
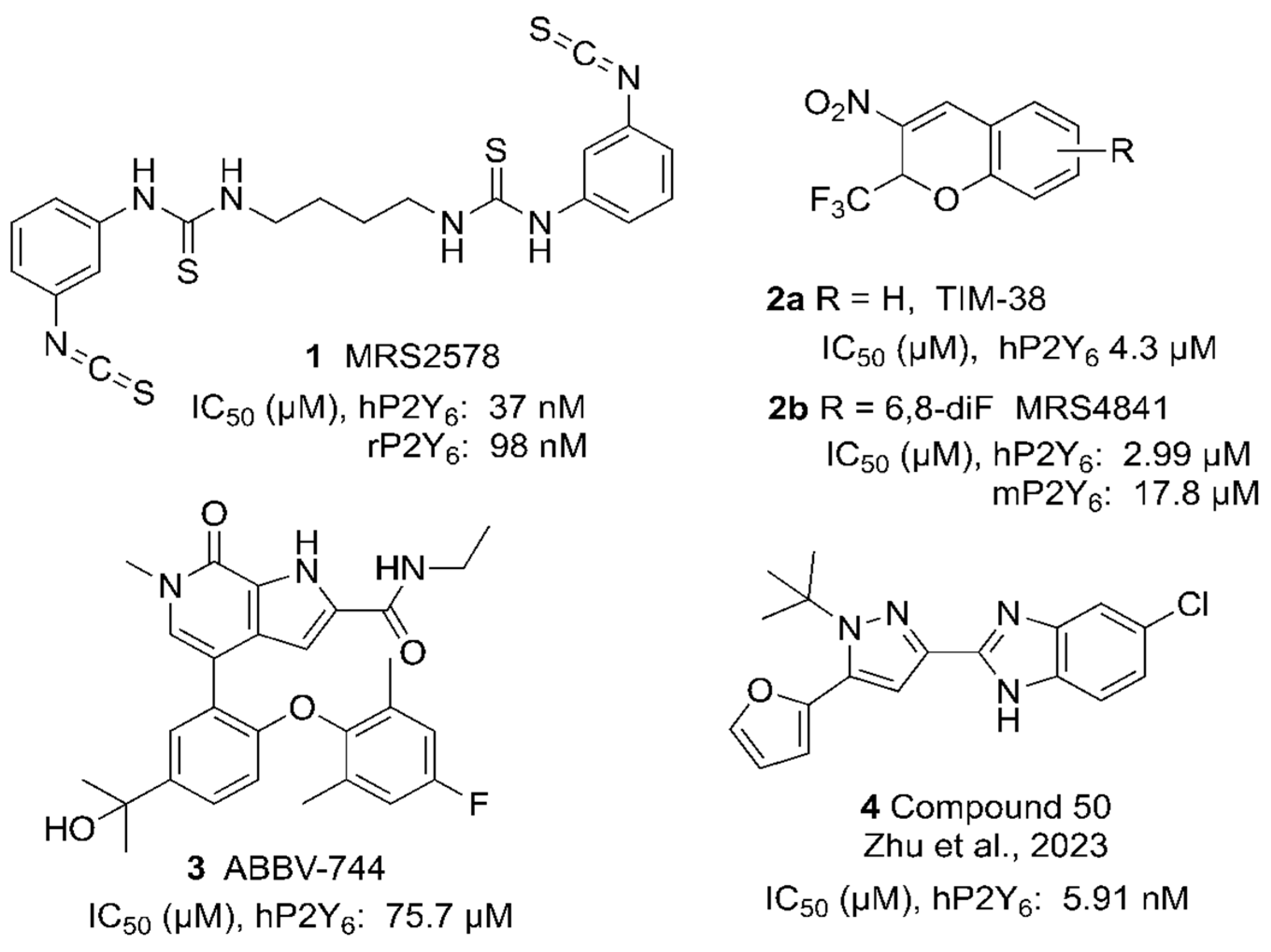
Functionalized Congeners of 2H-Chromene P2Y6 Receptor Antagonists
 ,
, 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Chemical Synthetic Procedures
2.3. Assay of hP2Y6R-Induced Ca2+ Transients
2.4. Assay of hP2Y14R Binding
2.5. Cell Culture and Cell Viability
2.6. Statistical Analysis
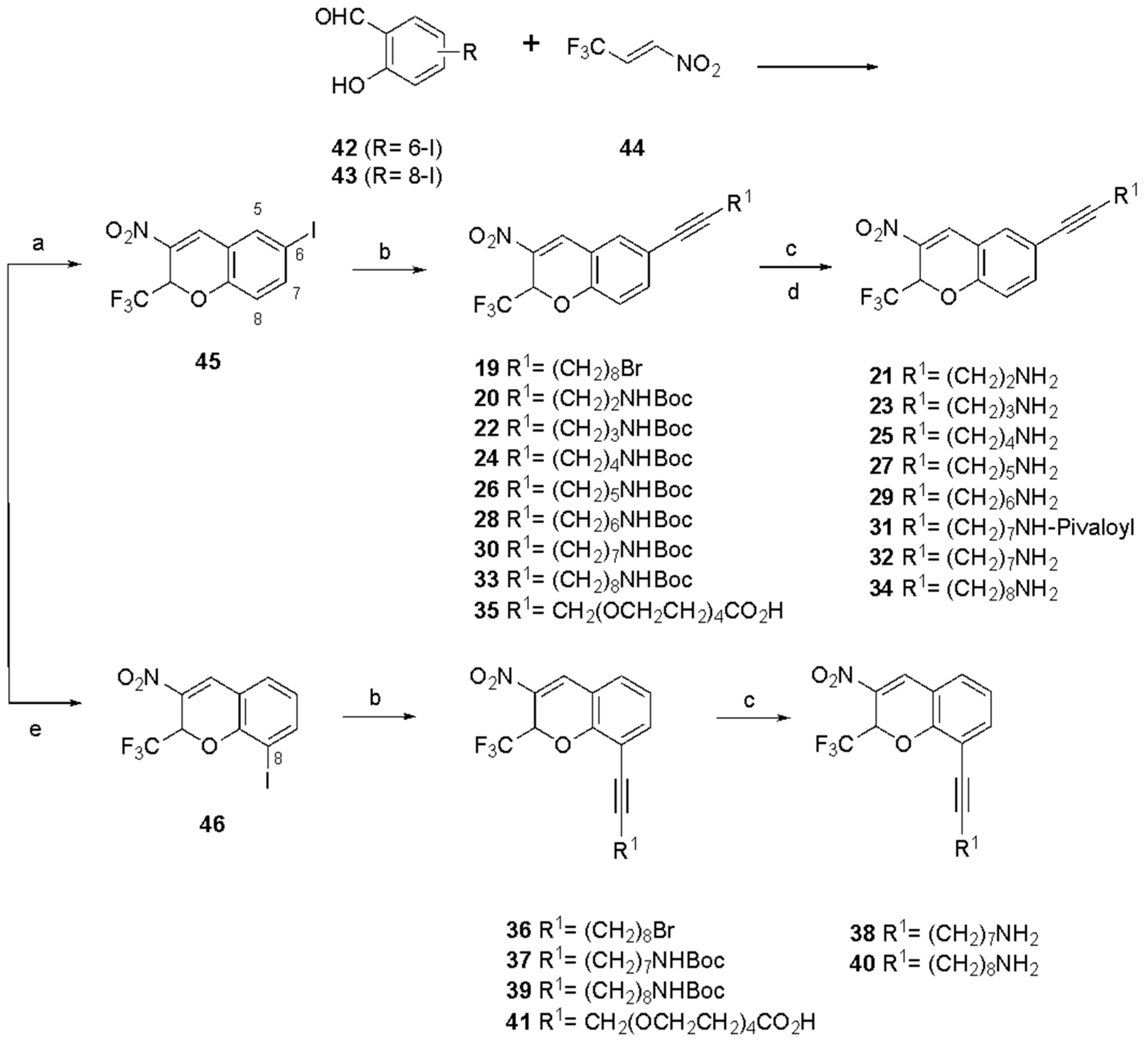
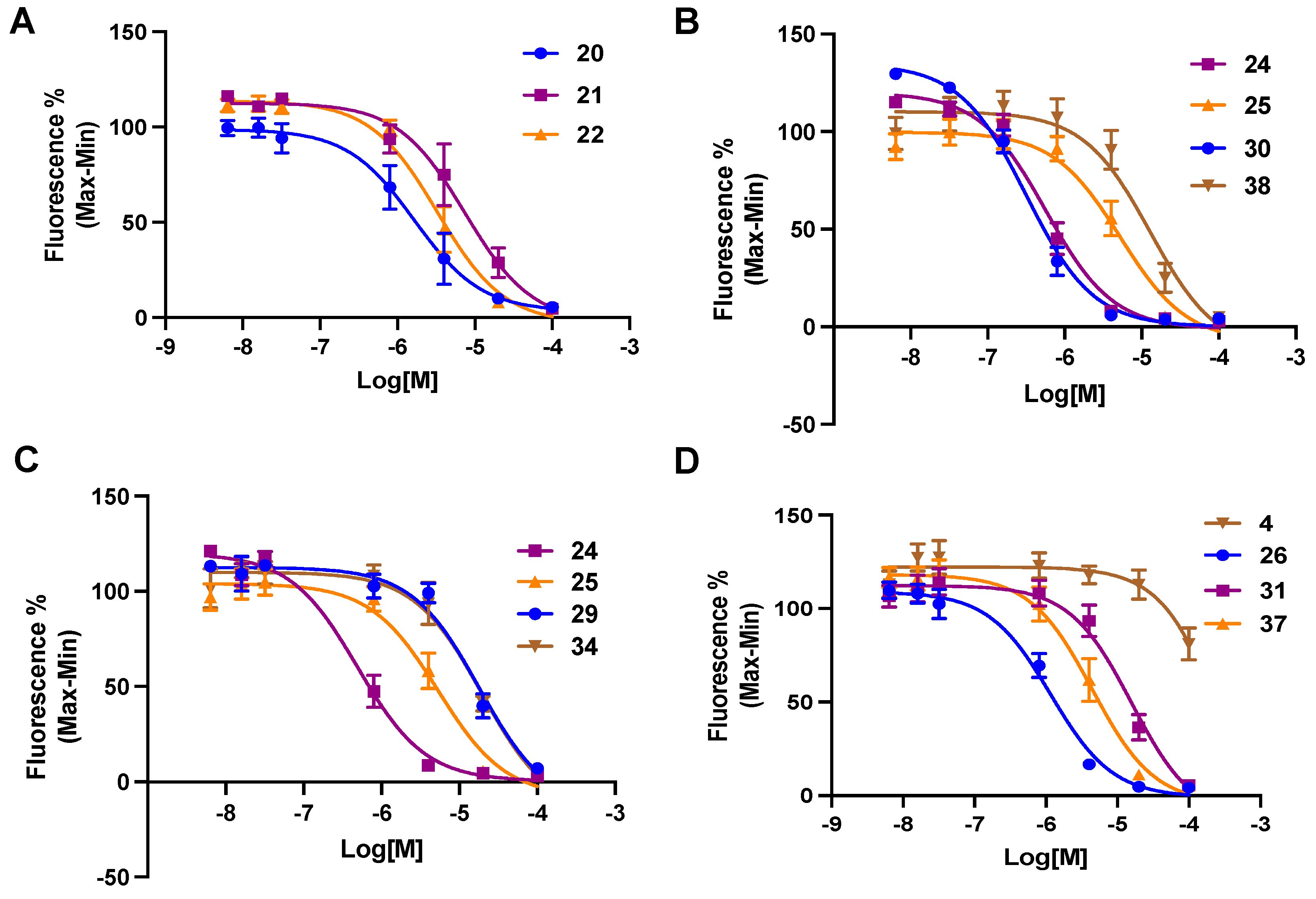
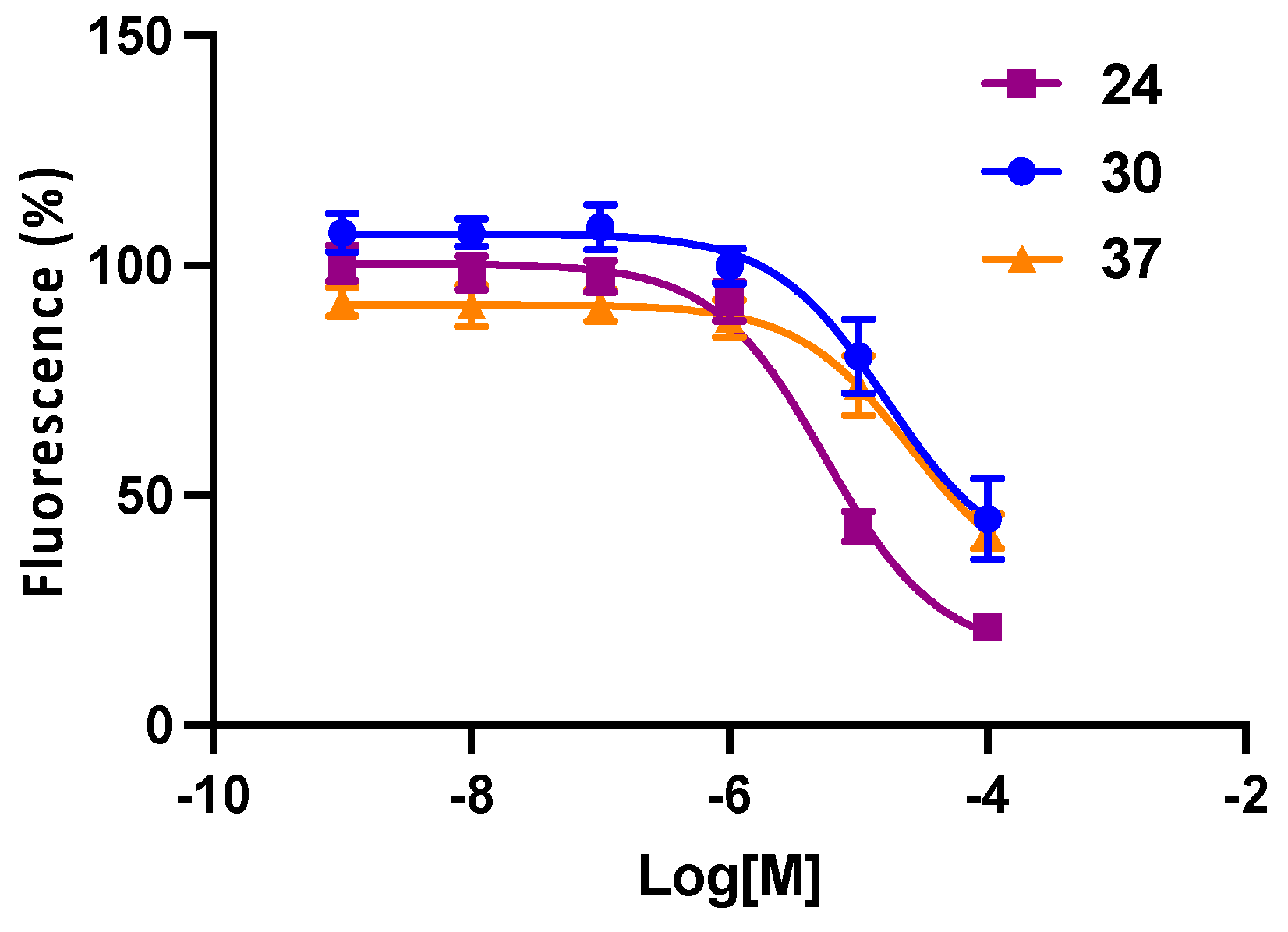
3. Results
Functional Antagonism
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jacobson, K.A.; Delicado, E.G.; Gachet, C.; Kennedy, C.; von Kügelgen, I.; Li, B.; Miras-Portugal, M.T.; Novak, I.; Schöneberg, T.; Perez-Sen, R.; et al. Update of P2Y receptor pharmacology: IUPHAR Review 27. Br. J. Pharmacol. 2020, 177, 2413–2433. [Google Scholar] [CrossRef] [PubMed]
- Al-Rashida, M.; Iqbal, J. Therapeutic potentials of ecto-nucleoside triphosphate diphosphohydrolase, ecto-nucleotide pyrophosphatase/phosphodiesterase, ecto-5′-nucleotidase, and alkaline phosphatase inhibitors. Med. Res. Rev. 2014, 34, 703–743. [Google Scholar] [CrossRef] [PubMed]
- Haas, C.B.; Lovászi, M.; Braganhol, E.; Pacher, P.; Haskó, G. Ectonucleotidases in inflammation, immunity, and cancer. J. Immunol. 2021, 206, 1983–1990. [Google Scholar] [CrossRef]
- Salem, M.; Lecka, J.; Pelletier, J.; Gomes Marconato, D.; Dumas, A.; Vallières, L.; Brochu, G.; Robaye, B.; Jobin, C.; Sévigny, J. NTPDase8 protects mice from intestinal inflammation by limiting P2Y6 receptor activation: Identification of a new pathway of inflammation for the potential treatment of IBD. Gut 2022, 71, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Wihlborg, A.K.; Balogh, J.; Wang, L.; Borna, C.; Dou, Y.; Joshi, B.V.; Lazarowski, E.; Jacobson, K.A.; Arner, A.; Erlinge, D. Positive inotropic effects by uridine triphosphate (UTP) and uridine diphosphate (UDP) via P2Y2 and P2Y6 receptors on cardiomyocytes and release of utp in man during myocardial infarction. Circ. Res. 2006, 98, 970–976. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, K.; Nishimura, A.; Shimoda, K.; Tanaka, T.; Kato, Y.; Shibata, T.; Tanaka, H.; Kurose, H.; Azuma, Y.T.; Ihara, H.; et al. Redox-dependent internalization of the purinergic P2Y6 receptor limits colitis progression. Sci. Signal. 2022, 15, eabj0644. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, S.; Shigemoto-Mogami, Y.; Nasu-Tada, K.; Shinozaki, Y.; Ohsawa, K.; Tsuda, M.; Joshi, B.V.; Jacobson, K.A.; Kohsaka, S.; Inoue, K. UDP acting at P2Y6 receptors is a mediator of microglial phagocytosis. Nature 2007, 446, 1091–1095. [Google Scholar] [CrossRef]
- Strassheim, D.; Verin, A.; Batori, R.; Nijmeh, H.; Burns, N.; Kovacs-Kasa, A.; Umapathy, N.S.; Kotamarthi, J.; Gokhale, Y.S.; Karoor, V.; et al. P2Y purinergic receptors, endothelial dysfunction, and cardiovascular diseases. Int. J. Mol. Sci. 2020, 21, 6855. [Google Scholar] [CrossRef] [PubMed]
- Kauffenstein, G.; Tamareille, S.; Prunier, F.; Roy, C.; Ayer, A.; Toutain, B.; Billaud, M.; Isakson, B.E.; Grimaud, L.; Loufrani, L.; et al. Central role of P2Y6 UDP receptor in arteriolar myogenic tone. Arterioscleros. Thromb. Vasc. Biol. 2016, 36, 1598–1606. [Google Scholar] [CrossRef]
- Wang, Q.; Li, Z.; Yang, G. P2Y6 receptor mediated cell proliferation and migration in human hepatocellular carcinoma cells. Purinergic Signal. 2011, 7, 413–420. [Google Scholar]
- Yin, L.; Zhang, E.; Mao, T.; Zhu, Y.; Ni, S.; Li, Y.; Liu, C.; Fang, Y.; Ni, K.; Lu, Y.; et al. Macrophage P2Y6R activation aggravates psoriatic inflammation through IL-27-mediated Th1 responses. Acta Pharmaceut. Sinica B, 2024; in press. [Google Scholar] [CrossRef]
- Mamedova, L.; Joshi, B.V.; Gao, Z.G.; von Kügelgen, I.; Jacobson, K.A. Diisothiocyanate derivatives as potent, insurmountable antagonists of P2Y6 nucleotide receptors. Biochem. Pharmacol. 2004, 67, 1763–1770. [Google Scholar] [CrossRef] [PubMed]
- Milde, S.; Brown, G.C. Knockout of the p2y6 receptor prevents peri-infarct neuronal loss after transient, focal ischemia in mouse brain. Int. J. Mol. Sci. 2022, 23, 2304. [Google Scholar] [CrossRef] [PubMed]
- Dundee, J.M.; Puigdellívol, M.; Butler, R.; Cockram, T.O.J.; Brown, G.C. p2y6 receptor-dependent microglial phagocytosis of synapses mediates synaptic and memory loss in aging. Aging Cell 2023, 22, e13761. [Google Scholar] [CrossRef] [PubMed]
- Clouet, S.; Di Pietrantonio, L.; Daskalopoulos, E.P.; Esfahani, H.; Horckmans, M.; Vanorlé, M.; Lemaire, A.; Balligand, J.L.; Beauloye, C.; Boeynaems, J.M.; et al. Loss of mouse P2Y6 nucleotide receptor is associated with physiological macrocardia and amplified pathological cardiac hypertrophy. J. Biol. Chem. 2016, 291, 15841–15852. [Google Scholar] [CrossRef] [PubMed]
- Shimoda, K.; Nishimura, A.; Sunggip, C.; Ito, T.; Nishiyama, K.; Kato, Y.; Tanaka, T.; Tozaki-Saitoh, H.; Tsuda, M.; Nishida, M. Modulation of P2Y6R expression exacerbates pressure overload-induced cardiac remodeling in mice. Sci. Rep. 2020, 10, 13926. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, M.; Shinozaki, Y.; Kashiwagi, K.; Shigetomi, E.; Robaye, B.; Koizumi, S. Müller cell-mediated neurite outgrowth of the retinal ganglion cells via P2Y6 receptor signals. J. Neurochem. 2016, 136, 741–751. [Google Scholar] [CrossRef] [PubMed]
- Jacob, T.F.; Singh, V.; Dixit, M.; Ginsburg-Shmuel, T.; Fonseca, B.; Pintor, J.; Youdim, M.B.H.; Fischer, B. A promising drug candidate for the treatment of glaucoma based on a P2Y6-receptor agonist. Purinergic Signal. 2018, 14, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Oliva, P.; Scortichini, M.; Dobelmann, C.; Jain, S.; Gopinatth, V.; Toti, K.S.; Phung, N.B.; Junker, A.; Jacobson, K.A. Structure-activity relationships of pyrimidine nucleotides containing a 5′-α, β-methylene diphosphonate at the P2Y6 receptor. Bioorg. Med. Chem. Lett. 2021, 45, 128137. [Google Scholar] [CrossRef]
- Steculorum, S.M.; Paeger, L.; Bremser, S.; Evers, N.; Hinze, Y.; Idzko, M.; Kloppenburg, P.; Brüning, J.C. Hypothalamic UDP increases in obesity and promotes feeding via P2Y6-dependent activation of AgRP neurons. Cell 2015, 162, 1404–1417. [Google Scholar] [CrossRef]
- Jain, S.; Pydi, S.P.; Toti, K.S.; Robaye, B.; Idzko, M.; Gavrilova, O.; Wess, J.; Jacobson, K.A. Lack of adipocyte purinergic P2Y6 receptor greatly improves whole body glucose homeostasis. Proc. Nat. Acad. Sci. USA 2020, 117, 30763–30774. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Wang, W.; Li, Y.; Zhang, Q.; Ji, H.; Li, H.; Hu, Q. The role of P2Y6R in cardiovascular diseases and recent development of P2Y6R antagonists. Drug Disc. Today 2020, 25, 568–573. [Google Scholar] [CrossRef]
- Milde, S.; van Tartwijk, F.W.; Vilalta, A.; Hornik, T.C.; Dundee, J.M.; Puigdellívol, M.; Brown, G.C. Inflammatory neuronal loss in the substantia nigra induced by systemic lipopolysaccharide is prevented by knockout of the P2Y6 receptor in mice. J. Neuroinflamm. 2021, 18, 225. [Google Scholar] [CrossRef] [PubMed]
- Bian, J.; Zhang, Y.; Liu, Y.; Li, Q.; Tang, H.; Liu, Q. P2Y6 receptor-mediated spinal microglial activation in neuropathic pain. Pain Res. Manag. 2019, 2019, 2612534. [Google Scholar] [CrossRef] [PubMed]
- Chetty, A.; Sharda, A.; Warburton, R.; Weinberg, E.O.; Dong, J.; Fang, M.; Sahagian, G.G.; Chen, T.; Xue, C.; Castellot, J.J.; et al. A purinergic P2Y6 receptor agonist prodrug modulates airway inflammation, remodeling, and hyperreactivity in a mouse model of asthma. J. Asthma Allergy 2018, 11, 159–171. [Google Scholar] [CrossRef] [PubMed]
- Placet, M.; Arguin, G.; Molle, C.M.; Babeu, J.P.; Jones, C.; Carrier, J.C.; Robaye, B.; Geha, S.; Boudreau, F.; Gendron, F.P. The G protein-coupled P2Y₆ receptor promotes colorectal cancer tumorigenesis by inhibiting apoptosis. Biochim. Biophys. Acta. Mol. Basis Dis. 2018, 1864 Pt A, 1539–1551. [Google Scholar] [CrossRef]
- Wan, H.; Xie, R.; Xu, J.; He, J.; Tang, B.; Liu, Q.; Wang, S.; Guo, Y.; Yang, X.; Dong, T.X.; et al. Anti-proliferative effects of nucleotides on gastric cancer via a novel P2Y6/SOCE/Ca2+/β-catenin pathway. Sci. Rep. 2017, 7, 2459. [Google Scholar] [CrossRef] [PubMed]
- Garcia, R.A.; Yan, M.; Search, D.; Zhang, R.; Carson, N.L.; Ryan, C.S.; Smith-Monroy, C.; Zheng, J.; Chen, J.; Kong, Y.; et al. P2Y6 receptor potentiates pro-inflammatory responses in macrophages and exhibits differential roles in atherosclerotic lesion development. PLoS ONE 2014, 9, e111385. [Google Scholar] [CrossRef]
- Ito, M.; Egashira, S.I.; Yoshida, K.; Mineno, T.; Kumagai, K.; Kojima, H.; Okabe, T.; Nagano, T.; Ui, M.; Matsuoka, I. Identification of novel selective P2Y6 receptor antagonists by high-throughput screening assay. Life Sci. 2017, 180, 137–142. [Google Scholar] [CrossRef]
- Jung, Y.H.; Jain, S.; Gopinatth, V.; Phung, N.B.; Gao, Z.G.; Jacobson, K.A. Structure activity relationship of 3-nitro-2-(trifluoromethyl)-2H-chromene derivatives as P2Y6 receptor antagonists. Bioorg. Med. Chem. Lett. 2021, 41, 128008. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.H.; Salmaso, V.; Wen, Z.; Bennett, J.M.; Phung, N.B.; Lieberman, D.I.; Gopinatth, V.; Randle, J.C.R.; Chen, Z.; Salvemini, D. Structure-activity relationships of heterocyclic P2Y14 receptor antagonists: Removal of the zwitterionic character with piperidine bioisosteres. J. Med. Chem. 2021, 64, 5099–5122. [Google Scholar] [CrossRef]
- Puhl, A.C.; Lewicki, S.A.; Gao, Z.G.; Pramanik, A.; Makarov, V.; Ekins, S.; Jacobson, K.A. Use of machine learning to search for ligands of P2Y6 and other P2Y receptors. Purinergic Signal. 2024, in press. [CrossRef]
- Zhu, Y.; Zhou, M.; Cheng, X.; Wang, H.; Li, Y.; Guo, Y.; Wang, Y.; Tian, S.; Mao, T.; Zhang, Z.; et al. Discovery of selective P2Y6R antagonists with high affinity and in vivo efficacy for inflammatory disease therapy. J. Med. Chem. 2023, 66, 6315–6332. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, K.A. Functionalized congener approach to the design of ligands for G protein–coupled receptors (GPCRs). Bioconjug. Chem. 2009, 20, 1816–1835. [Google Scholar] [CrossRef]
- Jacobson, K.A.; Ukena, D.; Padgett, W.; Kirk, K.L.; Daly, J.W. Molecular probes for extracellular adenosine receptors. Biochem. Pharmacol. 1987, 36, 1697–1707. [Google Scholar] [CrossRef] [PubMed]
- Doré, A.S.; Robertson, N.; Errey, J.C.; Ng, I.; Hollenstein, K.; Tehan, B.; Hurrell, E.; Bennett, K.; Congreve, M.; Magnani, F.; et al. Structure of the adenosine A2A receptor in complex with ZM241385 and the xanthines XAC and caffeine. Structure 2011, 19, 1283–1293. [Google Scholar] [CrossRef]
- Karton, Y.; Baumgold, J.; Handen, J.S.; Jacobson, K.A. Molecular probes for muscarinic receptors: Derivatives of the M1-antagonist telenzepine. Bioconjug. Chem. 1992, 3, 234–240. [Google Scholar] [CrossRef]
- Pradhan, B.; Pavan, M.; Fisher, C.L.; Salmaso, V.; Wan, T.C.; Keyes, R.F.; Rollison, N.; Suresh, R.R.; Kumar, T.S.; Gao, Z.G.; et al. Lipid trolling to optimize A3 adenosine receptor positive allosteric modulators (PAMs). J. Med. Chem. 2024, 67, 12221–12247. [Google Scholar] [CrossRef] [PubMed]
- Robaye, B.; Boeynaems, J.M.; Communi, D. Slow desensitization of the human P2Y6 receptor. Eur. J. Pharmacol. 1997, 329, 231–236. [Google Scholar] [CrossRef]
- Jung, Y.H.; Shah, Q.; Lewicki, S.A.; Pramanik, A.; Gopinatth, V.; Pelletier, J.; Sévigny, J.; Iqbal, J.; Jacobson, K.A. Synthesis and pharmacological characterization of multiply substituted 2H-chromene derivatives as P2Y6 receptor antagonists. Bioorg. Med. Chem. Lett. 2022, 75, 128981. [Google Scholar] [CrossRef]
- Segall, M.D. Multi-parameter optimization: Identifying high quality compounds with a balance of properties. Curr. Pharmaceut. Des. 2012, 18, 1292–1310. [Google Scholar] [CrossRef] [PubMed]
- Nepali, K.; Lee, H.Y.; Liou, J.P. Nitro-group-containing drugs. J. Med. Chem. 2019, 62, 2851–2893. [Google Scholar] [CrossRef] [PubMed]
- Besnard, J.; Ruda, G.F.; Setola, V.; Abecassis, K.; Rodriguiz, R.M.; Huang, X.P.; Norval, S.; Sassano, M.F.; Shin, A.I.; Webster, L.A.; et al. Automated design of ligands to polypharmacological profiles. Nature 2012, 492, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Meth. 1983, 65, 55–63. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
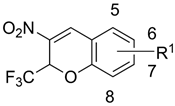 2, 5–41 | |||
| Compound | R1 = | cLogD (cLogS, pH7.4) | IC50, hP2Y6R (µM, Mean ± SEM) c or % inhib. |
| Halo substitution (reported compounds) | |||
| 2a b,c,d | H | 3.32 (2.10) | 2.91 ± 1.21 |
| 2b c,d | 6,8-diF | 3.52 (1.11) | 2.99 ± 0.56 |
| 4 | - | 5.22 (0.937) | >100 |
| 5 c | 5-Cl | 3.88 (1.92) | 15.7 ± 4.6 |
| 6 c | 6-Cl | 3.94 (2.10) | 1.79 ± 0.43 |
| 7 c | 7-Cl | 3.88 (1.92) | 7.98 ± 0.42 |
| 8 c | 8-Cl | 3.88 (1.92) | 3.69 ± 0.47 |
| 9 c,d | 6-Br | 4.17 (1.71) | 3.49 ± 1.54 |
| 10 c,d | 6-I | 4.52 (1.63) | 4.00 ± 1.59 |
| 11 c | 8-I | 4.40 (1.44) | 4.09 ± 1.04 |
| Alkyne substitution (reported compounds) | |||
| 12 c | 5-C≡C-Si(CH2CH3)3 | 6.47 (0.369) | 4.77 ± 2.73 |
| 13 | 6-C≡C-Si(CH2CH3)3 | 6.47 (0.369) | 0.604 ± 0.239 |
| 14 c,d | 8-C≡C-Si(CH2CH3)3 | 6.47 (0.369) | 0.461 ± 0.270 |
| 15 c | 6-C≡C-Ph-p-OH | 5.33 (0.670) | 5.36 ± 1.28 |
| 16 c,d | 6-C≡C-Ph-p-CH2OH | 4.70 (0.806) | 2.00 ± 0.30 |
| 17 b,c,d | 6-C≡C-Ph-p-CO2H | 1.34 (2.50) | 1.09 ± 0.42 |
| 18 c | 6-C≡C-Ph-p-CO2CH3 | 5.59 (0.0912) | 3.97 ± 1.58 |
| Newly synthesized 6-substituted compounds | |||
| 19 | 6-C≡C-(CH2)8Br | 6.06 (−0.0142) | 14.0 ± 4.7 |
| 20 d | 6-C≡C-(CH2)2-NH-Boc | 4.60 (0.679) | 0.587 ± 0.087 |
| 21 | 6-C≡C-(CH2)2-NH2 | 1.09 (2.38) | 22.6 ± 9.3 |
| 22 | 6-C≡C-(CH2)3-NH-Boc | 4.89 (0.507) | 1.87 ± 0.11 |
| 23 | 6-C≡C-(CH2)3-NH2 | 1.28 (2.30) | 9.98± 1.37 |
| 24 d | 6-C≡C-(CH2)4-NH-Boc | 5.16 (0.344) | 0.568 ± 0.163 |
| 25 | 6-C≡C-(CH2)4-NH2 | 1.50 (2.18) | 5.39 ± 1.49 |
| 26 | 6-C≡C-(CH2)5-NH-Boc | 5.45 (0.163) | 1.19± 0.25 |
| 27 | 6-C≡C-(CH2)5-NH2 | 1.68 (2.04) | 12.4± 1.3 |
| 28 | 6-C≡C-(CH2)6-NH-Boc | 5.66 (0.0289) | 10.5 ± 6.5 |
| 29 | 6-C≡C-(CH2)6-NH2 | 1.83 (1.90) | 20.0 ± 5.1 |
| 30 d | 6-C≡C-(CH2)7-NH-Boc | 5.84 (−0.064) | 0.162 ± 0.013 |
| 31 | 6-C≡C-(CH2)7-NH-pivaloyl | 5.69 (0.0417) | 17.3 ± 5.2 |
| 32 | 6-C≡C-(CH2)7-NH2 | 1.95 (1.77) | 6.37 ± 1.82 |
| 33 | 6-C≡C-(CH2)8-NH-Boc | 6.26 (−0.302) | 45.4 ± 17.7 |
| 34 | 6-C≡C-(CH2)8-NH2 | 2.06 (1.64) | 20.0 ± 5.2 |
| 35 | 6-C≡C-CH2(OCH2CH2)4-CO2H | 0.943 (2.53) | 63.3 ± 30.0 |
| Newly synthesized 8-substituted compounds | |||
| 36 | 8-C≡C-(CH2)8Br | 6.06 (−0.014) | 4.79 ± 1.45 |
| 37 | 8-C≡C-(CH2)7-NH-Boc | 5.84 (−0.064) | 4.96 ± 1.96 |
| 38 | 8-C≡C-(CH2)7-NH2 | 1.97 (1.69) | 13.0 ± 3.9 |
| 39 | 8-C≡C-(CH2)8-NH-Boc | 6.01 (−0.123) | 23.8 ± 6.8 |
| 40 | 8-C≡C-(CH2)8-NH2 | 2.08 (1.56) | 34.8 ± 16.3 |
| 41 | 8-C≡C-CH2(OCH2CH2)4-CO2H | 0.948 (2.42) | >50 |
| Compd | Structure | Receptor, Ki in µM c |
|---|---|---|
| 2b | 6,8-di-F | none |
| 13 a | 6-C≡C-Si(CH2CH3)3 | α2A, 2.0 |
| 16 a | 6-C≡C-Ph-p-CH2OH | D5, 0.60 |
| 17 a | 6-C≡C-Ph-p-CO2H | D1, 1.1; D5, 2.1; µ-opioid, 4.2; α2A, 1.8; α2B, 1.0; α2C, 2.6 |
| 20 b | 6-C≡C-(CH2)2-NH-Boc | α2A, 1.6; σ1, 6.5; σ2, 3.1; D5, 4.4 |
| 24 b | 6-C≡C-(CH2)4-NH-Boc | D1, 1.9; D5, 2.4; H1, 1.0; α2A, 0.16; α2B, 0.46; 5-HT1D, 3.2; σ2, 1.5 |
| 26 b | 6-C≡C-(CH2)5-NH-Boc | α2A, 0.75; α2B, 1.7; σ2, 3.6; 5-HT1D, 4.8 |
| 30 b | 6-C≡C-(CH2)7-NH-Boc | D4, 3.1; D5, 0.78; H1, 3.9; H2, 0.50; κ-opioid, 2.2; α2A, 0.086; α2B, 0.17 |
| 31 b | 6-C≡C-(CH2)7-NH-pivaloyl | α2A, 3.5; σ2, 3.3 |
| 37 b | 8-C≡C-(CH2)7-NH-Boc | α2A, 5.2; α2B, 4.8; σ2, 4.1; D5, 4.2; H1, 7.3; 5-HT1D, 3.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oliva, P.; Pramanik, A.; Jung, Y.-H.; Lewicki, S.A.; Mwendwa, J.M.; Park, J.H.; Jacobson, K.A. Functionalized Congeners of 2H-Chromene P2Y6 Receptor Antagonists. Cells 2024, 13, 1366. https://doi.org/10.3390/cells13161366
Oliva P, Pramanik A, Jung Y-H, Lewicki SA, Mwendwa JM, Park JH, Jacobson KA. Functionalized Congeners of 2H-Chromene P2Y6 Receptor Antagonists. Cells. 2024; 13(16):1366. https://doi.org/10.3390/cells13161366
Chicago/Turabian StyleOliva, Paola, Asmita Pramanik, Young-Hwan Jung, Sarah A. Lewicki, Jamie M. Mwendwa, Jong Hwan Park, and Kenneth A. Jacobson. 2024. "Functionalized Congeners of 2H-Chromene P2Y6 Receptor Antagonists" Cells 13, no. 16: 1366. https://doi.org/10.3390/cells13161366