Abstract
Melanoma is the most aggressive subtype of cancer, with a higher propensity to spread compared to most solid tumors. The application of OMICS approaches has revolutionized the field of melanoma research by providing comprehensive insights into the molecular alterations and biological processes underlying melanoma development and progression. This review aims to offer an overview of melanoma biology, covering its transition from primary to malignant melanoma, as well as the key genes and pathways involved in the initiation and progression of this disease. Utilizing online databases, we extensively explored the general expression profile of genes, identified the most frequently altered genes and gene mutations, and examined genetic alterations responsible for drug resistance. Additionally, we studied the mechanisms responsible for immune checkpoint inhibitor resistance in melanoma.
1. Introduction
The rising incidence of skin cancer is a severe threat to worldwide public health since it places a heavy demand on labor and the economy [1]. The epidermis and dermis are the two main layers that make up the skin. Melanocytes, keratinocytes, Langerhans cells, and Merkel cells are among the several cell types that make up the epidermis, the outermost layer. Cancer may result from a build-up of anomalies in these cells [1]. Skin cancer has a complicated and diverse etiology impacted by its phenotypic, genetic, and environmental variables. Ultraviolet radiation (UVR) is the most prevalent environmental risk factor. DNA damage caused by UVR may result in oxidative stress, the synthesis of pyrimidine dimers, gene mutations, and other cellular alterations that promote cancer [2]. Melanoma, caused by malfunctioning melanocytes, and non-melanoma skin cancers (NMSC), formed from cells derived from the epidermis, are the two primary forms of skin cancers routinely identified [3]. Globally, NMSC causes around a million new cases and 70,000 deaths, according to GLOBOCAN 2022. The incidence and mortality rates for men are roughly 1.5 times higher. Approximately 95% of skin malignancies are NMSCs or keratinocyte carcinomas. They are divided into two categories: squamous cell carcinoma (SCC) and basal cell carcinoma (BCC). On the other hand, because of its high propensity for metastasis, melanoma—which makes up 1.7% of all occurrences of skin cancer—is the deadliest type [4].
According to studies, UV is the cause of 90% of NMSC and approximately 65% of melanoma. Based on UV exposure or sun damage, the World Health Organization (WHO) 2018 divided melanoma into many kinds (Figure 1 and Table 1) [5]. The updated classification includes various types of melanoma, such as superficial spreading melanoma, nodular melanoma, lentigo maligna melanoma, and acral lentiginous melanoma. These types are distinguished based on their clinical presentation, histological characteristics, and UV-induced damage patterns [6]. Research indicates that UV radiation is responsible for about 90% of non-melanoma skin cancers, which include basal cell carcinoma (BCC) and squamous cell carcinoma (SCC). Long-term UV exposure, particularly from sunlight, is a major risk factor for skin cancer [7]. Superficial spreading melanoma (SSM) is one of the most prevalent forms of melanoma, characterized by its tendency to spread horizontally across the skin’s surface before invading deeper layers. This type of melanoma commonly appears on the trunk and extremities and is closely linked to UV exposure.
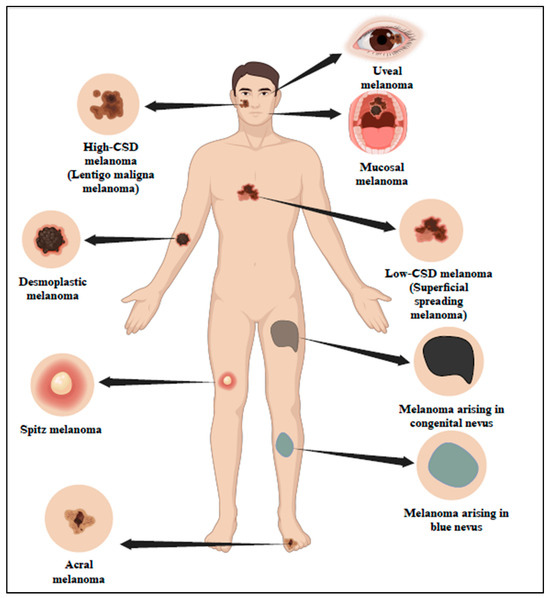
Figure 1.
Classification of melanoma in association with sun exposure as per WHO 2018. The 2018 classification by WHO has categorized melanoma into different types based on sun exposure or sun damage. Created with BioRender.com.

Table 1.
WHO 2018 classification of melanoma [5].
Guidelines for staging melanoma have been created by the American Joint Committee on Cancer (AJCC), with scores ranging from 0 (in situ) to IV (metastatic) [8]. Since primary melanoma has a 99% 5-year survival rate and metastatic melanoma has a 27% 5-year survival rate, early diagnosis is essential [9]. A considerable portion of patients (20–30%) receive a diagnosis at the metastatic stage, when the prognosis is noticeably worse [10]. A great deal of research has been performed in this area to learn more about the mechanisms and processes of metastasis and how it progresses [11]. The intricate chain of processes known as invasion, intravasation, circulation, extravasation, and colonization are what causes metastasis. Before going into circulation, malignant cells first invade the tissues around them as circulating tumor cells (CTCs). Disseminated tumor cells (DTCs) are those cells that make it to distant organs and have the ability to develop into secondary cancers [12]. Cancer cells, either as a group or as individual cells, invade the surrounding tissues during the local invasion. An epithelial-to-mesenchymal transition (EMT), which allows cells to pass through the basement membrane, is frequently involved in single-cell invasion. This shift is driven by important transcription factors such as TWIST, SNAIL, and ZEB 1 and 2. Tumor cells enter the stroma after breaching the basement membrane and interact with fibroblasts, endothelial cells, adipocytes, and macrophages, among other stromal cells, to support distinct stages of tumor progression [10].
When tumor cells penetrate endothelial and pericyte barriers to enter microvessels, this process is known as intravasation. Factors such as cyclooxygenase-2 (COX-2), metalloproteinase 1 (MMP1), metalloproteinase 2 (MMP2), vascular endothelial growth factors (VEGF), and others help tumors to create new blood vessels (neoangiogenesis). Hemodynamic forces and the body’s immune system create problems for CTCs in the bloodstream. For protection, they group together and communicate with platelets. A CTC’s final destination frequently relies on the body’s blood circulation patterns; it may go into dormancy or start colonizing a new place. When tumor cells enter microvessels, they have three options: they can grow intraluminally, break through the microvessels, and contact organ tissues, or they can enter through spaces between pericytes and endothelial cells. When cancer cells adjust to new surroundings, they can create micrometastases, some of which can grow into healthy secondary tumors [10].
Research has demonstrated that compared to normal skin cells, melanoma cells express and activate more significant quantities of MMP-2 and MMP-9. These enzymes degrade the extracellular matrix, which permits melanoma cells to metastasize—the spread of cancerous cells to other tissues, blood arteries, and lymphatic vessels. Furthermore, by releasing pro-angiogenic proteins and remodeling blood arteries within the tumor, MMP-2 and MMP-9 encourage angiogenesis. Weakening immune system components also aids in immunological evasion [13]. Patients with metastatic melanoma are receiving far better care and therapy due to the development of personalized medicine. Patient outcomes have significantly improved with immunotherapy [14]. Even though many patients have negative side effects or do not respond to treatment, precision medicine is essential when determining which treatment plan is best for a given patient. The OMICS-based approach has recently become more popular in the medical field. Researchers hope to thoroughly understand the molecular pathways driving cancer by integrating data from different OMICS-based methodologies [15]. This information may help to create personalized treatment plans and identify new biomarkers for the early identification of melanomas. This review provides an overview of immune checkpoint inhibitors (ICIs) and other therapies in treating melanoma and the genetic and immunological aspects that lead to melanoma resistance.
2. Genetics of Melanoma
A melanoma can have many gene alterations, with only a small subset of these mutations being true “drivers” of the tumor—these could be either gain-of-function (GOF)/activating or loss-of-function (LOF)/deleterious mutations that cause continuous proliferation of melanoma cells, leading to uncontrolled tumor growth. Similarly, tumor-suppressor genes are also susceptible to mutations, which, when altered, cease to function, potentially activating downstream growth pathways and enabling unregulated tumor growth [16]. In recent times, various genetic changes, including copy number variants (CNVs) and single-nucleotide variants (SNVs), have emerged. These mutations can be somatic or germline and may lead to either gain-of-function (GOF) or loss-of-function (LOF) [17]. GOF mutations, typically found in oncogenes, frequently influence essential cellular functions like proliferation, growth, metabolism, resistance to apoptosis, and cell cycle regulation, resulting in the abnormal activation of related signaling pathways.
Melanoma has the maximum mutational burden of all cancers due to UV-induced DNA damage and/or errors in DNA replication [9]. In primary melanoma, the most common mutations exist in proto-oncogene B-Raf or v-Raf murine sarcoma viral oncogene homolog B (BRAF), in the rat sarcoma gene (RAS). Mutations in other genes such as neurofibromin 1 (NF1), proto-oncogene receptor tyrosine kinase (KIT), telomerase reverse transcriptase (TERT) mutations, tumor protein p53 gene (TP53), cyclin-dependent kinase inhibitor 2A (CDKN2A), cyclin-dependent kinase inhibitor 2B (CDKN2B), Phosphatase and tensin homolog (PTEN), Ras-related C3 botulinum toxin substrate 1 (RAC1), and G Protein Subunit Alpha Q and 11 (GNAQ/GNA11), were reported [18]. A summary of the most frequently altered genes in somatic and germline mutations in melanoma and the pathway involved is listed in Table 2 [16,19]. Most frequently altered genes in melanoma subtypes are detailed in Table 3 [20].
Table 2.
Most frequently altered genes in melanoma [16,19,21,22].
Table 3.
Most frequently altered genes in melanoma subtypes.
According to the geographical distribution, the occurrence of cutaneous melanoma is higher in Caucasians when compared to Hispanic, African American, Indo-American, and Asian population groups [23]. The evaluation of various literature sources found that the following genetic mutations or gene signatures have been found specifically in some geographic regions. NRAS and KIT gene mutations are associated with the melanoma subtype in the Australian group [24]. There is an association between the prevalence of BRAF gene mutations in melanoma patients with respect to different ethnic groups. The highest prevalence is 70% in the USA, and the lowest is 41% in the Russian population [25]. Studies have also shown that the likelihood of developing melanoma in CDKN2A carriers varies across geographical areas and increases with age. According to a report by Bishop et al., carriers in Europe had the lowest penetrance of 13% at the age of 50, and the highest penetrance of 91% was in the Australian population at the mean age of 50 [26]. Several studies have also focused on the co-occurrence of the BRAF and NRAS mutations, suggesting that the concurrent presence of these mutations may complicate treatment options, as they may have distinct responses to targeted therapies [25,27]. The co-occurrence of BRAF and NRAS mutations in major geographical areas is depicted (Figure 2). A recent study focused on the positional identity linked to the anatomical location as a determining factor for the potential development of melanoma [28]. The RAS family of proteins comprises NRAS and BRAF, both integral in governing cell development, differentiation, and survival processes. RAS functions as a molecular switch that becomes activated by extracellular signals, subsequently triggering downstream signaling through MAP kinase pathways (Supplementary Materials Figure S1) [29]. The MAPK pathway interfaces multiple transcription factors and is triggered by receptor tyrosine kinases (RTKs) to turn on the transcription of several genes. In melanomas, the MAPK pathway is activated by mutations in BRAF and NRAS genes. BRAF mutations cause the long-term activation of the MAPK pathway, leading to uncontrolled cell growth and proliferation. BRAF inhibitors block downstream kinases, causing cell death and suppressing growth and proliferation [30].
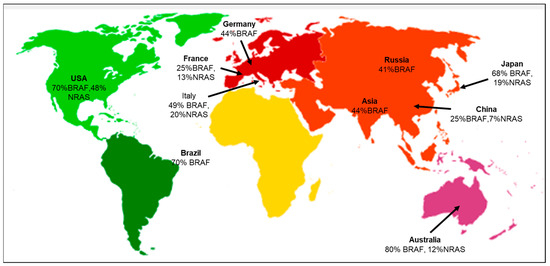
Figure 2.
Geographical distribution of co-occurring BRAF and NRAS mutations [31]. Co-occurrence of both BRAF and NRAS mutations specific to the various geographical areas, including Germany [32], USA [33,34], Italy [35], France [36], Brazil [37], Asia [38], Russia [39], China [40], Japan [41], and Australia [42].
All these variants accelerate the transition from primary to metastatic melanoma. In the breakthrough or initial phases, a normal melanocyte develops an initial driver mutation, causing melanocyte hyperplasia and the production of a melanocytic nevus. BRAF and NRAS mutations are the most common mutations found in melanocyte nevi, with the latter found mostly in congenital nevi [43,44,45]. In the subsequent step, called the expansion phase, certain melanocytic nevi advance into intermediate lesions that develop TERT promoter mutations and eventually develop into melanoma in situ [46]. Once the primary melanoma has accumulated several mutations in CDKN2A, TP53, PTEN, and other genes, it enters the invasive phase and transforms into malignant melanoma [45,47]. This is the phase with many genetic variants and a high mutational burden. The expression pattern of the top 20 most frequently altered genes in melanoma was explored using the UALCAN database [48,49] and is presented in Figure 3.
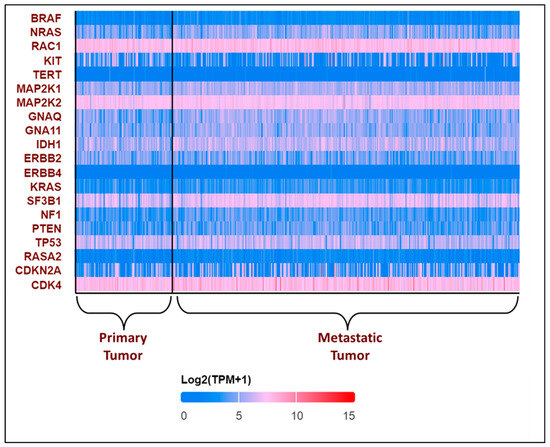
Figure 3.
Expression patterns of input genes in skin cutaneous melanoma: The heat map illustrates the expression profiles of various genes in primary and metastatic tumors in melanomas from the TCGA subset. Blue indicates lower expression, and red indicates higher expression on log 2 (TPM + 1) scale using UALCAN. The expression profile of genes associated with melanoma in melanoma patients was obtained from the UALCAN-UAB database [48]. The Y-axis represents the major genes associated with melanoma, and the X-axis represents the tumor type—primary tumor (n = 104) or metastatic tumor (n = 368). The image describes the differential expressions of the primary and the metastatic tumors. The above plot shows that RAC1, MAP2K2, and CDK4 consistently show high expression in all the patients, while TERT and ERBB4 show low expression. BRAF was found to have a lower expression, with log2(TPM + 1) values lying between 0 and 5, which is not consistent with the existing literature [50]. The expression profile of the other genes varies in each patient.
3. Understanding the Genetic Changes That Fuel Treatment Resistance: The Melanoma Code
3.1. BRAF and MEK Inhibitors
The mutations in the BRAF gene significantly impact more than 50% of melanoma cases. A mutation at position V600E results in the constant activation of the BRAF protein within the MAPK pathway [51]. The BRAF V600 mutation is the most common mutation in melanoma. Two FDA-approved drugs, vemurafenib and dabrafenib, target BRAF V600 mutations. However, the patient response to these BRAF inhibitors is very limited to certain patients. Treatment with MEK inhibitor trametinib as a first-line therapy yielded similar results as BRAF inhibitors [52]. Patients treated with BRAF or MEK inhibitors exhibited a fast antitumor response; the drivers of acquired antitumor resistance involve the reactivation of the MAPK pathway [52,53]. Additional mutations in the MAPK pathway, specifically in the MEK1 and MEK2 genes, are one of the most frequent and can reactivate the MAPK pathway, enabling the cancer cells to proliferate and advance [54]. Dabrafenib and trametinib were the first BRAF and MEK inhibitor combination and currently FDA-approved drug combination to treat advanced BRAFV600-mutated melanoma patients. The BRAF and MEK inhibitor combination exhibited better response and survival rates and fewer side effects compared to the standard monotherapy with BRAF inhibitors. A total of 15–20% of patients with BRAFV600E mutation do not respond to these drugs. Although the MAPK pathway is a successful approach for treating BRAF-mutated melanoma, the rewiring of signaling pathways inevitably leads to acquired resistance. Tumor cells can adapt to BRAF- and MEK-inhibitor treatments through dynamic alterations in signaling networks [18,52,55,56].
Hoogstrat et al., in 2015, showed that a BRAFL505H mutation in melanoma conferred resistance to vemurafenib treatment. This suggests that the BRAFL505H mutation may play a substantial role in therapy resistance [57]. Patients with metastatic BRAFV600-mutant melanoma develop resistance to selective RAF kinase inhibitors, which results in changes to the MAPK pathway that confer resistance. Additionally, the RAF inhibitor therapy results in various genetic resistance mechanisms, most notably the reactivation of the MAPK pathway [53]. There are several mechanisms by which BRAF inhibitors can acquire resistance. Additional mutations in the MAPK pathway, specifically in the MEK1 and MEK2 genes, are one of the most frequent. Even in the presence of the BRAF inhibitor, these alterations can reactivate the pathway, enabling the cancer cells to proliferate and advance [54].
Given the collaborative nature of the MEK and BRAF genes, drugs that hinder MEK proteins can be beneficial in addressing BRAF gene alterations present in melanomas. Notably, MEK inhibitors such as Trametinib (Mekinist), cobimetinib (Cotellic), and binimetinib (Mektovi) have been developed for this purpose [52,58].
BRAF protein reactivation can occur through various mechanisms, including the frequent occurrence of amplified mutated BRAF alleles, resulting in elevated BRAF protein expression [18]. Consequently, the administered dosage of BRAF inhibitors becomes insufficient to inhibit its activity effectively. This overexpression can also induce spontaneous dimerization of the mutated BRAFV600E protein, resulting in the revival of the ERK signal transduction pathway and subsequent resistance to inhibitors. Additionally, splice variants of BRAF, attributed to mutations or epigenetic changes, have been found in a subset of resistant melanomas, such as p61BRAFV600E, which forms dimers independently of RAS kinase activation, rendering BRAF inhibitors ineffective against BRAFV600E dimers [59,60,61]. Furthermore, BRAF gene amplification has been observed in a proportion of BRAF inhibitor resistant tumors, contributing to ERK reactivation [62]. Another resistance mechanism involves tumor microheterogeneity, where some cells carry the BRAF V600 mutation while others remain wild-type for BRAF.
3.2. Genetic Background of Therapy Resistance
Mitogen-activated protein kinase (MAPK) is a signal-transduction pathway involved in various physiological programs, such as cell proliferation, differentiation, development, migration, apoptosis, and transformation. The overall MAPK signaling is divided into three families: MAPK/ERK (extracellular signal-regulating kinase) family, c-Jun N-terminal kinase (JNK), and p38 MAPK signaling family [63]. Briefly, in MAPK-ERK, signaling occurs downstream of ligand binding to receptor tyrosine kinases (RTKs). This binding leads to the activation of signaling adapters GRB2 by activating RAS-GDP (inactive RAS bound GDP) to RAS-GTP (active form of RAS bound GTP). Activated RAS-GTP triggers a cascade of events by activating the protein kinase activity of RAF isoforms (RAF1, BRAF, and ARAF). Each RAF isoform activates MEK (BRAF is the strongest activator). MEK phosphorylates and activates ERK1 and ERK2. ERK can translocate to the nucleus and phosphorylate several transcription factors, which control the cell cycle progression and cell survival [64,65,66].
In melanoma, a dysregulated MAPK pathway can lead to abnormal cell proliferation, survival, invasion, metastasis, and angiogenesis. The BRAF (oncogenic driver mutation) is highly mutated in melanomas; approximately 93% of melanomas harbor BRAF mutations that have MAPK activation in melanoma development [64,65,66]. The BRAFV600E mutation is the most frequent mutation (70–88%), which is a GOF mutation with elevated BRAF kinase activity and constitutive activation of downstream targets [65,66]. The mutant KRASQ61, another common mutation of KRAS (Kirsten rat sarcoma viral oncogene homolog) in melanoma, can decrease its intrinsic hydrolytic activity and sustain the active state of KRAS. RAS overstimulation leads to LOF mutations in NF1 (Neurofibromin 1). In most melanomas, LOF mutations in NF1 lose their ability to inactivate RAS and promote the stimulation of the RAF and its downstream targets, leading to the activation of the MAPK pathway and subsequent cell proliferation and survival [66].
In melanoma, several genomic changes have been identified as drivers of acquired resistance to BRAF inhibitors. The most prevalent resistance mechanisms encompass BRAF splice variants, BRAF amplification, neuroblastoma RAS viral oncogene homolog (NRAS), and mutations in MEK1/2. Importantly, these mutations have been linked to distinct disease phenotypes, indicating that the specific genetic alteration can influence the course and characteristics of melanoma in response to treatment [67,68]. Table 4 illustrates the details of BRAF inhibitors leading to resistance in melanomas.
Splice variants of the BRAF gene play a role in causing resistance by impacting the process of dimerization. In cells containing the normal, or wild-type, BRAF gene, activation by RAS results in the formation of dimers, either as pairs of identical BRAF molecules (BRAF-BRAF) or mixed pairs with another protein called CRAF (BRAF-CRAF). Conversely, in cells with V600E mutations in the BRAF gene, dimerization does not happen, and MEK activation occurs through individual, unpaired BRAF molecules. Consequently, BRAF inhibitors are ineffective in treating melanomas that have the wild-type BRAF gene because the dimer pairs, whether they consist of identical or mixed molecules, maintain their signaling capabilities despite the presence of the inhibitors. On the other hand, these inhibitors effectively prevent the activity of single BRAF molecules. Interestingly, splice variants of the BRAFV600E gene possess the ability to form dimers, allowing them to activate the MEK pathway even when BRAF inhibitors are administered, thus contributing to resistance mechanisms [67,68].
Alterations in essential components within the NRAS/BRAF/MEK pathway result in the revival of previously inhibited MAPK pathways or the initiation of alternative signaling routes, such as the phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT) pathway. In certain cases of BRAF inhibitor resistance among patients, the activation of PI3K/AKT is triggered by the absence of phosphatase and tensin homolog (PTEN) expression [69,70]. BRAF inhibitor-resistant melanoma cells have an altered genetic makeup that leads to heightened cytoprotective autophagy, allowing uncontrolled tumor cell proliferation and increased melanoma cell invasion due to elevated adenosine triphosphate (ATP) secretion. However, certain mutations function autonomously without affecting downstream pathways, and frequently elevated gene expression in BRAF inhibitor-resistant cells is linked to growth factors, their receptors, cell-adhesion molecules, and extracellular matrix interactions, with typical mutations involving receptor tyrosine kinases (RTKs) like epidermal growth factor receptor (EGFR), platelet-derived growth factor receptor (PDGFR), hepatocyte growth factor (HGF), or insulin-like growth factor (IGF) receptor, thereby instigating parallel signaling pathways [67,68,71]. In cells that become resistant to BRAF inhibitors, research has found that multiple receptor tyrosine kinases (RTKs) become overactive. This occurs because inhibitors block the normal process of limiting RTK activity on the cell surface. As a result, there is an increase in RTK levels on the cell surface, which leads to the activation of alternative signaling pathways, contributing to drug resistance [67,68,71,72].
Table 4.
Genetic mutations causing resistance mechanism to BRAF inhibitors.
Table 4.
Genetic mutations causing resistance mechanism to BRAF inhibitors.
| Mutation | Mechanism |
|---|---|
| BRAF gene amplification and splicing | The BRAF gene was amplified, which significantly increased the expression (BRAF protein) and prompted the reactivation of ERK when BRAF inhibitors were present. The production of shortened BRAF proteins, which contain the kinase domain but lack the RAS-binding N-terminus region, can result through alternative splicing and form homodimers that are resistant to BRAF inhibitors [59,68,70,73,74,75]. |
| BRAF secondary mutations | Patients who were resistant to BRAF inhibitors showed secondary mutations in L505H or the single-nucleotide alteration V600E. The V600E mutation raises BRAF kinase activity and results in MEK inhibitor cross-resistance [57,76]. |
| MEK1/2 mutations | Without BRAF activation, MEK1/2 mutations could restart downstream ERK signaling [73,74,75,77]. |
| Receptor interaction proteins, RTKs, or membrane receptors are upregulated | Through the stimulation of parallel pathways or by directly inducing the RAS pathway, overexpression or hyperactivation of membrane receptors/RTKs, which is partially mediated by MITF copy gain, may promote acquired resistance [69,70,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92]. |
| Inconsistencies with in PI3K-AKT cascade | PI3K and AKT-activating mutations enhance AKT signaling by promoting anti-apoptotic signals and elevating expression of essential proliferative genes, enabling survival signals independent of BRAF [70,93,94,95,96,97,98,99,100,101,102]. |
In melanoma resistant to BRAF inhibitors, overexpressed tyrosine kinase receptors initiate signal transduction from the cell membrane to MAPK/ERK kinases, inducing cell division through ARAF and CRAF kinases rather than BRAF, leading to resistance to BRAF inhibitor/MEK inhibitor treatment as cells with the BRAFV600E mutation switch to different RAF isoforms (ARAF or CRAF), ultimately reactivating the ERK pathway.
Mutations in the RAS gene, a frequently mutated oncogene in human neoplasms, disrupt GTPase activity, maintain the protein in its active GTP-bound state, and mediate signal transduction from tyrosine kinase membrane receptors [18]. Mutations in the RAS gene can hyperactivate the MAPK/ERK pathway by phosphorylating ARAF and CRAF proteins, compensating for BRAF inhibition and inducing cell division, particularly when ARAF or CRAF is overexpressed while BRAF is inhibited; the mutated RAS protein, locked in its permanently activated GTP-bound state, also stimulates BRAFV600E dimerization, resulting in ERK pathway reactivation and resistance to BRAF inhibitors that exclusively target BRAFV600E monomers [78,103,104]. Dysregulated signaling through oncogenic BRAF, a crucial constituent of the RAS pathway, causes ongoing activation of downstream effectors involved in cell cycle progression, increasing cell division and tumor growth. Moreover, this RAS activation promotes enhanced cell survival and heightened resistance to apoptosis [105]. Melanomas can evade cell death mechanisms due to dysregulated signaling in the MAPK pathway, leading to increased signaling activity and promoting them to proliferate and survive. For instance, the MAPK pathway’s reactivation and activation of the PI3K/AKT pathway may lead to the emergence of resistance to MAPK inhibitors, which can elevate the expression of the RAS/RAF/MEK/ERK network’s genes. However, the activation of other signaling pathways, such as the PI3K/AKT pathway, might result in the resistance to MAPK inhibitors. Various cytokines and growth factors that activate the PI3K/AKT pathway are crucial for regulating cell survival and growth [106]. The MAPK pathway can be reactivated and become resistant to MAPK inhibitors if the PI3K/AKT pathway is engaged, which can promote the production of genes implicated in the process. The MAPK pathway upstream of the RAS/RAF/MEK/ERK pathway can be stimulated by PI3K/AKT pathway activation. For RAF, MEK, and ERK to be activated, one of these genes, RAS, must be present. RAS and RAF activity can override the MAPK pathway’s inhibition by BRAF inhibitors and revive it, leading to resistance [107]. An additional 20% of melanomas resistant to BRAF inhibitors develop modifications that activate both the MAPK cascade and the adaptive PI3K/AKT survival pathway, which encompass gain-of-function mutations in NRAS and KRAS, along with an increased expression of receptor tyrosine kinases such as EGFR and PDGFRß [108]. Additionally, the induction of angiogenesis further feeds the developing tumor with nutrition and oxygen, thereby aiding in its survival and growth. Furthermore, the increased invasion and metastasis observed in melanoma cells may be caused by dysregulated RAS signaling.
Another gene activation that transcribes neurofibromin is the NF1 gene, a GTPase-activating protein that negatively regulates RAS, the initial component of the MAPK signaling pathway, by facilitating the conversion of RAS-GTP to RAS-GDP through hydrolysis. When neurofibromin is non-functional, it leads to the activation of multiple signaling pathways like MAPK and PI3K, consequently stimulating cell proliferation and survival [109,110]. Mutations in these proteins can lead to their constant activation, contributing to oncogenic signaling pathways [111]. The epidermal growth factor receptor (EGFR) is closely associated with melanoma reoccurrence and progression [112]. Further, by elevating protein synthesis and glucose metabolism, the PI3K/AKT pathway can support cancer cells’ survival and development (Figure 4) [113].
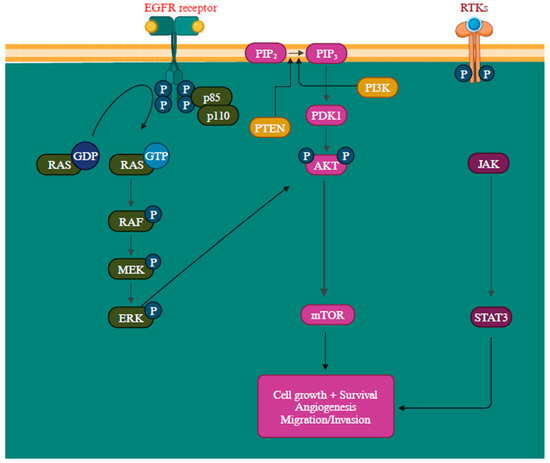
Figure 4.
Overview of PI3K/AKT, RAS/MAPK, and JAK (Janus Kinases)/STAT (signal transducer and activator of transcription) signaling pathways promoting melanoma metastasis [114,115]. The MAPK, PI3K/AKT, and JAK/STAT pathways are all regulated by the receptor tyrosine kinases (RTKs) [113]. Created with BioRender.com.
The PTEN functions as a suppressor gene by regulating the cell cycle through catalyzing PIP3 dephosphorylation at the 3′ position of the inositol ring, thereby inhibiting the PI3K/AKT signaling pathway and, consequently, halting cellular proliferation [116]. The inactivation of the PTEN gene is detected in approximately 10% to 35% of melanoma and is a prevalent factor contributing to the resistance to BRAF inhibitors [18,117,118]. When PTEN protein expression is diminished, it leads to continuous PI3K/AKT signaling pathway activation, promoting cell proliferation growth and suppressing apoptosis [118]. The intricacy of the PI3K/AKT/mTOR signaling network entails multiple feedback mechanisms, extensive interactions with other signaling pathways, and alternative pathways, creating numerous avenues to bypass the impact of PI3K inhibition [119]. The precise mechanism remains incompletely characterized, but recent research has outlined several potential resistance mechanisms, such as the reactivation of PI3K, the activation of parallel pathways, and the influence of the tumor microenvironment. The RAS-RAF-MEK-ERK pathway exhibits extensive interconnections with PI3K signaling. Simultaneous blocking of PI3K and mTOR has been observed to trigger a favorable feedback reaction, resulting in the heightened activation of JAK/STAT, consequently contributing to the development of drug resistance (Figure 4) [113].
3.3. MicroRNA and Melanoma
MicroRNAs (miRNAs) are small, non-coding RNA molecules that regulate gene expression and have emerged as critical players in cancer biology, especially in melanoma. Since their discovery, miRNAs have been found to influence molecular and cellular processes. In melanoma, specific miRNAs regulate key signaling pathways that drive tumor growth, metastasis, and treatment resistance [120]. miRNA profiling studies in melanoma identified a network of over 20 miRNAs dysregulated by aberrant B-RAF/MKK/ERK signaling. Similarly, 19 novel miRNA candidates dysregulated in clinical cutaneous melanoma samples [121,122]. For instance, dysregulation of miR-21 was detected in melanoma, with an increased copy number in some melanoma cell lines and upregulated in several highly invasive cell lines [123,124]. miR-21 expression in melanoma leads to the inhibition of PTEN and PDCD4 targets [125,126]. The dysregulation of several miRNAs has been influenced by transcription factors, epigenetic mechanisms, and DNA copy number alterations. In melanomas, few miRNAs act as tumor suppressors or as oncogenes. These miRNAs add complexity to the signaling networks and are seen as a potential therapeutic target for melanoma treatment [126].
4. Mechanisms of Resistance to Immune Therapy/Checkpoint Inhibitors
Immune checkpoint inhibitors (ICIs) are drugs that block immune checkpoints such as PD-1 (anti-programmed cell death protein 1), PD-L1 (ligand for PD-1 receptor), and CTLA-4 (cytotoxic T lymphocyte-associated antigen-4), which typically restrain the immune system’s activity. Cancer cells can manipulate the PD-1/PD-L1 pathway to evade immune recognition. CTLA-4 and PD-1 are T-cell surface receptors associated with immune suppression and dysfunction. Currently, seven ICIs have been approved by the U.S. FDA, of which one is a CTLA-4 inhibitor (ipilimumab), three are PD-1 inhibitors (nivolumab, pembrolizumab, and cemiplimab), and three are PD-L1 inhibitors (atezolizumab, durvalumab, and avelumab). ICIs have changed the approach of treatment strategy in numerous cancer types. The rapid FDA approval of ICIs can be attributed to clinical trial data demonstrating their superior anti-melanoma effects compared to traditional therapies. Many groups around the world have studied the approach of ICIs in melanomas; most approaches were successful, while in advanced-stage melanoma patients’ application of ICIs, monoclonal antibodies CTLA-4, PD-1, and PD-L1 have produced a good response (a median survival of 5 years). However, most patients show no response to the treatment. Identifying the predictive biomarkers to differentiate between responders and non-responder patients for these therapies is necessary for choosing the treatment regime. The circulating cytokine IL-6 level is a potential biomarker to distinguish between advanced stage and poor prognosis in patients with different cancer types. A recent study suggests that cytokine IL-6 could be the biomarker of disease progression and poor prognosis in BRAF wild-type advanced melanoma treated with pembrolizumab. However, more studies are required to establish the definitive role of IL-6 levels as potential prognostic and predictive biomarkers in various groups of melanoma patients [127,128,129,130,131,132,133,134]. Besides the lack of definitive biomarkers, the underlying mechanism for the resistance remains elusive.
The major concern with ICIs is deciphering the intricate resistance mechanisms and to developing novel drug combinations to optimize treatment approaches to overcome the resistance. The resistance mechanism can be primary or acquired. Primary resistance is an inherent lack of response to the treatment; acquired resistance emerges during treatment. An overview of primary and acquired resistance mechanisms to ICI therapy is summarized in Table 5. The mechanism of resistance is categorized as intrinsic or extrinsic to tumor cells. Intrinsic resistances are related to the mechanism specific to the tumor cells involved in immune responses, cell signaling, gene expression, and DNA damage response. Extrinsic resistances are associated externally with the tumor cells throughout the T-cell activation [127].
Table 5.
Mechanisms of resistance against immune checkpoint inhibitors (ICIs).
Table 5.
Mechanisms of resistance against immune checkpoint inhibitors (ICIs).
| Drivers | Immune-Evasion Mechanism | References |
|---|---|---|
| Primary resistance | ||
| VEGF and ANG2 overexpression | TME infiltration by TILs | [135] |
| CXCR3 | Restores the cytotoxicity of CD8+ in TME | [136,137] |
| TMB | Regulates immunotherapy response | [138,139,140,141] |
| IL-6 and IL-10 levels in TME | Impairs DC maturation | [142,143,144] |
| Acquired resistance | ||
| B2M | Regulates MHC class I-mediated tumor antigen presentation in tumor lesions | [127,145] |
| JAK1 | Regulates transcription of the IFN- γ-inducible genes and T-cell infiltration | [146] |
| EZH2 |
| [147,148] |
| KDM5B | Overexpression of SETDB1 (H3K9 methyl transferase) | [149,150,151,152] |
| SETDB1 | Regulates the expression of immune-related gene clusters that encode MHC I antigens | [134,150] |
| HDAC6 | Regulates IL-10 and PD-L1 expression | [148] |
| FTO | Regulates PD-1 expression | [153] |
| LAG3 | Regulates the activity of T-cells | [154,155,156,157] |
| TIM-3 | Regulates the activity of T-cells | [134,158,159] |
| SK1 | Regulates S1P, which in turn regulates lymphocyte trafficking and differentiation | [160,161] |
| FCRL6 | Regulates cytotoxic NK cells and effector T-cells | [162] |
| NLRP3 | Regulates the recruitment into tumor tissues | [163] |
| Microbiome | Regulates macrophage polarization and DCs activation and CD8+tumor recruitment | [164,165,166,167,168] |
VEGF—vascular endothelial growth factor; ANG2—angiopoietin 2; TME—tumor microenvironment; TILs—tumor-infiltrating lymphocytes; CXCR3—C-X-C Motif Chemokine Receptor 3; TMB—Tumor Mutational Burden; DC-dendritic cell; B2M—Beta 2 microglobulin; JAK1—Janus Kinase 1; EZH2—histone methyltransferase; KDM5B—H3K4 demethylase; FTO—m6A RNA demethylase); LAG3—lymphocyte-activation gene 3; TIM-3—T-cell immunoglobulin and mucin domain 3; SK1—Sphingosine kinase 1; S1P—sphingosine-1-phosphate; FCRL6—Fc receptor-like 6 protein; NLRP3—Nucleotide-Binding Domain, Leucine-Rich Containing Family, Pyrin Domain-Containing-3; PMN-MDSCs—granulocytic myeloid-derived suppressor cells.
Transcriptomic analysis from the melanoma patients reveals that responsiveness to the pretreatment with anti-CTLA-4 showed a positive correlation with increased tumor mutational burden (TMB) and increased expression of neoantigen and cytolytic markers in the immune microenvironment [169]. In the case of anti-PD-1 pretreatment, responsive melanoma exhibited elevated levels of CD8+ T-cell infiltration and expression of PD-L1 on tumor cells or immune cells; thus, these particular signatures might act as a potential biomarker for treatment responsiveness [170,171,172]. In the melanoma mice model, more infiltration of intratumoral follicular Treg cells reduced responsiveness to anti-PD1 treatment [173,174]. In melanoma patients, MHC-II expression on tumor cells correlates with a more favorable response to anti-PD1/PDL1 treatment [175]. In certain individuals, due to immunoediting, the immune system selects subclones of tumor cells lacking expression of neoantigens, causing poor immunogenicity and resistance to ICIs [127]. Altogether, high TMB, increased expression of MHC-II, and depleted levels of Tregs improve the efficacy of anti-PD1 treatment [173].
4.1. Clinical Predictors of Immune Therapy in Metastatic Melanoma
In a multi-institutional retrospective analysis of 229 melanoma patients, 60 patients (26%) had NRAS G12/G13/Q61 mutations, 53 patients (23%) had BRAFV600 mutations, and 116 (51%) had neither NRAS/BRAF mutations. In response to first-line immune therapy (IL2, ipilimumab, and anti-PD-1/PD-L1), 28% of the NRAS-mutant cohort showed a complete response/partial response (CR/PR). In contrast, the NRAS/BRAF wild-type cohort exhibited a 16% response (28% vs. 16%, p = 0.04), and the best response to any line of immunotherapy was 32% and 20%, respectively (32% vs. 20%, p = 0.07). The patients with NRAS-mutant melanoma exhibited a heightened response rate and experienced clinical benefit from immune therapy (Table 6). This retrospective study indicates that advanced melanoma with NRAS mutations exhibits better immune-based treatment outcomes than non-NRAS mutations [176].
Table 6.
Overall response rate and clinical benefit to immune therapy [176].
We evaluated OS and PFS for patients with BRAF mutations from the ICI cohort (Miao_Melanoma-OS and Miao_Melanoma-PFS datasets) to generate survival curves using Kaplan–Meier analysis. We observed an improved OS and PFS trend for patients with BRAF mutations (Figure 5).
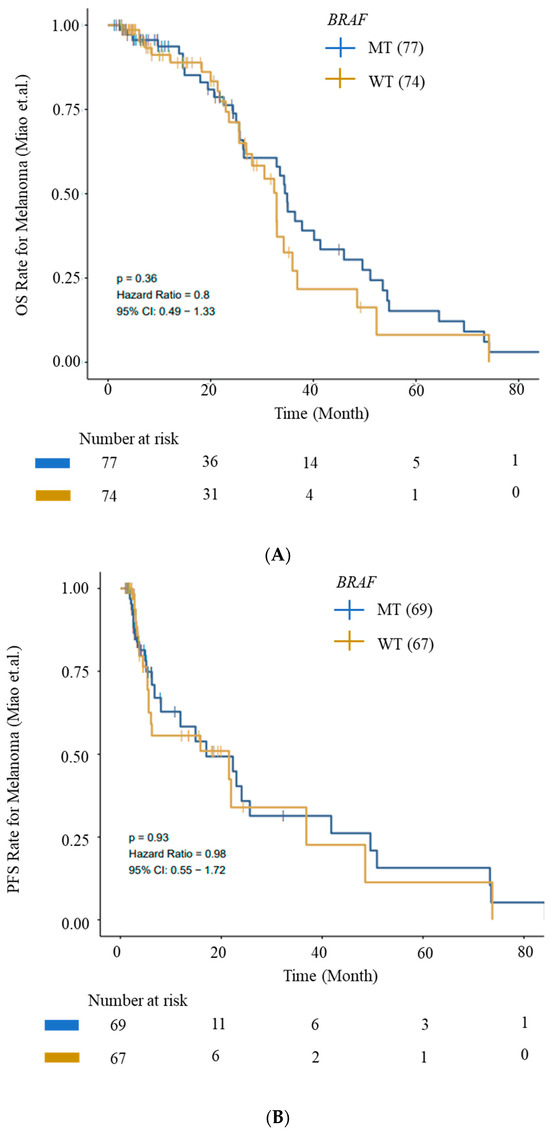
Figure 5.
Kaplan–Meier survival curves for OS and PFS. (A) OS and (B) PFS from ICI Cohort for BRAF (wild-type—WT) and BRAF-mutant (MT) cohorts. Improved OS and PFS trends for patients with BRAF mutations are depicted. Data were generated using the CAMOIP tool.
Besides somatic mutations, CNVs might also aid in the selective response to ICIs. Data from small cohorts of melanoma patients treated with ICIs suggest that the integrity of the IFN-γ pathway is essential for the responsiveness to anti-PD1 and anti-CTLA-4 treatment. This indicates that a loss of IFN-γ signaling in tumor cells may promote resistance to immune checkpoint therapies [177,178].
A high mutational load (nonsynonymous mutations per exome) also exhibited a better clinical benefit from ipilimumab treatment. However, the mutational load alone does not effectively indicate CTLA-4 blockade therapy response. The therapeutic advantages of ipilimumab were observed in correlation with tumor-specific neoantigens. The tumor-specific expression of somatic neoepitopes increased the overall antigenicity trend. Patients with sustained clinical benefits demonstrated the expression of a tetrapeptide neoantigen signature. Similarly, the presence of this tetrapeptide signature correlated strongly with survival. Mutations resulting in the presentation of specific neoepitopes enhance MHC class I binding, eliciting an intensified antitumor response augmented by CTLA-4 blockade [179].
In a parallel study, a transcriptomic analysis of tumor biopsies from 40 melanoma patients revealed a connection between improved immune therapy response and factors such as a higher mutational load, increased neoantigen load, and elevated expression of cytolytic markers within the immune microenvironment [169]. Single-cell RNA sequencing and computational analyses on 33 melanomas identified a distinct resistance program unique to tumor cells. This program is linked to T-cell exclusion and immune evasion. CDK4 is one of the key master regulators involved in the resistance program. Counteracting this program through CDK4/6-inhibition enhances the responsiveness of melanoma to ICIs in mouse models [180].
4.2. Predictive Features of Response to Immune Checkpoint Blockade (ICB)
Auslander et al. developed an immuno-predictive score (IMPRES), a predictor of ICB response in melanoma patients. IMPRES is constructed based on pair-wise relations between the expressions of 28 checkpoint genes with co-stimulatory or co-inhibitory effects. The above study identified seven immune-related consistently differentially expressed pathways (termed CDPs) that are common in all anti-PD-1 datasets and four CDPs common across all anti-CTLA-4 datasets. The correlation between each IMPRES feature and the expression of each of the CDPs was computed. Subsequently, IMPRES was used to predict the response to ICB among melanoma patients. While IMPRES can predict all the true responders, it misses half of the nonresponders. Elevated IMPRES scores correlate with enhanced OS and PFS in melanoma patients treated with ICB [181].
An analysis of copy number variations using whole-exome sequences (WES) from 469 melanoma cases did not identify any specific recurrent variation to either responders or non-responders to immune therapy treatment. BRAC2 with nsSNVs (6 of 21 tumors) are better responders. These BRAC2 loss-of-function mutations might lead to a defect in homologous recombination, double-strand DNA break repair, or some unknown effects that add to responsiveness to anti-PD-1 treatment. Transcriptomic analysis was performed on anti-PD-1 responding and non-responding tumors to analyze the differentially expressed genes (DEGs). A total of 693 genes were differentially expressed, and relative gene up-expression events were higher in non-responding tumors than in responding tumors. DEGs that are expressed in higher levels in pre-treatment tumors that do not respond encompass genes linked to mesenchymal transition, immunosuppression, chemotaxis of monocytes and macrophages, as well as genes associated with wound healing and angiogenesis. Transcriptomic signatures derived from perturbation-based analysis displayed co-enrichment patterns (9 of 13 non-responding vs. 1 of 15 responding pretreated anti-PD-1 tumors). These collective signatures are termed as the innate anti-PD-1 resistance (IPRES) signature. Innately resistant tumors exhibit IPRES, indicating upregulation of events involved in regulating mesenchymal transition, cell adhesion, remodeling of the extracellular matrix (ECM), wound healing, and angiogenesis. Treatment with mitogen-activated protein kinase (MAPK) inhibitors causes comparable alterations in residual melanoma. This observation implies that these signatures might negatively impact the responsiveness to anti-PD-1 therapy [182].
5. Gender Differences in Melanoma
Melanoma exhibits notable differences in molecular mechanisms between sexes. These differences can influence disease susceptibility, progression, and response to treatment. Understanding these disparities is crucial for developing tailored prevention and treatment strategies [183]. Recent studies indicate that men and women experience melanoma differently. Males have a higher risk of developing melanoma and a higher risk of mortality compared to female counterparts [184]. In the retrospective study performed on a population of 1023 cutaneous melanoma patients, female melanoma patients showed statistical differences in disease-free survival (DFS) and overall survival (OS) compared to male patients. Men showed a significantly lower median DFS than women (22 vs. 104 months; p < 0.001) and OS (20.7 vs. 104 months; p < 0.001). Similarly, subgroup analysis revealed a statistically significant difference in DFS and OS favoring females compared to male patients with TNM stages I and II. However, no significant differences were observed in TNM stages III and IV. These data were examined without any consideration given to various therapies undergone by the patients [185]. In general, mortality rates are higher among men than women [186].
A population-based cohort study of 11,774 cutaneous melanoma cases from the Munich Cancer Registry found that females showed a 38% lower risk of death compared to males. Female patients, in general, exhibited thinner tumors and less disease progression and metastasis compared to male patients. These differences might be attributed to differences in tumor–host interaction across genders [184,187,188]. The mechanisms responsible for the gender disparity in melanoma treatment outcomes are not well understood. But several factors, such as hormonal differences, signaling pathways, immune function, oxidative stress response, and gene expression, are likely to contribute to the significant differences in melanoma [189,190].
In a retrospective, multicohort analysis of patients with metastatic melanoma, obese male patients showed improved PFS and OS compared to male patients with a normal BMI treated with targeted or immune therapy [191].
6. Conclusions
Given its location, melanoma is a reasonably easy malignancy to collect and one that is potentially lethal. The scientific community has learned a great deal about the disease and its progression because of the ease with which patient samples can now be accessed. Melanoma, characterized by its molecular complexity and heterogeneity, poses significant challenges in understanding its pathogenesis and effectively treating it. The disease is driven by genetic mutations, such as those in the BRAF, NRAS, and KIT genes, contributing to its aggressive nature and variability in clinical outcomes [192]. Also, epigenetic alterations, including DNA methylation and histone modifications, play critical roles in melanoma development and therapy resistance [193]. The regulatory impact of microRNAs further complicates the molecular landscape, with certain miRNAs acting as either oncogenes or tumor suppressors, influencing tumor progression, metastasis, and treatment response [121,122,125,126]. We have succeeded in determining the most common mutations in the BRAF, NRAS, and TERT genes that cause melanoma [194].
Genomic sequencing of cancer patient tumor samples has helped researchers analyze gene abundance and identify treatment strategies. Melanoma has a high mutation burden, with 70–80% of melanomas having BRAF mutations. These mutations activate the MAPK pathway, which is essential for tumor growth. Survival analysis varies in males and females, with BRAF mutations having a higher survival rate than NRAS. Patients with BRAF mutations have an average of 80 months of OS and 70 months of PFS when using the CAMOIP tool.
The use of OMICS-based approaches has been a boon to researchers in handling large amounts of data, interpreting them, and easily analyzing various mutations, co-factors, and biomarker identification with the latest use of AI in the identification of treatment strategies that benefit a high proportion of patients for the precision targeted therapy. Certain mutations are exclusive to a given region, the co-occurrence of some mutations has been noted, and the survival analysis for the BRAF and NRAS oncogenes differs significantly in males and females.
Skin cancer is treated using a variety of techniques, such as photodynamic treatment, radiation, cryotherapy, and immunotherapy. An innovative, cost-effective, and effective treatment for skin cancer is needed due to its rising severity and numerous treatment restrictions. Over the last ten years, targeted therapy and immunotherapy have emerged as the two main approaches that have transformed the landscape of systemic treatment for metastatic melanoma.
Several somatic and germline alterations, such as SNVs and CNVs, are linked with melanoma. The oncogenes acquire GOF mutations in the process, as well as LOF mutations, which silence the tumor-suppressor genes. UALCAN is one of the beneficial open-source OMICS instruments used to examine the expression of these tumor-suppressor and oncogene genes. Notably, the finding that BRAF mutations are common in about 40–50% of melanoma patients has spurred a renaissance of interest in this sector. This genetic mutation causes the downstream MAPK pathway (RAS/RAF/MEK/ERK proteins) to be constitutively activated, which is essential for tumor growth. The predominant mutation is BRAFV600E, which accounts for over 80% of all BRAF alterations. This mutation replaces valine with glutamate through a single nucleotide change (GTG to GAG). Moreover, V600K mutations, which involve a two-fold nucleotide change (GTG to AAG) and the substitution of lysine for valine, account for approximately 16% of BRAF alterations. These oncogenic alterations significantly influence tumor growth and metastasis initiation and promotion. Novel BRAF small-molecule inhibitors, including vemurafenib, dabrafenib, and encorafenib, have been developed as therapeutic approaches for treating melanoma in response to this insight. These inhibitors provide a promising method of treating this difficult condition by focusing on faulty BRAF signaling.
ICI treatment has been successful in treating melanoma, but a significant percentage of patients show resistance. Factors influencing resistance include elevated neoantigen, cytolytic markers, PD-L1, and MHC-II expression. New therapeutic approaches should consider the tumor microenvironment and its elements. Advanced melanoma patients with NRAS and BRAF mutations respond better to treatment. Recent studies have established IMPRES predictors for better treatment outcomes, but finding definitive indicators is challenging due to resistance systems.
The transcriptome analyses of several melanoma patients show that different factors may influence the resistance to immune checkpoint inhibitor treatment, such as elevated expression of neoantigen, cytolytic markers, PD-L1, and MHC-II. New therapeutic approaches could be evaluated with a deeper comprehension of the tumor microenvironment, its elements, and the biochemical makeup and metabolic profile of its constituent cells. A small-cohort, multi-institutional retrospective investigation indicates that advanced melanoma patients with NRAS mutations respond better to first-line immunotherapy. BRAF mutations from the ICI cohort showed a greater response to the treatment in a similar type of analysis. This suggests that the mutational load, mutations in NRAS and BRAF, and DEGs of specific targets involved in immunological responses can function as a prognostic marker for the treatment. The methods for predicting characteristics that lead to better treatment outcomes have been established in recent studies. IMPRES is one such predictor; a higher IMPRES score corresponds to increased OS and PFS in melanoma patients receiving ICI treatment. IPRES signatures also suggest the influence of treatment resistance in patients with melanoma. However, it would be difficult to definitively find predictive indicators because of the many systems behind resistance. However, extensive cohorts must be used to validate the discovered predictive markers.
Further, sex-based differences in melanoma susceptibility and treatment responses highlight the need for gender-specific approaches to melanoma management. Variations in hormonal influences, genetic predispositions, and immune responses between sexes can affect disease progression and therapeutic efficacy [183,184]. For example, estrogen has been shown to impact melanoma progression in women, while androgens may influence disease behavior in men. Furthermore, differences in mutation prevalence, such as the higher incidence of BRAF mutations in women, underscore the need for personalized treatment strategies [195].
Despite advancements in targeted therapies and immunotherapies, including BRAF inhibitors and checkpoint inhibitors, challenges remain in managing resistance and relapse. Resistance mechanisms are multifaceted, involving changes in tumor biology, such as alterations in signaling pathways and immune-evasion strategies [196,197]. The emergence of resistant melanoma clones and the development of secondary mutations necessitate ongoing research to identify novel therapeutic targets and combination strategies.
Future progress in melanoma treatment relies on integrating comprehensive molecular insights into clinical practice. By utilizing advanced genomic, epigenomic, and proteomic analyses, researchers and clinicians can enhance the precision of melanoma treatments. This personalized approach aims to improve patient outcomes through tailored therapies that address the specific molecular characteristics of each patient’s tumor. Continued exploration of the molecular mechanisms underlying melanoma will be crucial in overcoming the existing treatment challenges and achieving better therapeutic success.
New therapeutic approaches could be evaluated with a deeper comprehension of the tumor microenvironment, its elements, and the biochemical makeup and metabolic profile of its constituent cells.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/cells13161383/s1, Figure S1. The downstream effect of the RAS signaling pathway.
Author Contributions
Conceptualization, D.M.; writing—original draft preparation, K.K.K., R.N. and P.V.S.; writing—review and editing R.N., K.K.K., P.V.S., Y.M., S.M.V., R.S.A. and D.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data sharing is not applicable.
Acknowledgments
We would like to acknowledge the team of the Global Cancer Consortium (GCC) for organizing the Cancer OMICS Training Workshop. We would also like to acknowledge Sooryanarayana Varambally for providing comprehensive insight on UALCAN-UAB. PVS has received Intra Mural Funding (IMF) from the Manipal Academy of Higher Education. Figure 1, Figure 2, Figure 3 and Figure 4 and the Supplementary Figure were created with BioRender.com.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Khan, N.H.; Mir, M.; Qian, L.; Baloch, M.; Ali Khan, M.F.; Rehman, A.-U.; Ngowi, E.E.; Wu, D.-D.; Ji, X.-Y. Skin Cancer Biology and Barriers to Treatment: Recent Applications of Polymeric Micro/Nanostructures. J. Adv. Res. 2022, 36, 223–247. [Google Scholar] [CrossRef]
- Vink, A.A.; Roza, L. Biological Consequences of Cyclobutane Pyrimidine Dimers. J. Photochem. Photobiol. B Biol. 2001, 65, 101–104. [Google Scholar] [CrossRef]
- Esteva, A.; Kuprel, B.; Novoa, R.A.; Ko, J.; Swetter, S.M.; Blau, H.M.; Thrun, S. Dermatologist-Level Classification of Skin Cancer with Deep Neural Networks. Nature 2017, 542, 115–118. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Ferrara, G.; Argenziano, G. The WHO 2018 Classification of Cutaneous Melanocytic Neoplasms: Suggestions from Routine Practice. Front. Oncol. 2021, 11, 675296. [Google Scholar] [CrossRef]
- Bobos, M. Histopathologic Classification and Prognostic Factors of Melanoma: A 2021 Update. Ital. J. Dermatol. Venereol. 2021, 156, 300–321. [Google Scholar] [CrossRef] [PubMed]
- Savoye, I.; Olsen, C.M.; Whiteman, D.C.; Bijon, A.; Wald, L.; Dartois, L.; Clavel-Chapelon, F.; Boutron-Ruault, M.-C.; Kvaskoff, M. Patterns of Ultraviolet Radiation Exposure and Skin Cancer Risk: The E3N-SunExp Study. J. Epidemiol. 2018, 28, 27–33. [Google Scholar] [CrossRef]
- Gershenwald, J.E.; Scolyer, R.A.; Hess, K.R.; Sondak, V.K.; Long, G.V.; Ross, M.I.; Lazar, A.J.; Faries, M.B.; Kirkwood, J.M.; McArthur, G.A.; et al. Melanoma Staging: Evidence-Based Changes in the American Joint Committee on Cancer Eighth Edition Cancer Staging Manual. CA Cancer J. Clin. 2017, 67, 472–492. [Google Scholar] [CrossRef]
- Eddy, K.; Shah, R.; Chen, S. Decoding Melanoma Development and Progression: Identification of Therapeutic Vulnerabilities. Front. Oncol. 2020, 10, 626129. [Google Scholar] [CrossRef] [PubMed]
- Timis, T.; Bergthorsson, J.T.; Greiff, V.; Cenariu, M.; Cenariu, D. Pathology and Molecular Biology of Melanoma. Curr. Issues Mol. Biol. 2023, 45, 5575–5597. [Google Scholar] [CrossRef]
- Rebecca, V.W.; Sondak, V.K.; Smalley, K.S.M. A Brief History of Melanoma: From Mummies to Mutations. Melanoma Res. 2012, 22, 114–122. [Google Scholar] [CrossRef]
- Fares, J.; Fares, M.Y.; Khachfe, H.H.; Salhab, H.A.; Fares, Y. Molecular Principles of Metastasis: A Hallmark of Cancer Revisited. Signal Transduct. Target. Ther. 2020, 5, 28. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, U.B.; Westphal, J.R.; Waas, E.T.; Zendman, A.J.; Cornelissen, I.M.; Ruiter, D.J.; van Muijen, G.N. Matrix Metalloproteinases in Human Melanoma Cell Lines and Xenografts: Increased Expression of Activated Matrix Metalloproteinase-2 (MMP-2) Correlates with Melanoma Progression. Br. J. Cancer 1999, 81, 774–782. [Google Scholar] [CrossRef] [PubMed]
- Valenti, F.; Falcone, I.; Ungania, S.; Desiderio, F.; Giacomini, P.; Bazzichetto, C.; Conciatori, F.; Gallo, E.; Cognetti, F.; Ciliberto, G.; et al. Precision Medicine and Melanoma: Multi-Omics Approaches to Monitoring the Immunotherapy Response. Int. J. Mol. Sci. 2021, 22, 3837. [Google Scholar] [CrossRef]
- Jurisic, V. Multiomic Analysis of Cytokines in Immuno-Oncology. Expert Rev. Proteom. 2020, 17, 663–674. [Google Scholar] [CrossRef]
- Scatena, C.; Murtas, D.; Tomei, S. Cutaneous Melanoma Classification: The Importance of High-Throughput Genomic Technologies. Front. Oncol. 2021, 11, 635488. [Google Scholar] [CrossRef]
- Bruno, W.; Martinuzzi, C.; Dalmasso, B.; Andreotti, V.; Pastorino, L.; Cabiddu, F.; Gualco, M.; Spagnolo, F.; Ballestrero, A.; Queirolo, P.; et al. Combining Molecular and Immunohistochemical Analyses of Key Drivers in Primary Melanomas: Interplay between Germline and Somatic Variations. Oncotarget 2018, 9, 5691–5702. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Czarnecka, A.M.; Bartnik, E.; Fiedorowicz, M.; Rutkowski, P. Targeted Therapy in Melanoma and Mechanisms of Resistance. Int. J. Mol. Sci. 2020, 21, 4576. [Google Scholar] [CrossRef] [PubMed]
- Leonardi, G.C.; Falzone, L.; Salemi, R.; Zanghì, A.; Spandidos, D.A.; Mccubrey, J.A.; Candido, S.; Libra, M. Cutaneous Melanoma: From Pathogenesis to Therapy (Review). Int. J. Oncol. 2018, 52, 1071–1080. [Google Scholar] [CrossRef]
- Davis, E.J.; Johnson, D.B.; Sosman, J.A.; Chandra, S. Melanoma: What Do All the Mutations Mean? Cancer 2018, 124, 3490–3499. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The CBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the CBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Cormier, J.N.; Xing, Y.; Ding, M.; Lee, J.E.; Mansfield, P.F.; Gershenwald, J.E.; Ross, M.I.; Du, X.L. Ethnic Differences among Patients with Cutaneous Melanoma. Arch. Intern. Med. 2006, 166, 1907–1914. [Google Scholar] [CrossRef]
- Devitt, B.; Liu, W.; Salemi, R.; Wolfe, R.; Kelly, J.; Tzen, C.-Y.; Dobrovic, A.; McArthur, G. Clinical Outcome and Pathological Features Associated with NRAS Mutation in Cutaneous Melanoma. Pigment Cell Melanoma Res. 2011, 24, 666–672. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Castañeda, L.D.; Gamboa, M.; Nova, J.A.; Pulido, L.; Tovar-Parra, J.D. Mutations in the BRAF, NRAS, and C-KIT Genes of Patients Diagnosed with Melanoma in Colombia Population. BioMed Res. Int. 2020, 2020, 2046947. [Google Scholar] [CrossRef] [PubMed]
- Bishop, D.T.; Demenais, F.; Goldstein, A.M.; Bergman, W.; Bishop, J.N.; Bressac-de Paillerets, B.; Chompret, A.; Ghiorzo, P.; Gruis, N.; Hansson, J.; et al. Geographical Variation in the Penetrance of CDKN2A Mutations for Melanoma. J. Natl. Cancer Inst. 2002, 94, 894–903. [Google Scholar] [CrossRef]
- Raaijmakers, M.I.G.; Widmer, D.S.; Narechania, A.; Eichhoff, O.; Freiberger, S.N.; Wenzina, J.; Cheng, P.F.; Mihic-Probst, D.; Desalle, R.; Dummer, R.; et al. Co-Existence of BRAF and NRAS Driver Mutations in the Same Melanoma Cells Results in Heterogeneity of Targeted Therapy Resistance. Oncotarget 2016, 7, 77163–77174. [Google Scholar] [CrossRef]
- Weiss, J.M.; Hunter, M.V.; Cruz, N.M.; Baggiolini, A.; Tagore, M.; Ma, Y.; Misale, S.; Marasco, M.; Simon-Vermot, T.; Campbell, N.R.; et al. Anatomic Position Determines Oncogenic Specificity in Melanoma. Nature 2022, 604, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Fox, D.B.; Ebright, R.Y.; Hong, X.; Russell, H.C.; Guo, H.; LaSalle, T.J.; Wittner, B.S.; Poux, N.; Vuille, J.A.; Toner, M.; et al. Downregulation of KEAP1 in Melanoma Promotes Resistance to Immune Checkpoint Blockade. NPJ Precis. Oncol. 2023, 7, 25. [Google Scholar] [CrossRef]
- Loftus, S.K.; Baxter, L.L.; Cronin, J.C.; Fufa, T.D.; Pavan, W.J. Hypoxia-Induced HIF1α Targets in Melanocytes Reveal a Molecular Profile Associated with Poor Melanoma Prognosis. Pigment Cell Melanoma Res. 2017, 30, 339–352. [Google Scholar] [CrossRef]
- Gutiérrez-Castañeda, L.D.; Nova, J.A.; Tovar-Parra, J.D. Frequency of Mutations in BRAF, NRAS, and KIT in Different Populations and Histological Subtypes of Melanoma: A Systemic Review. Melanoma Res. 2020, 30, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Schlaak, M.; Bajah, A.; Podewski, T.; Kreuzberg, N.; von Bartenwerffer, W.; Wardelmann, E.; Merkelbach-Bruse, S.; Büttner, R.; Mauch, C.; Kurschat, P. Assessment of Clinical Parameters Associated with Mutational Status in Metastatic Malignant Melanoma: A Single-Centre Investigation of 141 Patients. Br. J. Dermatol. 2013, 168, 708–716. [Google Scholar] [CrossRef]
- Jakob, J.A.; Bassett, R.L.J.; Ng, C.S.; Curry, J.L.; Joseph, R.W.; Alvarado, G.C.; Rohlfs, M.L.; Richard, J.; Gershenwald, J.E.; Kim, K.B.; et al. NRAS Mutation Status Is an Independent Prognostic Factor in Metastatic Melanoma. Cancer 2012, 118, 4014–4023. [Google Scholar] [CrossRef]
- Goel, V.K.; Lazar, A.J.F.; Warneke, C.L.; Redston, M.S.; Haluska, F.G. Examination of Mutations in BRAF, NRAS, and PTEN in Primary Cutaneous Melanoma. J. Investig. Dermatol. 2006, 126, 154–160. [Google Scholar] [CrossRef]
- Turri-Zanoni, M.; Medicina, D.; Lombardi, D.; Ungari, M.; Balzarini, P.; Rossini, C.; Pellegrini, W.; Battaglia, P.; Capella, C.; Castelnuovo, P.; et al. Sinonasal Mucosal Melanoma: Molecular Profile and Therapeutic Implications from a Series of 32 Cases. Head Neck 2013, 35, 1066–1077. [Google Scholar] [CrossRef] [PubMed]
- Pracht, M.; Mogha, A.; Lespagnol, A.; Fautrel, A.; Mouchet, N.; Le Gall, F.; Paumier, V.; Lefeuvre-Plesse, C.; Rioux-Leclerc, N.; Mosser, J.; et al. Prognostic and Predictive Values of Oncogenic BRAF, NRAS, c-KIT and MITF in Cutaneous and Mucous Melanoma. J. Eur. Acad. Dermatol. Venereol. JEADV 2015, 29, 1530–1538. [Google Scholar] [CrossRef]
- Inumaru, J.S.S.; Gordo, K.I.F.; Fraga Junior, A.C.; Silva, A.M.T.C.; Leal, C.B.Q.S.; Ayres, F.M.; Wastowski, I.J.; Borges, N.F.; Saddi, V.A. Analysis of the BRAF V600E Mutation in Primary Cutaneous Melanoma. Genet. Mol. Res. GMR 2014, 13, 2840–2848. [Google Scholar] [CrossRef] [PubMed]
- Massad, C.; Loya, A.; Taraif, S.; Saroufim, M.; Kibbi, A.G.; Habib, R.; Novy, M.; Rauscher, B.; Oberkanins, C.; Khalifeh, I. BRAF Mutation Status in Primary and Metastatic Melanomas in Two Regions with Differing Potential Ultraviolet Radiation Exposure. Clin. Exp. Dermatol. 2014, 39, 932–943. [Google Scholar] [CrossRef]
- Aksenenko, M.B.; Kirichenko, A.K.; Ruksha, T.G. Russian Study of Morphological Prognostic Factors Characterization in BRAF-Mutant Cutaneous Melanoma. Pathol. Res. Pract. 2015, 211, 521–527. [Google Scholar] [CrossRef]
- Si, L.; Kong, Y.; Xu, X.; Flaherty, K.T.; Sheng, X.; Cui, C.; Chi, Z.; Li, S.; Mao, L.; Guo, J. Prevalence of BRAF V600E Mutation in Chinese Melanoma Patients: Large Scale Analysis of BRAF and NRAS Mutations in a 432-Case Cohort. Eur. J. Cancer 2012, 48, 94–100. [Google Scholar] [CrossRef]
- Sakaizawa, K.; Ashida, A.; Uchiyama, A.; Ito, T.; Fujisawa, Y.; Ogata, D.; Matsushita, S.; Fujii, K.; Fukushima, S.; Shibayama, Y.; et al. Clinical Characteristics Associated with BRAF, NRAS and KIT Mutations in Japanese Melanoma Patients. J. Dermatol. Sci. 2015, 80, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Thomas, N.E.; Edmiston, S.N.; Alexander, A.; Groben, P.A.; Parrish, E.; Kricker, A.; Armstrong, B.K.; Anton-Culver, H.; Gruber, S.B.; From, L.; et al. Association between NRAS and BRAF Mutational Status and Melanoma-Specific Survival among Patients with Higher-Risk Primary Melanoma. JAMA Oncol. 2015, 1, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Pollock, P.M.; Harper, U.L.; Hansen, K.S.; Yudt, L.M.; Stark, M.; Robbins, C.M.; Moses, T.Y.; Hostetter, G.; Wagner, U.; Kakareka, J.; et al. High Frequency of BRAF Mutations in Nevi. Nat. Genet. 2003, 33, 19–20. [Google Scholar] [CrossRef] [PubMed]
- Poynter, J.N.; Elder, J.T.; Fullen, D.R.; Nair, R.P.; Soengas, M.S.; Johnson, T.M.; Redman, B.; Thomas, N.E.; Gruber, S.B. BRAF and NRAS Mutations in Melanoma and Melanocytic Nevi. Melanoma Res. 2006, 16, 267–273. [Google Scholar] [CrossRef]
- Sensi, M.; Nicolini, G.; Petti, C.; Bersani, I.; Lozupone, F.; Molla, A.; Vegetti, C.; Nonaka, D.; Mortarini, R.; Parmiani, G.; et al. Mutually Exclusive NRASQ61R and BRAFV600E Mutations at the Single-Cell Level in the Same Human Melanoma. Oncogene 2006, 25, 3357–3364. [Google Scholar] [CrossRef] [PubMed]
- Chiba, K.; Lorbeer, F.K.; Shain, A.H.; McSwiggen, D.T.; Schruf, E.; Oh, A.; Ryu, J.; Darzacq, X.; Bastian, B.C.; Hockemeyer, D. Mutations in the Promoter of the Telomerase Gene TERT Contribute to Tumorigenesis by a Two-Step Mechanism. Science 2017, 357, 1416–1420. [Google Scholar] [CrossRef] [PubMed]
- Shain, A.H.; Bastian, B.C. From Melanocytes to Melanomas. Nat. Rev. Cancer 2016, 16, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.V.S.K.; Varambally, S. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Chandrashekar, D.S.; Karthikeyan, S.K.; Korla, P.K.; Patel, H.; Shovon, A.R.; Athar, M.; Netto, G.J.; Qin, Z.S.; Kumar, S.; Manne, U.; et al. UALCAN: An Update to the Integrated Cancer Data Analysis Platform. Neoplasia 2022, 25, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Tanami, H.; Imoto, I.; Hirasawa, A.; Yuki, Y.; Sonoda, I.; Inoue, J.; Yasui, K.; Misawa-Furihata, A.; Kawakami, Y.; Inazawa, J. Involvement of Overexpressed Wild-Type BRAF in the Growth of Malignant Melanoma Cell Lines. Oncogene 2004, 23, 8796–8804. [Google Scholar] [CrossRef]
- Guo, W.; Wang, H.; Li, C. Signal Pathways of Melanoma and Targeted Therapy. Signal Transduct. Target. Ther. 2021, 6, 424. [Google Scholar] [CrossRef] [PubMed]
- Eroglu, Z.; Ribas, A. Combination Therapy with BRAF and MEK Inhibitors for Melanoma: Latest Evidence and Place in Therapy. Ther. Adv. Med. Oncol. 2016, 8, 48–56. [Google Scholar] [CrossRef]
- Van Allen, E.M.; Wagle, N.; Sucker, A.; Treacy, D.J.; Johannessen, C.M.; Goetz, E.M.; Place, C.S.; Taylor-Weiner, A.; Whittaker, S.; Kryukov, G.V. The Genetic Landscape of Clinical Resistance to RAF Inhibition in Metastatic MelanomaGenetic Resistance Mechanisms to RAF Inhibition in Melanoma. Cancer Discov. 2014, 4, 94–109. [Google Scholar] [CrossRef]
- Gerosa, L.; Chidley, C.; Fröhlich, F.; Sanchez, G.; Lim, S.K.; Muhlich, J.; Chen, J.-Y.; Vallabhaneni, S.; Baker, G.J.; Schapiro, D.; et al. Receptor-Driven ERK Pulses Reconfigure MAPK Signaling and Enable Persistence of Drug-Adapted BRAF-Mutant Melanoma Cells. Cell Syst. 2020, 11, 478–494.e9. [Google Scholar] [CrossRef]
- Wang, J.; Kong, P.-F.; Wang, H.-Y.; Song, D.; Wu, W.-Q.; Zhou, H.-C.; Weng, H.-Y.; Li, M.; Kong, X.; Meng, B.; et al. Identification of a Gene-Related Risk Signature in Melanoma Patients Using Bioinformatic Profiling. J. Oncol. 2020, 2020, 7526204. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.T.; Infante, J.R.; Daud, A.; Gonzalez, R.; Kefford, R.F.; Sosman, J.; Hamid, O.; Schuchter, L.; Cebon, J.; Ibrahim, N.; et al. Combined BRAF and MEK Inhibition in Melanoma with BRAF V600 Mutations. N. Engl. J. Med. 2012, 367, 1694–1703. [Google Scholar] [CrossRef]
- Hoogstraat, M.; Gadellaa-van Hooijdonk, C.G.; Ubink, I.; Besselink, N.J.M.; Pieterse, M.; Veldhuis, W.; van Stralen, M.; Meijer, E.F.J.; Willems, S.M.; Hadders, M.A.; et al. Detailed Imaging and Genetic Analysis Reveal a Secondary BRAF(L505H) Resistance Mutation and Extensive Intrapatient Heterogeneity in Metastatic BRAF Mutant Melanoma Patients Treated with Vemurafenib. Pigment Cell Melanoma Res. 2015, 28, 318–323. [Google Scholar] [CrossRef]
- Sanchez, J.N.; Wang, T.; Cohen, M.S. BRAF and MEK Inhibitors: Use and Resistance in BRAF-Mutated Cancers. Drugs 2018, 78, 549–566. [Google Scholar] [CrossRef] [PubMed]
- Pupo, G.M.; Boyd, S.C.; Fung, C.; Carlino, M.S.; Menzies, A.M.; Pedersen, B.; Johansson, P.; Hayward, N.K.; Kefford, R.F.; Scolyer, R.A.; et al. Clinical Significance of Intronic Variants in BRAF Inhibitor Resistant Melanomas with Altered BRAF Transcript Splicing. Biomark. Res. 2017, 5, 17. [Google Scholar] [CrossRef]
- Vido, M.J.; Le, K.; Hartsough, E.J.; Aplin, A.E. BRAF Splice Variant Resistance to RAF Inhibitor Requires Enhanced MEK Association. Cell Rep. 2018, 25, 1501–1510.e3. [Google Scholar] [CrossRef]
- Johnson, D.B.; Menzies, A.M.; Zimmer, L.; Eroglu, Z.; Ye, F.; Zhao, S.; Rizos, H.; Sucker, A.; Scolyer, R.A.; Gutzmer, R.; et al. Acquired BRAF Inhibitor Resistance: A Multicenter Meta-Analysis of the Spectrum and Frequencies, Clinical Behaviour, and Phenotypic Associations of Resistance Mechanisms. Eur. J. Cancer 2015, 51, 2792–2799. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.; Cohen, M.S. The Discovery of Vemurafenib for the Treatment of BRAF-Mutated Metastatic Melanoma. Expert Opin. Drug Discov. 2016, 11, 907–916. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, H.T. MAPK Signal Pathways in the Regulation of Cell Proliferation in Mammalian Cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Peyssonnaux, C.; Eychène, A. The Raf/MEK/ERK Pathway: New Concepts of Activation. Biol. Cell 2001, 93, 53–62. [Google Scholar] [CrossRef]
- Castellani, G.; Buccarelli, M.; Arasi, M.B.; Rossi, S.; Pisanu, M.E.; Bellenghi, M.; Lintas, C.; Tabolacci, C. BRAF Mutations in Melanoma: Biological Aspects, Therapeutic Implications, and Circulating Biomarkers. Cancers 2023, 15, 4026. [Google Scholar] [CrossRef]
- Teixido, C.; Castillo, P.; Martinez-Vila, C.; Arance, A.; Alos, L. Molecular Markers and Targets in Melanoma. Cells 2021, 10, 2320. [Google Scholar] [CrossRef] [PubMed]
- Molina-Arcas, M.; Downward, J. How to Fool a Wonder Drug: Truncate and Dimerize. Cancer Cell 2012, 21, 7–9. [Google Scholar] [CrossRef] [PubMed]
- Poulikakos, P.I.; Persaud, Y.; Janakiraman, M.; Kong, X.; Ng, C.; Moriceau, G.; Shi, H.; Atefi, M.; Titz, B.; Gabay, M.T.; et al. RAF Inhibitor Resistance Is Mediated by Dimerization of Aberrantly Spliced BRAF(V600E). Nature 2011, 480, 387–390. [Google Scholar] [CrossRef]
- Atzori, M.G.; Ceci, C.; Ruffini, F.; Trapani, M.; Barbaccia, M.L.; Tentori, L.; D’Atri, S.; Lacal, P.M.; Graziani, G. Role of VEGFR-1 in Melanoma Acquired Resistance to the BRAF Inhibitor Vemurafenib. J. Cell. Mol. Med. 2020, 24, 465–475. [Google Scholar] [CrossRef]
- Louveau, B.; Delyon, J.; De Moura, C.R.; Battistella, M.; Jouenne, F.; Golmard, L.; Sadoux, A.; Podgorniak, M.-P.; Chami, I.; Marco, O. A Targeted Genomic Alteration Analysis Predicts Survival of Melanoma Patients under BRAF Inhibitors. Oncotarget 2019, 10, 1669. [Google Scholar] [CrossRef]
- Florent, L.; Saby, C.; Slimano, F.; Morjani, H. BRAF V600-Mutated Metastatic Melanoma and Targeted Therapy Resistance: An Update of the Current Knowledge. Cancers 2023, 15, 2607. [Google Scholar] [CrossRef]
- Winge, M.C.G.; Kellman, L.N.; Guo, K.; Tang, J.Y.; Swetter, S.M.; Aasi, S.Z.; Sarin, K.Y.; Chang, A.L.S.; Khavari, P.A. Advances in Cutaneous Squamous Cell Carcinoma. Nat. Rev. Cancer 2023, 23, 430–449. [Google Scholar] [CrossRef]
- Carlino, M.S.; Fung, C.; Shahheydari, H.; Todd, J.R.; Boyd, S.C.; Irvine, M.; Nagrial, A.M.; Scolyer, R.A.; Kefford, R.F.; Long, G.V.; et al. Preexisting MEK1P124 Mutations Diminish Response to BRAF Inhibitors in Metastatic Melanoma Patients. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 98–105. [Google Scholar] [CrossRef]
- Villanueva, J.; Infante, J.R.; Krepler, C.; Reyes-Uribe, P.; Samanta, M.; Chen, H.-Y.; Li, B.; Swoboda, R.K.; Wilson, M.; Vultur, A.; et al. Concurrent MEK2 Mutation and BRAF Amplification Confer Resistance to BRAF and MEK Inhibitors in Melanoma. Cell Rep. 2013, 4, 1090–1099. [Google Scholar] [CrossRef]
- Wagle, M.-C.; Kirouac, D.; Klijn, C.; Liu, B.; Mahajan, S.; Junttila, M.; Moffat, J.; Merchant, M.; Huw, L.; Wongchenko, M.; et al. A Transcriptional MAPK Pathway Activity Score (MPAS) Is a Clinically Relevant Biomarker in Multiple Cancer Types. NPJ Precis. Oncol. 2018, 2, 7. [Google Scholar] [CrossRef] [PubMed]
- Wagenaar, T.R.; Ma, L.; Roscoe, B.; Park, S.M.; Bolon, D.N.; Green, M.R. Resistance to Vemurafenib Resulting from a Novel Mutation in the BRAFV600E Kinase Domain. Pigment Cell Melanoma Res. 2014, 27, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Fung, C.; Menzies, A.M.; Pupo, G.M.; Carlino, M.S.; Hyman, J.; Shahheydari, H.; Tembe, V.; Thompson, J.F.; Saw, R.P.; et al. Increased MAPK Reactivation in Early Resistance to Dabrafenib/Trametinib Combination Therapy of BRAF-Mutant Metastatic Melanoma. Nat. Commun. 2014, 5, 5694. [Google Scholar] [CrossRef]
- Nazarian, R.; Shi, H.; Wang, Q.; Kong, X.; Koya, R.C.; Lee, H.; Chen, Z.; Lee, M.-K.; Attar, N.; Sazegar, H.; et al. Melanomas Acquire Resistance to B-RAF(V600E) Inhibition by RTK or N-RAS Upregulation. Nature 2010, 468, 973–977. [Google Scholar] [CrossRef] [PubMed]
- Ruggiero, C.F.; Malpicci, D.; Fattore, L.; Madonna, G.; Vanella, V.; Mallardo, D.; Liguoro, D.; Salvati, V.; Capone, M.; Bedogni, B.; et al. ErbB3 Phosphorylation as Central Event in Adaptive Resistance to Targeted Therapy in Metastatic Melanoma: Early Detection in CTCs during Therapy and Insights into Regulation by Autocrine Neuregulin. Cancers 2019, 11, 1425. [Google Scholar] [CrossRef] [PubMed]
- Sabbatino, F.; Wang, Y.; Wang, X.; Flaherty, K.T.; Yu, L.; Pepin, D.; Scognamiglio, G.; Pepe, S.; Kirkwood, J.M.; Cooper, Z.A.; et al. PDGFRα Up-Regulation Mediated by Sonic Hedgehog Pathway Activation Leads to BRAF Inhibitor Resistance in Melanoma Cells with BRAF Mutation. Oncotarget 2014, 5, 1926–1941. [Google Scholar] [CrossRef]
- Straussman, R.; Morikawa, T.; Shee, K.; Barzily-Rokni, M.; Qian, Z.R.; Du, J.; Davis, A.; Mongare, M.M.; Gould, J.; Frederick, D.T.; et al. Tumour Micro-Environment Elicits Innate Resistance to RAF Inhibitors through HGF Secretion. Nature 2012, 487, 500–504. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Wang, L.; Huang, S.; Heynen, G.J.J.E.; Prahallad, A.; Robert, C.; Haanen, J.; Blank, C.; Wesseling, J.; Willems, S.M.; et al. Reversible and Adaptive Resistance to BRAF(V600E) Inhibition in Melanoma. Nature 2014, 508, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Xue, G.; Kohler, R.; Tang, F.; Hynx, D.; Wang, Y.; Orso, F.; Prêtre, V.; Ritschard, R.; Hirschmann, P.; Cron, P.; et al. MTORC1/Autophagy-Regulated MerTK in Mutant BRAFV600 Melanoma with Acquired Resistance to BRAF Inhibition. Oncotarget 2017, 8, 69204–69218. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yadav, V.; Zhang, X.; Liu, J.; Estrem, S.; Li, S.; Gong, X.-Q.; Buchanan, S.; Henry, J.R.; Starling, J.J.; Peng, S.-B. Reactivation of Mitogen-Activated Protein Kinase (MAPK) Pathway by FGF Receptor 3 (FGFR3)/Ras Mediates Resistance to Vemurafenib in Human B-RAF V600E Mutant Melanoma. J. Biol. Chem. 2012, 287, 28087–28098. [Google Scholar] [CrossRef] [PubMed]
- Caenepeel, S.; Cooke, K.; Wadsworth, S.; Huang, G.; Robert, L.; Moreno, B.H.; Parisi, G.; Cajulis, E.; Kendall, R.; Beltran, P.; et al. MAPK Pathway Inhibition Induces MET and GAB1 Levels, Priming BRAF Mutant Melanoma for Rescue by Hepatocyte Growth Factor. Oncotarget 2017, 8, 17795–17809. [Google Scholar] [CrossRef] [PubMed]
- Dugo, M.; Nicolini, G.; Tragni, G.; Bersani, I.; Tomassetti, A.; Colonna, V.; Del Vecchio, M.; De Braud, F.; Canevari, S.; Anichini, A.; et al. A Melanoma Subtype with Intrinsic Resistance to BRAF Inhibition Identified by Receptor Tyrosine Kinases Gene-Driven Classification. Oncotarget 2015, 6, 5118–5133. [Google Scholar] [CrossRef] [PubMed]
- Janostiak, R.; Malvi, P.; Wajapeyee, N. Anaplastic Lymphoma Kinase Confers Resistance to BRAF Kinase Inhibitors in Melanoma. iScience 2019, 16, 453–467. [Google Scholar] [CrossRef]
- Krayem, M.; Aftimos, P.; Najem, A.; van den Hooven, T.; van den Berg, A.; Hovestad-Bijl, L.; de Wijn, R.; Hilhorst, R.; Ruijtenbeek, R.; Sabbah, M.; et al. Kinome Profiling to Predict Sensitivity to MAPK Inhibition in Melanoma and to Provide New Insights into Intrinsic and Acquired Mechanism of Resistance. Cancers 2020, 12, 512. [Google Scholar] [CrossRef]
- Lehraiki, A.; Cerezo, M.; Rouaud, F.; Abbe, P.; Allegra, M.; Kluza, J.; Marchetti, P.; Imbert, V.; Cheli, Y.; Bertolotto, C.; et al. Increased CD271 Expression by the NF-κB Pathway Promotes Melanoma Cell Survival and Drives Acquired Resistance to BRAF Inhibitor Vemurafenib. Cell Discov. 2015, 1, 15030. [Google Scholar] [CrossRef] [PubMed]
- Lei, F.X.; Jin, L.; Liu, X.Y.; Lai, F.; Yan, X.G.; Farrelly, M.; Guo, S.T.; Zhao, X.H.; Zhang, X.D. RIP1 Protects Melanoma Cells from Apoptosis Induced by BRAF/MEK Inhibitors. Cell Death Dis. 2018, 9, 679. [Google Scholar] [CrossRef]
- Molnár, E.; Garay, T.; Donia, M.; Baranyi, M.; Rittler, D.; Berger, W.; Tímár, J.; Grusch, M.; Hegedűs, B. Long-Term Vemurafenib Exposure Induced Alterations of Cell Phenotypes in Melanoma: Increased Cell Migration and Its Association with EGFR Expression. Int. J. Mol. Sci. 2019, 20, 4484. [Google Scholar] [CrossRef]
- Müller, J.; Krijgsman, O.; Tsoi, J.; Robert, L.; Hugo, W.; Song, C.; Kong, X.; Possik, P.A.; Cornelissen-Steijger, P.D.M.; Geukes Foppen, M.H.; et al. Low MITF/AXL Ratio Predicts Early Resistance to Multiple Targeted Drugs in Melanoma. Nat. Commun. 2014, 5, 5712. [Google Scholar] [CrossRef]
- Abd Elmageed, Z.Y.; Moore, R.F.; Tsumagari, K.; Lee, M.M.; Sholl, A.B.; Friedlander, P.; Al-Qurayshi, Z.; Hassan, M.; Wang, A.R.; Boulares, H.A.; et al. Prognostic Role of BRAF(V600E) Cellular Localization in Melanoma. J. Am. Coll. Surg. 2018, 226, 526–537. [Google Scholar] [CrossRef]
- Perna, D.; Karreth, F.A.; Rust, A.G.; Perez-Mancera, P.A.; Rashid, M.; Iorio, F.; Alifrangis, C.; Arends, M.J.; Bosenberg, M.W.; Bollag, G.; et al. BRAF Inhibitor Resistance Mediated by the AKT Pathway in an Oncogenic BRAF Mouse Melanoma Model. Proc. Natl. Acad. Sci. USA 2015, 112, E536–E545. [Google Scholar] [CrossRef] [PubMed]
- Anastas, J.N.; Kulikauskas, R.M.; Tamir, T.; Rizos, H.; Long, G.V.; von Euw, E.M.; Yang, P.-T.; Chen, H.-W.; Haydu, L.; Toroni, R.A.; et al. WNT5A Enhances Resistance of Melanoma Cells to Targeted BRAF Inhibitors. J. Clin. Investig. 2014, 124, 2877–2890. [Google Scholar] [CrossRef] [PubMed]
- Atefi, M.; von Euw, E.; Attar, N.; Ng, C.; Chu, C.; Guo, D.; Nazarian, R.; Chmielowski, B.; Glaspy, J.A.; Comin-Anduix, B.; et al. Reversing Melanoma Cross-Resistance to BRAF and MEK Inhibitors by Co-Targeting the AKT/MTOR Pathway. PLoS ONE 2011, 6, e28973. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.-P.; Mori, M.; Pan, B.; Yau, C.; Wolf, D.M.; Ruiz-Saenz, A.; Brunen, D.; Prahallad, A.; Cornelissen-Steijger, P.; Kemper, K.; et al. Mapping Phospho-Catalytic Dependencies of Therapy-Resistant Tumours Reveals Actionable Vulnerabilities. Nat. Cell Biol. 2019, 21, 778–790. [Google Scholar] [CrossRef]
- Hu, W.; Jin, L.; Jiang, C.C.; Long, G.V.; Scolyer, R.A.; Wu, Q.; Zhang, X.D.; Mei, Y.; Wu, M. AEBP1 Upregulation Confers Acquired Resistance to BRAF (V600E) Inhibition in Melanoma. Cell Death Dis. 2013, 4, e914. [Google Scholar] [CrossRef]
- Inozume, T.; Tsunoda, T.; Morisaki, T.; Harada, K.; Shirasawa, S.; Kawamura, T. Acquisition of Resistance to Vemurafenib Leads to Interleukin-10 Production through an Aberrant Activation of Akt in a Melanoma Cell Line. J. Dermatol. 2018, 45, 1434–1439. [Google Scholar] [CrossRef]
- Jain, A.; Tripathi, R.; Turpin, C.P.; Wang, C.; Plattner, R. Abl Kinase Regulation by BRAF/ERK and Cooperation with Akt in Melanoma. Oncogene 2017, 36, 4585–4596. [Google Scholar] [CrossRef][Green Version]
- Shi, H.; Hong, A.; Kong, X.; Koya, R.C.; Song, C.; Moriceau, G.; Hugo, W.; Yu, C.C.; Ng, C.; Chodon, T.; et al. A Novel AKT1 Mutant Amplifies an Adaptive Melanoma Response to BRAF Inhibition. Cancer Discov. 2014, 4, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Shull, A.Y.; Latham-Schwark, A.; Ramasamy, P.; Leskoske, K.; Oroian, D.; Birtwistle, M.R.; Buckhaults, P.J. Novel Somatic Mutations to PI3K Pathway Genes in Metastatic Melanoma. PLoS ONE 2012, 7, e43369. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, R.B.; Dias-Santagata, D.; Bergethon, K.; Iafrate, A.J.; Settleman, J.; Engelman, J.A. BRAF Gene Amplification Can Promote Acquired Resistance to MEK Inhibitors in Cancer Cells Harboring the BRAF V600E Mutation. Sci. Signal. 2010, 3, ra84. [Google Scholar] [CrossRef] [PubMed]
- Romano, E.; Pradervand, S.; Paillusson, A.; Weber, J.; Harshman, K.; Muehlethaler, K.; Speiser, D.; Peters, S.; Rimoldi, D.; Michielin, O. Identification of Multiple Mechanisms of Resistance to Vemurafenib in a Patient with BRAFV600E-Mutated Cutaneous Melanoma Successfully Rechallenged after Progression. Clin. Cancer Res. 2013, 19, 5749–5757. [Google Scholar] [CrossRef] [PubMed]
- Broussard, L.; Howland, A.; Ryu, S.; Song, K.; Norris, D.; Armstrong, C.A.; Song, P.I. Melanoma Cell Death Mechanisms. Chonnam Med. J. 2018, 54, 135–142. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Corrales, E.; Levit-Zerdoun, E.; Metzger, P.; Mertes, R.; Lehmann, A.; Münch, J.; Lemke, S.; Kowar, S.; Boerries, M. PI3K/AKT Signaling Allows for MAPK/ERK Pathway Independency Mediating Dedifferentiation-Driven Treatment Resistance in Melanoma. Cell Commun. Signal. 2022, 20, 187. [Google Scholar] [CrossRef] [PubMed]
- Patterson, A.; Auslander, N. Mutated Processes Predict Immune Checkpoint Inhibitor Therapy Benefit in Metastatic Melanoma. Nat. Commun. 2022, 13, 5151. [Google Scholar] [CrossRef] [PubMed]
- Irvine, M.; Stewart, A.; Pedersen, B.; Boyd, S.; Kefford, R.; Rizos, H. Oncogenic PI3K/AKT Promotes the Step-Wise Evolution of Combination BRAF/MEK Inhibitor Resistance in Melanoma. Oncogenesis 2018, 7, 72. [Google Scholar] [CrossRef] [PubMed]
- Peltonen, S.; Kallionpää, R.A.; Peltonen, J. Neurofibromatosis Type 1 (NF1) Gene: Beyond Café Au Lait Spots and Dermal Neurofibromas. Exp. Dermatol. 2017, 26, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Al Mahi, A.; Ablain, J. RAS Pathway Regulation in Melanoma. Dis. Models Mech. 2022, 15, dmm049229. [Google Scholar] [CrossRef] [PubMed]
- Alan, J.K.; Lundquist, E.A. Mutationally Activated Rho GTPases in Cancer. Small GTPases 2013, 4, 159–163. [Google Scholar] [CrossRef]
- Sibilia, M.; Kroismayr, R.; Lichtenberger, B.M.; Natarajan, A.; Hecking, M.; Holcmann, M. The Epidermal Growth Factor Receptor: From Development to Tumorigenesis. Differentiation 2007, 75, 770–787. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in Cancer: Mechanisms and Advances in Clinical Trials. Mol. Cancer 2019, 18, 26. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.-H.; Liu, D.-Y.; Lai, Y.-C.; Chen, Y.-Y.; Yu, L.-Z.; Shao, M.; Liu, J.-S. Inhibition of the STAT3 Signaling Pathway Contributes to the Anti-Melanoma Activities of Shikonin. Front. Pharmacol. 2020, 11, 748. [Google Scholar] [CrossRef] [PubMed]
- Swoboda, A.; Soukup, R.; Eckel, O.; Kinslechner, K.; Wingelhofer, B.; Schörghofer, D.; Sternberg, C.; Pham, H.T.T.; Vallianou, M.; Horvath, J.; et al. STAT3 Promotes Melanoma Metastasis by CEBP-Induced Repression of the MITF Pathway. Oncogene 2021, 40, 1091–1105. [Google Scholar] [CrossRef] [PubMed]
- Aguissa-Touré, A.-H.; Li, G. Genetic Alterations of PTEN in Human Melanoma. Cell. Mol. Life Sci. 2012, 69, 1475–1491. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Guo, W. A Review of the Molecular Pathways Involved in Resistance to BRAF Inhibitors in Patients with Advanced-Stage Melanoma. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2020, 26, e920957. [Google Scholar] [CrossRef]
- Paraiso, K.H.T.; Xiang, Y.; Rebecca, V.W.; Abel, E.V.; Chen, Y.A.; Munko, A.C.; Wood, E.; Fedorenko, I.V.; Sondak, V.K.; Anderson, A.R.A.; et al. PTEN Loss Confers BRAF Inhibitor Resistance to Melanoma Cells through the Suppression of BIM Expression. Cancer Res. 2011, 71, 2750–2760. [Google Scholar] [CrossRef] [PubMed]
- Glaviano, A.; Foo, A.S.C.; Lam, H.Y.; Yap, K.C.H.; Jacot, W.; Jones, R.H.; Eng, H.; Nair, M.G.; Makvandi, P.; Geoerger, B.; et al. PI3K/AKT/MTOR Signaling Transduction Pathway and Targeted Therapies in Cancer. Mol. Cancer 2023, 22, 138. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Croce, C.M. MicroRNA: Trends in Clinical Trials of Cancer Diagnosis and Therapy Strategies. Exp. Mol. Med. 2023, 55, 1314–1321. [Google Scholar] [CrossRef] [PubMed]
- Couts, K.L.; Anderson, E.M.; Gross, M.M.; Sullivan, K.; Ahn, N.G. Oncogenic B-Raf Signaling in Melanoma Cells Controls a Network of MicroRNAs with Combinatorial Functions. Oncogene 2013, 32, 1959–1970. [Google Scholar] [CrossRef]
- Sand, M.; Skrygan, M.; Sand, D.; Georgas, D.; Gambichler, T.; Hahn, S.A.; Altmeyer, P.; Bechara, F.G. Comparative Microarray Analysis of MicroRNA Expression Profiles in Primary Cutaneous Malignant Melanoma, Cutaneous Malignant Melanoma Metastases, and Benign Melanocytic Nevi. Cell Tissue Res. 2013, 351, 85–98. [Google Scholar] [CrossRef]
- Grignol, V.; Fairchild, E.T.; Zimmerer, J.M.; Lesinski, G.B.; Walker, M.J.; Magro, C.M.; Kacher, J.E.; Karpa, V.I.; Clark, J.; Nuovo, G.; et al. MiR-21 and MiR-155 Are Associated with Mitotic Activity and Lesion Depth of Borderline Melanocytic Lesions. Br. J. Cancer 2011, 105, 1023–1029. [Google Scholar] [CrossRef]
- Mueller, D.W.; Rehli, M.; Bosserhoff, A.K. MiRNA Expression Profiling in Melanocytes and Melanoma Cell Lines Reveals MiRNAs Associated with Formation and Progression of Malignant Melanoma. J. Investig. Dermatol. 2009, 129, 1740–1751. [Google Scholar] [CrossRef]
- Talotta, F.; Cimmino, A.; Matarazzo, M.R.; Casalino, L.; De Vita, G.; D’Esposito, M.; Di Lauro, R.; Verde, P. An Autoregulatory Loop Mediated by MiR-21 and PDCD4 Controls the AP-1 Activity in RAS Transformation. Oncogene 2009, 28, 73–84. [Google Scholar] [CrossRef]
- Luo, C.; Weber, C.E.M.; Osen, W.; Bosserhoff, A.-K.; Eichmüller, S.B. The Role of MicroRNAs in Melanoma. Eur. J. Cell Biol. 2014, 93, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Fares, C.M.; Van Allen, E.M.; Drake, C.G.; Allison, J.P.; Hu-Lieskovan, S. Mechanisms of Resistance to Immune Checkpoint Blockade: Why Does Checkpoint Inhibitor Immunotherapy Not Work for All Patients? Am. Soc. Clin. Oncol. Educ. Book. Am. Soc. Clin. Oncol. Annu. Meet. 2019, 39, 147–164. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Allison, J.P. The Future of Immune Checkpoint Therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef]
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front. Oncol. 2018, 8, 86. [Google Scholar] [CrossRef] [PubMed]
- Luke, J.J.; Flaherty, K.T.; Ribas, A.; Long, G. V Targeted Agents and Immunotherapies: Optimizing Outcomes in Melanoma. Nat. Rev. Clin. Oncol. 2017, 14, 463–482. [Google Scholar] [CrossRef]
- Nebhan, C.A.; Johnson, D.B. Predictive Biomarkers of Response to Immune Checkpoint Inhibitors in Melanoma. Expert Rev. Anticancer Ther. 2020, 20, 137–145. [Google Scholar] [CrossRef]
- Martinović, K.M.; Vuletić, A.; Miletić, N.T.; Matković, S.; Gavrilović, D.; Ninković, A.; Jurišić, V.; Babović, N. Circulating IL-6 Is Associated with Disease Progression in BRAFwt Metastatic Melanoma Patients Receiving Anti-PD-1 Therapy. J. Clin. Pathol. 2024, 77, 343–351. [Google Scholar] [CrossRef]
- Lippitz, B.E.; Harris, R.A. Cytokine Patterns in Cancer Patients: A Review of the Correlation between Interleukin 6 and Prognosis. OncoImmunology 2016, 5, e1093722. [Google Scholar] [CrossRef] [PubMed]
- Thornton, J.; Chhabra, G.; Singh, C.K.; Guzmán-Pérez, G.; Shirley, C.A.; Ahmad, N. Mechanisms of Immunotherapy Resistance in Cutaneous Melanoma: Recognizing a Shapeshifter. Front. Oncol. 2022, 12, 880876. [Google Scholar] [CrossRef] [PubMed]
- Fukumura, D.; Kloepper, J.; Amoozgar, Z.; Duda, D.G.; Jain, R.K. Enhancing Cancer Immunotherapy Using Antiangiogenics: Opportunities and Challenges. Nat. Rev. Clin. Oncol. 2018, 15, 325–340. [Google Scholar] [CrossRef] [PubMed]
- Chow, M.T.; Ozga, A.J.; Servis, R.L.; Frederick, D.T.; Lo, J.A.; Fisher, D.E.; Freeman, G.J.; Boland, G.M.; Luster, A.D. Intratumoral Activity of the CXCR3 Chemokine System Is Required for the Efficacy of Anti-PD-1 Therapy. Immunity 2019, 50, 1498–1512.e5. [Google Scholar] [CrossRef]
- Han, X.; Wang, Y.; Sun, J.; Tan, T.; Cai, X.; Lin, P.; Tan, Y.; Zheng, B.; Wang, B.; Wang, J.; et al. Role of CXCR3 Signaling in Response to Anti-PD-1 Therapy. EBioMedicine 2019, 48, 169–177. [Google Scholar] [CrossRef]
- Olbryt, M.; Rajczykowski, M.; Widłak, W. Biological Factors behind Melanoma Response to Immune Checkpoint Inhibitors. Int. J. Mol. Sci. 2020, 21, 4071. [Google Scholar] [CrossRef]
- Hodi, F.S.; Wolchok, J.D.; Schadendorf, D.; Larkin, J.; Long, G.V.; Qian, X.; Saci, A.; Young, T.C.; Srinivasan, S.; Chang, H.; et al. TMB and Inflammatory Gene Expression Associated with Clinical Outcomes Following Immunotherapy in Advanced Melanoma. Cancer Immunol. Res. 2021, 9, 1202–1213. [Google Scholar] [CrossRef]
- Huang, T.; Chen, X.; Zhang, H.; Liang, Y.; Li, L.; Wei, H.; Sun, W.; Wang, Y. Prognostic Role of Tumor Mutational Burden in Cancer Patients Treated with Immune Checkpoint Inhibitors: A Systematic Review and Meta-Analysis. Front. Oncol. 2021, 11, 706652. [Google Scholar] [CrossRef]
- Valero, C.; Lee, M.; Hoen, D.; Zehir, A.; Berger, M.F.; Seshan, V.E.; Chan, T.A.; Morris, L.G.T. Response Rates to Anti-PD-1 Immunotherapy in Microsatellite-Stable Solid Tumors with 10 or More Mutations per Megabase. JAMA Oncol. 2021, 7, 739–743. [Google Scholar] [CrossRef] [PubMed]
- Bandola-Simon, J.; Roche, P.A. Dysfunction of Antigen Processing and Presentation by Dendritic Cells in Cancer. Mol. Immunol. 2019, 113, 31–37. [Google Scholar] [CrossRef]
- Konjević, G.M.; Vuletić, A.M.; Mirjačić Martinović, K.M.; Larsen, A.K.; Jurišić, V.B. The Role of Cytokines in the Regulation of NK Cells in the Tumor Environment. Cytokine 2019, 117, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Mirjačić Martinović, K.; Vuletić, A.; Mališić, E.; Srdić-Rajić, T.; Tišma Miletić, N.; Babović, N.; Jurišić, V. Increased Circulating TGF-Β1 Is Associated with Impairment in NK Cell Effector Functions in Metastatic Melanoma Patients. Growth Factors 2022, 40, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, Z.; Yao, T.; Zhou, J.; Wang, Z. The Immune-Related Role of Beta-2-Microglobulin in Melanoma. Front. Oncol. 2022, 12, 944722. [Google Scholar] [CrossRef] [PubMed]
- Draghi, A.; Chamberlain, C.A.; Furness, A.; Donia, M. Acquired Resistance to Cancer Immunotherapy. Semin. Immunopathol. 2019, 41, 31–40. [Google Scholar] [CrossRef]
- Xu, T.; Dai, J.; Tang, L.; Yang, L.; Si, L.; Sheng, X.; Cui, C.; Chi, Z.; Kong, Y.; Guo, J. EZH2 Inhibitor Enhances the STING Agonist-Induced Antitumor Immunity in Melanoma. J. Investig. Dermatol. 2022, 142, 1158–1170.e8. [Google Scholar] [CrossRef]
- Gracia-Hernandez, M.; Munoz, Z.; Villagra, A. Enhancing Therapeutic Approaches for Melanoma Patients Targeting Epigenetic Modifiers. Cancers 2021, 13, 6180. [Google Scholar] [CrossRef]
- Zhang, S.-M.; Cai, W.L.; Liu, X.; Thakral, D.; Luo, J.; Chan, L.H.; McGeary, M.K.; Song, E.; Blenman, K.R.M.; Micevic, G.; et al. KDM5B Promotes Immune Evasion by Recruiting SETDB1 to Silence Retroelements. Nature 2021, 598, 682–687. [Google Scholar] [CrossRef]
- Griffin, G.K.; Wu, J.; Iracheta-Vellve, A.; Patti, J.C.; Hsu, J.; Davis, T.; Dele-Oni, D.; Du, P.P.; Halawi, A.G.; Ishizuka, J.J.; et al. Epigenetic Silencing by SETDB1 Suppresses Tumour Intrinsic Immunogenicity. Nature 2021, 595, 309–314. [Google Scholar] [CrossRef]
- Lazaro-Camp, V.J.; Salari, K.; Meng, X.; Yang, S. SETDB1 in Cancer: Overexpression and Its Therapeutic Implications. Am. J. Cancer Res. 2021, 11, 1803–1827. [Google Scholar] [PubMed]
- Strepkos, D.; Markouli, M.; Klonou, A.; Papavassiliou, A.G.; Piperi, C. Histone Methyltransferase SETDB1: A Common Denominator of Tumorigenesis with Therapeutic Potential. Cancer Res. 2021, 81, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Wei, J.; Cui, Y.-H.; Park, G.; Shah, P.; Deng, Y.; Aplin, A.E.; Lu, Z.; Hwang, S.; He, C.; et al. M6A MRNA Demethylase FTO Regulates Melanoma Tumorigenicity and Response to Anti-PD-1 Blockade. Nat. Commun. 2019, 10, 2782. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, V.; Khattak, A.; Haydon, A.; Eastgate, M.; Roy, A.; Prithviraj, P.; Mueller, C.; Brignone, C.; Triebel, F. Eftilagimod Alpha, a Soluble Lymphocyte Activation Gene-3 (LAG-3) Protein plus Pembrolizumab in Patients with Metastatic Melanoma. J. Immunother. Cancer 2020, 8, e001681. [Google Scholar] [CrossRef] [PubMed]
- Shen, R.; Postow, M.A.; Adamow, M.; Arora, A.; Hannum, M.; Maher, C.; Wong, P.; Curran, M.A.; Hollmann, T.J.; Jia, L.; et al. LAG-3 Expression on Peripheral Blood Cells Identifies Patients with Poorer Outcomes after Immune Checkpoint Blockade. Sci. Transl. Med. 2021, 13, eabf5107. [Google Scholar] [CrossRef] [PubMed]
- Sidaway, P. LAG3 Inhibition Improves Outcomes. Nat. Rev. Clin. Oncol. 2022, 19, 149. [Google Scholar] [CrossRef] [PubMed]
- Gide, T.N.; Wilmott, J.S.; Scolyer, R.A.; Long, G. V Primary and Acquired Resistance to Immune Checkpoint Inhibitors in Metastatic Melanoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 1260–1270. [Google Scholar] [CrossRef]
- Das, M.; Zhu, C.; Kuchroo, V.K. Tim-3 and Its Role in Regulating Anti-Tumor Immunity. Immunol. Rev. 2017, 276, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Vukadin, S.; Khaznadar, F.; Kizivat, T.; Vcev, A.; Smolic, M. Molecular Mechanisms of Resistance to Immune Checkpoint Inhibitors in Melanoma Treatment: An Update. Biomedicines 2021, 9, 835. [Google Scholar] [CrossRef] [PubMed]
- Cartier, A.; Hla, T. Sphingosine 1-Phosphate: Lipid Signaling in Pathology and Therapy. Science 2019, 366, eaar5551. [Google Scholar] [CrossRef]
- Imbert, C.; Montfort, A.; Fraisse, M.; Marcheteau, E.; Gilhodes, J.; Martin, E.; Bertrand, F.; Marcellin, M.; Burlet-Schiltz, O.; de Peredo, A.G.; et al. Resistance of Melanoma to Immune Checkpoint Inhibitors Is Overcome by Targeting the Sphingosine Kinase-1. Nat. Commun. 2020, 11, 437. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.B.; Nixon, M.J.; Wang, Y.; Wang, D.Y.; Castellanos, E.; Estrada, M.V.; Ericsson-Gonzalez, P.I.; Cote, C.H.; Salgado, R.; Sanchez, V.; et al. Tumor-Specific MHC-II Expression Drives a Unique Pattern of Resistance to Immunotherapy via LAG-3/FCRL6 Engagement. JCI Insight 2018, 3, e120360. [Google Scholar] [CrossRef] [PubMed]
- Theivanthiran, B.; Evans, K.S.; DeVito, N.C.; Plebanek, M.; Sturdivant, M.; Wachsmuth, L.P.; Salama, A.K.; Kang, Y.; Hsu, D.; Balko, J.M.; et al. A Tumor-Intrinsic PD-L1/NLRP3 Inflammasome Signaling Pathway Drives Resistance to Anti-PD-1 Immunotherapy. J. Clin. Investig. 2020, 130, 2570–2586. [Google Scholar] [CrossRef] [PubMed]
- Lam, K.C.; Araya, R.E.; Huang, A.; Chen, Q.; Di Modica, M.; Rodrigues, R.R.; Lopès, A.; Johnson, S.B.; Schwarz, B.; Bohrnsen, E.; et al. Microbiota Triggers STING-Type I IFN-Dependent Monocyte Reprogramming of the Tumor Microenvironment. Cell 2021, 184, 5338–5356.e21. [Google Scholar] [CrossRef] [PubMed]
- Si, W.; Liang, H.; Bugno, J.; Xu, Q.; Ding, X.; Yang, K.; Fu, Y.; Weichselbaum, R.R.; Zhao, X.; Wang, L. Lactobacillus Rhamnosus GG Induces CGAS/STING-Dependent Type I Interferon and Improves Response to Immune Checkpoint Blockade. Gut 2022, 71, 521–533. [Google Scholar] [CrossRef] [PubMed]
- Baruch, E.N.; Youngster, I.; Ben-Betzalel, G.; Ortenberg, R.; Lahat, A.; Katz, L.; Adler, K.; Dick-Necula, D.; Raskin, S.; Bloch, N.; et al. Fecal Microbiota Transplant Promotes Response in Immunotherapy-Refractory Melanoma Patients. Science 2021, 371, 602–609. [Google Scholar] [CrossRef] [PubMed]
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Lei, Y.M.; Jabri, B.; Alegre, M.-L.; et al. Commensal Bifidobacterium Promotes Antitumor Immunity and Facilitates Anti-PD-L1 Efficacy. Science 2015, 350, 1084–1089. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut Microbiome Modulates Response to Anti-PD-1 Immunotherapy in Melanoma Patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Van Allen, E.M.; Miao, D.; Schilling, B.; Shukla, S.A.; Blank, C.; Zimmer, L.; Sucker, A.; Hillen, U.; Foppen, M.H.G.; Goldinger, S.M.; et al. Genomic Correlates of Response to CTLA-4 Blockade in Metastatic Melanoma. Science 2015, 350, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Weiss, S.A.; Wolchok, J.D.; Sznol, M. Immunotherapy of Melanoma: Facts and Hopes. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 5191–5201. [Google Scholar] [CrossRef]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.M.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 Blockade Induces Responses by Inhibiting Adaptive Immune Resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, Activity, and Immune Correlates of Anti-PD-1 Antibody in Cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Machiraju, D.; Schäfer, S.; Hassel, J.C. Potential Reasons for Unresponsiveness to Anti-PD1 Immunotherapy in Young Patients with Advanced Melanoma. Life 2021, 11, 1318. [Google Scholar] [CrossRef]
- Eschweiler, S.; Clarke, J.; Ramírez-Suástegui, C.; Panwar, B.; Madrigal, A.; Chee, S.J.; Karydis, I.; Woo, E.; Alzetani, A.; Elsheikh, S.; et al. Intratumoral Follicular Regulatory T Cells Curtail Anti-PD-1 Treatment Efficacy. Nat. Immunol. 2021, 22, 1052–1063. [Google Scholar] [CrossRef]
- Johnson, D.B.; Estrada, M.V.; Salgado, R.; Sanchez, V.; Doxie, D.B.; Opalenik, S.R.; Vilgelm, A.E.; Feld, E.; Johnson, A.S.; Greenplate, A.R.; et al. Melanoma-Specific MHC-II Expression Represents a Tumour-Autonomous Phenotype and Predicts Response to Anti-PD-1/PD-L1 Therapy. Nat. Commun. 2016, 7, 10582. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.B.; Lovly, C.M.; Flavin, M.; Panageas, K.S.; Ayers, G.D.; Zhao, Z.; Iams, W.T.; Colgan, M.; DeNoble, S.; Terry, C.R.; et al. Impact of NRAS Mutations for Patients with Advanced Melanoma Treated with Immune Therapies. Cancer Immunol. Res. 2015, 3, 288–295. [Google Scholar] [CrossRef]
- Gao, J.; Shi, L.Z.; Zhao, H.; Chen, J.; Xiong, L.; He, Q.; Chen, T.; Roszik, J.; Bernatchez, C.; Woodman, S.E.; et al. Loss of IFN-γ Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell 2016, 167, 397–404.e9. [Google Scholar] [CrossRef]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic Basis for Clinical Response to CTLA-4 Blockade in Melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef]
- Jerby-Arnon, L.; Shah, P.; Cuoco, M.S.; Rodman, C.; Su, M.-J.; Melms, J.C.; Leeson, R.; Kanodia, A.; Mei, S.; Lin, J.-R.; et al. A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell 2018, 175, 984–997.e24. [Google Scholar] [CrossRef]
- Auslander, N.; Zhang, G.; Lee, J.S.; Frederick, D.T.; Miao, B.; Moll, T.; Tian, T.; Wei, Z.; Madan, S.; Sullivan, R.J.; et al. Robust Prediction of Response to Immune Checkpoint Blockade Therapy in Metastatic Melanoma. Nat. Med. 2018, 24, 1545–1549. [Google Scholar] [CrossRef] [PubMed]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Bellenghi, M.; Puglisi, R.; Pontecorvi, G.; De Feo, A.; Carè, A.; Mattia, G. Sex and Gender Disparities in Melanoma. Cancers 2020, 12, 1819. [Google Scholar] [CrossRef]
- Schwartz, M.R.; Luo, L.; Berwick, M. Sex Differences in Melanoma. Curr. Epidemiol. Rep. 2019, 6, 112–118. [Google Scholar] [CrossRef]
- Morgese, F.; Sampaolesi, C.; Torniai, M.; Conti, A.; Ranallo, N.; Giacchetti, A.; Serresi, S.; Onofri, A.; Burattini, M.; Ricotti, G.; et al. Gender Differences and Outcomes in Melanoma Patients. Oncol. Ther. 2020, 8, 103–114. [Google Scholar] [CrossRef]
- Conforti, C.; Zalaudek, I. Epidemiology and Risk Factors of Melanoma: A Review. Dermatol. Pract. Concept. 2021, 11, e2021161S. [Google Scholar] [CrossRef]
- Joosse, A.; de Vries, E.; Eckel, R.; Nijsten, T.; Eggermont, A.M.M.; Hölzel, D.; Coebergh, J.W.W.; Engel, J. Gender Differences in Melanoma Survival: Female Patients Have a Decreased Risk of Metastasis. J. Investig. Dermatol. 2011, 131, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Joosse, A.; Collette, S.; Suciu, S.; Nijsten, T.; Lejeune, F.; Kleeberg, U.R.; Coebergh, J.W.W.; Eggermont, A.M.M.; de Vries, E. Superior Outcome of Women with Stage I/II Cutaneous Melanoma: Pooled Analysis of Four European Organisation for Research and Treatment of Cancer Phase III Trials. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2012, 30, 2240–2247. [Google Scholar] [CrossRef]
- Nosrati, A.; Wei, M.L. Sex Disparities in Melanoma Outcomes: The Role of Biology. Arch. Biochem. Biophys. 2014, 563, 42–50. [Google Scholar] [CrossRef]
- Samarkina, A.; Youssef, M.K.; Ostano, P.; Ghosh, S.; Ma, M.; Tassone, B.; Proust, T.; Chiorino, G.; Levesque, M.P.; Goruppi, S.; et al. Androgen Receptor Is a Determinant of Melanoma Targeted Drug Resistance. Nat. Commun. 2023, 14, 6498. [Google Scholar] [CrossRef]
- McQuade, J.L.; Daniel, C.R.; Hess, K.R.; Mak, C.; Wang, D.Y.; Rai, R.R.; Park, J.J.; Haydu, L.E.; Spencer, C.; Wongchenko, M.; et al. Association of Body-Mass Index and Outcomes in Patients with Metastatic Melanoma Treated with Targeted Therapy, Immunotherapy, or Chemotherapy: A Retrospective, Multicohort Analysis. Lancet Oncol. 2018, 19, 310–322. [Google Scholar] [CrossRef] [PubMed]
- Atkins, M.B.; Curiel-Lewandrowski, C.; Fisher, D.E.; Swetter, S.M.; Tsao, H.; Aguirre-Ghiso, J.A.; Soengas, M.S.; Weeraratna, A.T.; Flaherty, K.T.; Herlyn, M.; et al. The State of Melanoma: Emergent Challenges and Opportunities. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2021, 27, 2678–2697. [Google Scholar] [CrossRef] [PubMed]
- Sadida, H.Q.; Abdulla, A.; Al Marzooqi, S.; Hashem, S.; Macha, M.A.; Akil, A.S.A.-S.; Bhat, A.A. Epigenetic Modifications: Key Players in Cancer Heterogeneity and Drug Resistance. Transl. Oncol. 2024, 39, 101821. [Google Scholar] [CrossRef]
- Wagstaff, W.; Mwamba, R.N.; Grullon, K.; Armstrong, M.; Zhao, P.; Hendren-Santiago, B.; Qin, K.H.; Li, A.J.; Hu, D.A.; Youssef, A.; et al. Melanoma: Molecular Genetics, Metastasis, Targeted Therapies, Immunotherapies, and Therapeutic Resistance. Genes Dis. 2022, 9, 1608–1623. [Google Scholar] [CrossRef]
- Alqathama, A. BRAF in Malignant Melanoma Progression and Metastasis: Potentials and Challenges. Am. J. Cancer Res. 2020, 10, 1103–1114. [Google Scholar] [PubMed]
- Guo, C.-X.; Huang, X.; Xu, J.; Zhang, X.-Z.; Shen, Y.-N.; Liang, T.-B.; Bai, X.-L. Combined Targeted Therapy and Immunotherapy for Cancer Treatment. World J. Clin. Cases 2021, 9, 7643–7652. [Google Scholar] [CrossRef]
- Haas, L.; Elewaut, A.; Gerard, C.L.; Umkehrer, C.; Leiendecker, L.; Pedersen, M.; Krecioch, I.; Hoffmann, D.; Novatchkova, M.; Kuttke, M.; et al. Acquired Resistance to Anti-MAPK Targeted Therapy Confers an Immune-Evasive Tumor Microenvironment and Cross-Resistance to Immunotherapy in Melanoma. Nat. Cancer 2021, 2, 693–708. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).