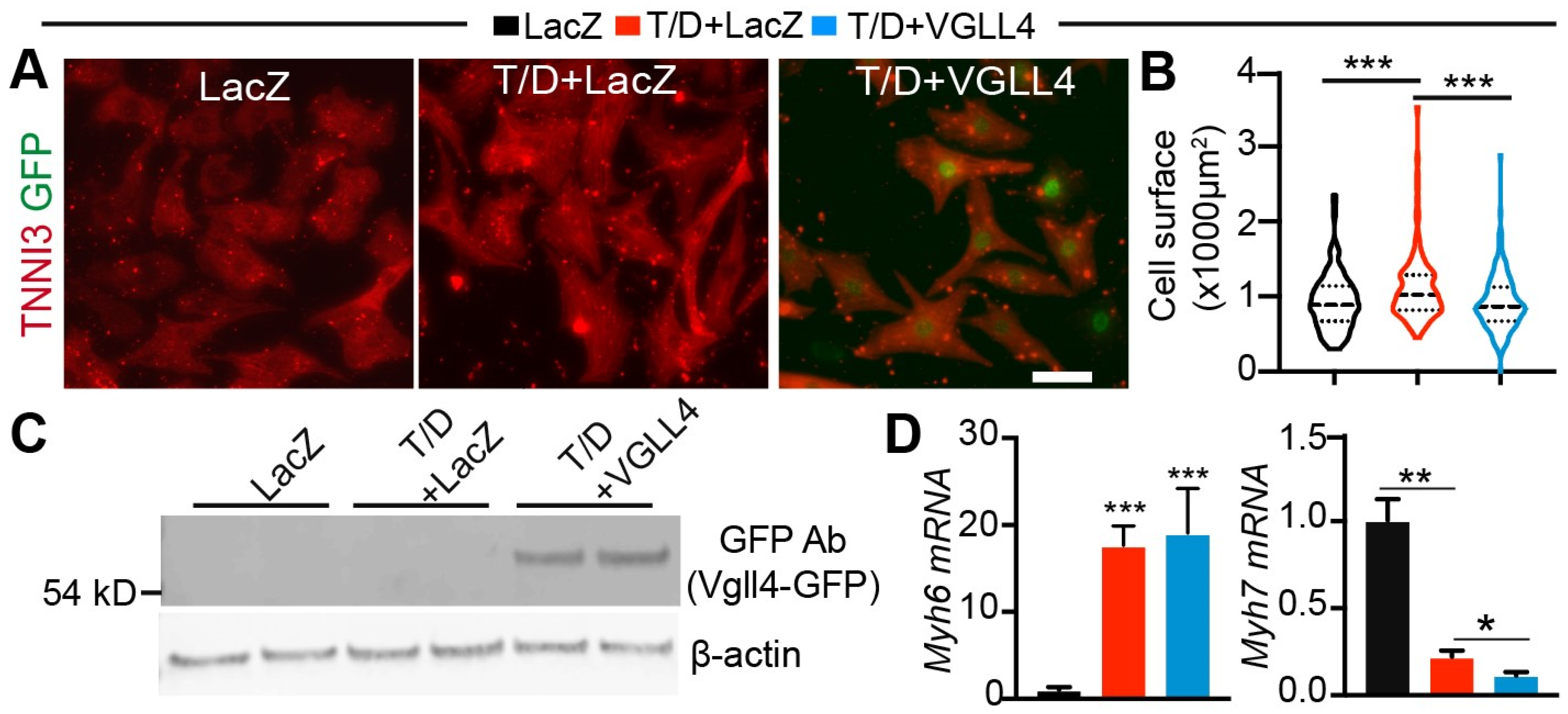
3.6. Activation of VGLL4 Suppresses CM Hypertrophic Growth through TEAD
3.7. VGLL4 Suppresses PI3K-AKT Pathway
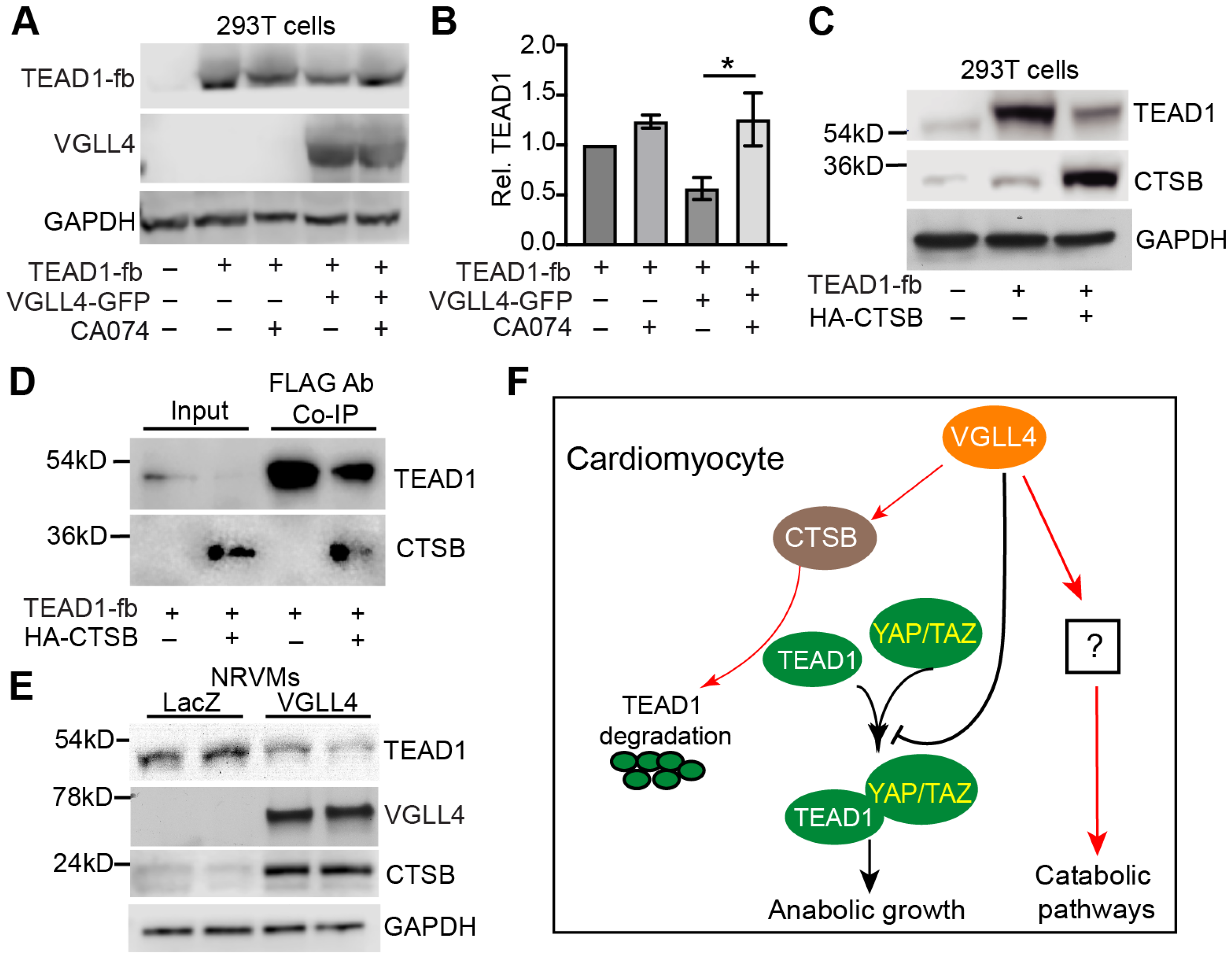
3.8. VGLL4 Induces TEAD1 Degradation through CTSB
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Von Gise, A.; Lin, Z.; Schlegelmilch, K.; Honor, L.B.; Pan, G.M.; Buck, J.N.; Ma, Q.; Ishiwata, T.; Zhou, B.; Camargo, F.D.; et al. YAP1, the nuclear target of Hippo signaling, stimulates heart growth through cardiomyocyte proliferation but not hypertrophy. Proc. Natl. Acad. Sci. USA 2012, 109, 2394–2399. [Google Scholar] [CrossRef] [PubMed]
- Maillet, M.; van Berlo, J.H.; Molkentin, J.D. Molecular basis of physiological heart growth: Fundamental concepts and new players. Nat. Rev. Mol. Cell Biol. 2013, 14, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Rog-Zielinska, E.A.; Thomson, A.; Kenyon, C.J.; Brownstein, D.G.; Moran, C.M.; Szumska, D.; Michailidou, Z.; Richardson, J.; Owen, E.; Watt, A.; et al. Glucocorticoid receptor is required for foetal heart maturation. Hum. Mol. Genet. 2013, 22, 3269–3282. [Google Scholar] [CrossRef] [PubMed]
- Chattergoon, N.N.; Giraud, G.D.; Louey, S.; Stork, P.; Fowden, A.L.; Thornburg, K.L. Thyroid hormone drives fetal cardiomyocyte maturation. FASEB J. 2012, 26, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Li, L.; Zhao, B.; Guan, K.L. The hippo pathway in heart development, regeneration, and diseases. Circ. Res. 2015, 116, 1431–1447. [Google Scholar] [CrossRef] [PubMed]
- Mauviel, A.; Nallet-Staub, F.; Varelas, X. Integrating developmental signals: A Hippo in the (path)way. Oncogene 2012, 31, 1743–1756. [Google Scholar] [CrossRef] [PubMed]
- Koontz, L.M.; Liu-Chittenden, Y.; Yin, F.; Zheng, Y.; Yu, J.; Huang, B.; Chen, Q.; Wu, S.; Pan, D. The Hippo effector Yorkie controls normal tissue growth by antagonizing scalloped-mediated default repression. Dev. Cell 2013, 25, 388–401. [Google Scholar] [CrossRef]
- Lin, Z.; Guo, H.; Cao, Y.; Zohrabian, S.; Zhou, P.; Ma, Q.; VanDusen, N.; Guo, Y.; Zhang, J.; Stevens, S.M.; et al. Acetylation of VGLL4 Regulates Hippo-YAP Signaling and Postnatal Cardiac Growth. Dev. Cell 2016, 39, 466–479. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, C.; Farley, A.; Ma, Q.; Pu, W.T.; Lin, Z. Depletion of VGLL4 Causes Perinatal Lethality without Affecting Myocardial Development. Cells 2022, 11, 2832. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Zhou, P.; von Gise, A.; Gu, F.; Ma, Q.; Chen, J.; Guo, H.; van Gorp, P.R.; Wang, D.Z.; Pu, W.T. Pi3kcb links Hippo-YAP and PI3K-AKT signaling pathways to promote cardiomyocyte proliferation and survival. Circ. Res. 2015, 116, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Xin, M.; Kim, Y.; Sutherland, L.B.; Qi, X.; McAnally, J.; Schwartz, R.J.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Regulation of insulin-like growth factor signaling by Yap governs cardiomyocyte proliferation and embryonic heart size. Sci. Signal 2011, 4, ra70. [Google Scholar] [CrossRef] [PubMed]
- Shioi, T.; Kang, P.M.; Douglas, P.S.; Hampe, J.; Yballe, C.M.; Lawitts, J.; Cantley, L.C.; Izumo, S. The conserved phosphoinositide 3-kinase pathway determines heart size in mice. EMBO J. 2000, 19, 2537–2548. [Google Scholar] [CrossRef] [PubMed]
- Nasci, V.L.; Chuppa, S.; Griswold, L.; Goodreau, K.A.; Dash, R.K.; Kriegel, A.J. miR-21-5p regulates mitochondrial respiration and lipid content in H9C2 cells. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H710–H721. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Pu, W.T. Cardiomyocyte Maturation: New Phase in Development. Circ. Res. 2020, 126, 1086–1106. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, A.; Alrob, O.A.; Zhang, L.; Wagg, C.S.; Altamimi, T.; Rawat, S.; Rebeyka, I.M.; Kantor, P.F.; Lopaschuk, G.D. Acetylation and succinylation contribute to maturational alterations in energy metabolism in the newborn heart. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H347–H363. [Google Scholar] [CrossRef] [PubMed]
- Akerberg, B.N.; Gu, F.; VanDusen, N.J.; Zhang, X.; Dong, R.; Li, K.; Zhang, B.; Zhou, B.; Sethi, I.; Ma, Q.; et al. A reference map of murine cardiac transcription factor chromatin occupancy identifies dynamic and conserved enhancers. Nat. Commun. 2019, 10, 4907. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, R.E.; Tokuyama, T.; Anzai, T.; Chanthra, N.; Uosaki, H. Sarcomere maturation: Function acquisition, molecular mechanism, and interplay with other organelles. Philos. Trans. R. Soc. B 2022, 377, 20210325. [Google Scholar] [CrossRef] [PubMed]
- Paul, D.M.; Morris, E.P.; Kensler, R.W.; Squire, J.M. Structure and orientation of troponin in the thin filament. J. Biol. Chem. 2009, 284, 15007–15015. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Pi, Y.; Lee, K.J.; Henkel, A.S.; Gregg, R.G.; Powers, P.A.; Walker, J.W. Cardiac troponin I gene knockout: A mouse model of myocardial troponin I deficiency. Circ. Res. 1999, 84, 1–8. [Google Scholar] [CrossRef]
- Sasse, S.; Brand, N.J.; Kyprianou, P.; Dhoot, G.K.; Wade, R.; Arai, M.; Periasamy, M.; Yacoub, M.H.; Barton, P.J. Troponin I gene expression during human cardiac development and in end-stage heart failure. Circ. Res. 1993, 72, 932–938. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Wu, B.; Feng, X.; Cheng, W.; Kitsis, R.N.; Zhou, B. Cardiac Myosin Heavy Chain Reporter Mice to Study Heart Development and Disease. Circ. Res. 2022, 131, 364–366. [Google Scholar] [CrossRef] [PubMed]
- Miyata, S.; Minobe, W.; Bristow, M.R.; Leinwand, L.A. Myosin heavy chain isoform expression in the failing and nonfailing human heart. Circ. Res. 2000, 86, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Hirose, K.; Payumo, A.Y.; Cutie, S.; Hoang, A.; Zhang, H.; Guyot, R.; Lunn, D.; Bigley, R.B.; Yu, H.; Wang, J.; et al. Evidence for hormonal control of heart regenerative capacity during endothermy acquisition. Science 2019, 364, 184–188. [Google Scholar] [CrossRef] [PubMed]
- Parikh, S.S.; Blackwell, D.J.; Gomez-Hurtado, N.; Frisk, M.; Wang, L.; Kim, K.; Dahl, C.P.; Fiane, A.; Tønnessen, T.; Kryshtal, D.O.; et al. Thyroid and Glucocorticoid Hormones Promote Functional T-Tubule Development in Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Circ. Res. 2017, 121, 1323–1330. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Martin, J.F. The regulation and function of the Hippo pathway in heart regeneration. Wiley Interdiscip. Rev. Dev. Biol. 2019, 8, e335. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Jagannathan, R.; Li, F.; Lee, J.; Balasubramanyam, N.; Kim, B.S.; Yang, P.; Yechoor, V.K.; Moulik, M. Tead1 is required for perinatal cardiomyocyte proliferation. PLoS ONE 2019, 14, e0212017. [Google Scholar] [CrossRef] [PubMed]
- Tsika, R.W.; Ma, L.; Kehat, I.; Schramm, C.; Simmer, G.; Morgan, B.; Fine, D.M.; Hanft, L.M.; McDonald, K.S.; Molkentin, J.D.; et al. TEAD-1 overexpression in the mouse heart promotes an age-dependent heart dysfunction. J. Biol. Chem. 2010, 285, 13721–13735. [Google Scholar] [CrossRef] [PubMed]
- Xin, M.; Kim, Y.; Sutherland, L.B.; Murakami, M.; Qi, X.; McAnally, J.; Porrello, E.R.; Mahmoud, A.I.; Tan, W.; Shelton, J.M.; et al. Hippo pathway effector Yap promotes cardiac regeneration. Proc. Natl. Acad. Sci. USA 2013, 110, 13839–13844. [Google Scholar] [CrossRef] [PubMed]
- Aoyagi, T.; Matsui, T. Phosphoinositide-3 kinase signaling in cardiac hypertrophy and heart failure. Curr. Pharm. Des. 2011, 17, 1818–1824. [Google Scholar] [CrossRef] [PubMed]
- Porrello, E.R.; Mahmoud, A.I.; Simpson, E.; Hill, J.A.; Richardson, J.A.; Olson, E.N.; Sadek, H.A. Transient regenerative potential of the neonatal mouse heart. Science 2011, 331, 1078–1080. [Google Scholar] [CrossRef] [PubMed]
- Jiao, S.; Wang, H.; Shi, Z.; Dong, A.; Zhang, W.; Song, X.; He, F.; Wang, Y.; Zhang, Z.; Wang, W.; et al. A peptide mimicking VGLL4 function acts as a YAP antagonist therapy against gastric cancer. Cancer Cell 2014, 25, 166–180. [Google Scholar] [CrossRef]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Osaki, M.; Oshimura, M.; Ito, H. PI3K-Akt pathway: Its functions and alterations in human cancer. Apoptosis 2004, 9, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Barrett, A.J.; Kembhavi, A.A.; Brown, M.A.; Kirschke, H.; Knight, C.G.; Tamai, M.; Hanada, K. L-trans-Epoxysuccinyl-leucylamido (4-guanidino) butane (E-64) and its analogues as inhibitors of cysteine proteinases including cathepsins B, H and L. Biochem. J. 1982, 201, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, X.; Huang, J.; Feng, L.; Dolinta, K.G.; Chen, J. Defining the protein-protein interaction network of the human hippo pathway. Mol. Cell Proteom. 2014, 13, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Steverding, D. The cathepsin B-selective inhibitors CA-074 and CA-074Me inactivate cathepsin L under reducing conditions. Open Enzym. Inhib. J. 2011, 4, 11–16. [Google Scholar] [CrossRef]
- Chopra, I.J.; Williams, D.E.; Orgiazzi, J.; Solomon, D.H. Opposite effects of dexamethasone on serum concentrations of 3, 3′, 5′-triiodothyronine (reverse T3) and 3, 3′, 5′-triiodothyronine (T3). J. Clin. Endocrinol. Metab. 1975, 41, 911–920. [Google Scholar] [CrossRef] [PubMed]
- Montesinos, M.M.; Alamino, V.A.; Mascanfroni, I.D.; Susperreguy, S.; Gigena, N.; Masini-Repiso, A.M.; Rabinovich, G.A.; Pellizas, C.G. Dexamethasone counteracts the immunostimulatory effects of triiodothyronine (T3) on dendritic cells. Steroids 2012, 77, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Rodriguez, M.L.; Leonard, A.; Sun, L.; Fischer, K.A.; Wang, Y.; Ritterhoff, J.; Zhao, L.; Kolwicz, S.C.; Pabon, L. Fatty acids enhance the maturation of cardiomyocytes derived from human pluripotent stem cells. Stem Cell Rep. 2019, 13, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Haemmerle, G.; Moustafa, T.; Woelkart, G.; Büttner, S.; Schmidt, A.; Van De Weijer, T.; Hesselink, M.; Jaeger, D.; Kienesberger, P.C.; Zierler, K. ATGL-mediated fat catabolism regulates cardiac mitochondrial function via PPAR-α and PGC-1. Nat. Med. 2011, 17, 1076–1085. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, N. Multiple roles of vestigial-like family members in tumor development. Front. Oncol. 2020, 10, 1266. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Li, Y.; Heims-Waldron, D.; Bezzerides, V.; Guatimosim, S.; Guo, Y.; Gu, F.; Zhou, P.; Lin, Z.; Ma, Q. Mitochondrial cardiomyopathy caused by elevated reactive oxygen species and impaired cardiomyocyte proliferation. Circ. Res. 2018, 122, 74–87. [Google Scholar] [CrossRef] [PubMed]
- Quignodon, L.; Vincent, S.; Winter, H.; Samarut, J.; Flamant, F. A point mutation in the activation function 2 domain of thyroid hormone receptor alpha1 expressed after CRE-mediated recombination partially recapitulates hypothyroidism. Mol. Endocrinol. 2007, 21, 2350–2360. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farley, A.; Gao, Y.; Sun, Y.; Zohrabian, S.; Pu, W.T.; Lin, Z. Activation of VGLL4 Suppresses Cardiomyocyte Maturational Hypertrophic Growth. Cells 2024, 13, 1342. https://doi.org/10.3390/cells13161342
Farley A, Gao Y, Sun Y, Zohrabian S, Pu WT, Lin Z. Activation of VGLL4 Suppresses Cardiomyocyte Maturational Hypertrophic Growth. Cells. 2024; 13(16):1342. https://doi.org/10.3390/cells13161342
Chicago/Turabian StyleFarley, Aaron, Yunan Gao, Yan Sun, Sylvia Zohrabian, William T. Pu, and Zhiqiang Lin. 2024. "Activation of VGLL4 Suppresses Cardiomyocyte Maturational Hypertrophic Growth" Cells 13, no. 16: 1342. https://doi.org/10.3390/cells13161342