
De Novo Cancer Mutations Frequently Associate with Recurrent Chromosomal Abnormalities during Long-Term Human Pluripotent Stem Cell Culture

 , , , , ,
, , , , ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. hESCs Lines and Cell Culture
2.2. Fingerprinting
2.3. Whole-Genome Shallow Sequencing
2.4. Gene Panels for Cancer-Associated Genes
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kobold, S.; Bultjer, N.; Stacey, G.; Mueller, S.C.; Kurtz, A.; Mah, N. History and Current Status of Clinical Studies Using Human Pluripotent Stem Cells. Stem Cell Rep. 2023, 18, 1592–1598. [Google Scholar] [CrossRef] [PubMed]
- Draper, J.S.; Smith, K.; Gokhale, P.; Moore, H.D.; Maltby, E.; Johnson, J.; Meisner, L.; Zwaka, T.P.; Thomson, J.A.; Andrews, P.W. Recurrent Gain of Chromosomes 17q and 12 in Cultured Human Embryonic Stem Cells. Nat. Biotechnol. 2004, 22, 53–54. [Google Scholar] [CrossRef]
- Amps, K.; Andrews, P.W.; Anyfantis, G.; Armstrong, L.; Avery, S.; Baharvand, H.; Baker, J.; Baker, D.; Munoz, M.B.; Beil, S.; et al. Screening Ethnically Diverse Human Embryonic Stem Cells Identifies a Chromosome 20 Minimal Amplicon Conferring Growth Advantage. Nat. Biotechnol. 2011, 29, 1132–1144. [Google Scholar] [CrossRef]
- Baker, D.; Hirst, A.J.; Gokhale, P.J.; Juarez, M.A.; Williams, S.; Wheeler, M.; Bean, K.; Allison, T.F.; Moore, H.D.; Andrews, P.W.; et al. Detecting Genetic Mosaicism in Cultures of Human Pluripotent Stem Cells. Stem Cell Rep. 2016, 7, 998–1012. [Google Scholar] [CrossRef] [PubMed]
- Merkle, F.T.; Ghosh, S.; Genovese, G.; Handsaker, R.E.; Kashin, S.; Meyer, D.; Karczewski, K.J.; O’Dushlaine, C.; Pato, C.; Pato, M.; et al. Whole-Genome Analysis of Human Embryonic Stem Cells Enables Rational Line Selection Based on Genetic Variation. Cell Stem Cell 2022, 29, 472–486. [Google Scholar] [CrossRef] [PubMed]
- Andrews, P.W.; Barbaric, I.; Benvenisty, N.; Draper, J.S.; Ludwig, T.; Merkle, F.T.; Sato, Y.; Spits, C.; Stacey, G.N.; Wang, H.; et al. The Consequences of Recurrent Genetic and Epigenetic Variants in Human Pluripotent Stem Cells. Cell Stem Cell 2022, 29, 1624–1636. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, J.; Barbaric, I.; Andrews, P.W. Acquired Genetic Changes in Human Pluripotent Stem Cells: Origins and Consequences. Nat. Rev. Mol. Cell Biol. 2020, 21, 715–728. [Google Scholar] [CrossRef]
- Vallabhaneni, H.; Lynch, P.J.; Chen, G.; Park, K.; Liu, Y.; Goehe, R.; Mallon, B.S.; Boehm, M.; Hursh, D.A. High Basal Levels of ΓH2AX in Human Induced Pluripotent Stem Cells Are Linked to Replication-Associated DNA Damage and Repair. Stem Cells 2018, 36, 1501–1513. [Google Scholar] [CrossRef]
- Becker, K.A.; Ghule, P.N.; Therrien, J.A.; Lian, J.B.; Stein, J.L.; van Wijnen, A.J.; Stein, G.S. Self-Renewal of Human Embryonic Stem Cells Is Supported by a Shortened G1 Cell Cycle Phase. J. Cell. Physiol. 2006, 209, 883–893. [Google Scholar] [CrossRef]
- Desmarais, J.A.; Hoffmann, M.J.; Bingham, G.; Gagou, M.E.; Meuth, M.; Andrews, P.W. Human Embryonic Stem Cells Fail to Activate CHK1 and Commit to Apoptosis in Response to DNA Replication Stress. Stem Cells 2012, 30, 1385–1393. [Google Scholar] [CrossRef]
- Mantel, C.; Guo, Y.; Lee, M.R.; Kim, M.-K.; Han, M.-K.; Shibayama, H.; Fukuda, S.; Yoder, M.C.; Pelus, L.M.; Kim, K.-S.; et al. Checkpoint-Apoptosis Uncoupling in Human and Mouse Embryonic Stem Cells: A Source of Karyotpic Instability. Blood 2007, 109, 4518–4527. [Google Scholar] [CrossRef] [PubMed]
- Padgett, J.; Santos, S.D.M. From Clocks to Dominoes: Lessons on Cell Cycle Remodelling from Embryonic Stem Cells. FEBS Lett. 2020, 594, 2031–2045. [Google Scholar] [CrossRef] [PubMed]
- Milagre, I.; Pereira, C.; Oliveira, R.A. Compromised Mitotic Fidelity in Human Pluripotent Stem Cells. Int. J. Mol. Sci. 2023, 24, 11933. [Google Scholar] [CrossRef] [PubMed]
- Mucci, S.; Isaja, L.; Rodríguez-Varela, M.S.; Ferriol-Laffouillere, S.L.; Marazita, M.; Videla-Richardson, G.A.; Sevlever, G.E.; Scassa, M.E.; Romorini, L. Acute Severe Hypoxia Induces Apoptosis of Human Pluripotent Stem Cells by a HIF-1α and P53 Independent Mechanism. Sci. Rep. 2022, 12, 18803. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Ren, Z.; Lu, K.; Song, C.; Cheung, E.C.W.; Zhou, Z.; Chen, G. The Suppression of Medium Acidosis Improves the Maintenance and Differentiation of Human Pluripotent Stem Cells at High Density in Defined Cell Culture Medium. Int. J. Biol. Sci. 2018, 14, 485–496. [Google Scholar] [CrossRef]
- Jacobs, K.; Zambelli, F.; Mertzanidou, A.; Smolders, I.; Geens, M.; Nguyen, H.T.; Sermon, K.; Spits, C.; Barbé, L.; Sermon, K.; et al. Higher-Density Culture in Human Embryonic Stem Cells Results in DNA Damage and Genome Instability. Stem Cell Rep. 2016, 6, 330–341. [Google Scholar] [CrossRef]
- Thompson, O.; Von Meyenn, F.; Hewitt, Z.; Alexander, J.; Wood, A.; Weightman, R.; Gregory, S.; Krueger, F.; Andrews, S.; Barbaric, I.; et al. Low Rates of Mutation in Clinical Grade Human Pluripotent Stem Cells under Different Culture Conditions. Nat. Commun. 2020, 11, 1528. [Google Scholar] [CrossRef]
- Fu, X.; Cui, K.; Yi, Q.; Yu, L.; Xu, Y. DNA Repair Mechanisms in Embryonic Stem Cells. Cell. Mol. Life Sci. 2017, 74, 487–493. [Google Scholar] [CrossRef]
- Milholland, B.; Dong, X.; Zhang, L.; Hao, X.; Suh, Y.; Vijg, J. Differences between Germline and Somatic Mutation Rates in Humans and Mice. Nat. Commun. 2017, 8, 15183. [Google Scholar] [CrossRef]
- Hasaart, K.A.L.; Manders, F.; Ubels, J.; Verheul, M.; van Roosmalen, M.J.; Groenen, N.M.; Oka, R.; Kuijk, E.; Lopes, S.M.C.d.S.; van Boxtel, R. Human Induced Pluripotent Stem Cells Display a Similar Mutation Burden as Embryonic Pluripotent Cells in Vivo. iScience 2022, 25, 103736. [Google Scholar] [CrossRef]
- Jacobs, K.; Mertzanidou, A.; Geens, M.; Thi Nguyen, H.; Staessen, C.; Spits, C. Low-Grade Chromosomal Mosaicism in Human Somatic and Embryonic Stem Cell Populations. Nat. Commun. 2014, 5, 4227. [Google Scholar] [CrossRef] [PubMed]
- Keller, A.; Tilleman, L.; Dziedzicka, D.; Zambelli, F.; Sermon, K.; Van Nieuwerburgh, F.; Spits, C.; Geens, M. Uncovering Low-Level Mosaicism in Human Embryonic Stem Cells Using High Throughput Single Cell Shallow Sequencing. Sci. Rep. 2019, 9, 14844. [Google Scholar] [CrossRef]
- Olariu, V.; Harrison, N.J.; Coca, D.; Gokhale, P.J.; Baker, D.; Billings, S.; Kadirkamanathan, V.; Andrews, P.W. Modeling the Evolution of Culture-Adapted Human Embryonic Stem Cells. Stem Cell Res. 2010, 4, 50–56. [Google Scholar] [CrossRef]
- Barbaric, I.; Biga, V.; Gokhale, P.J.; Jones, M.; Stavish, D.; Glen, A.; Coca, D.; Andrews, P.W. Time-Lapse Analysis of Human Embryonic Stem Cells Reveals Multiple Bottlenecks Restricting Colony Formation and Their Relief upon Culture Adaptation. Stem Cell Rep. 2014, 3, 142–155. [Google Scholar] [CrossRef]
- Stavish, D.; Price, C.J.; Gelezauskaite, G.; Alsehli, H.; Leonhard, K.A.; Taapken, S.M.; McIntire, E.M.; Laing, O.; James, B.M.; Riley, J.J.; et al. Feeder-Free Culture of Human Pluripotent Stem Cells Drives MDM4-Mediated Gain of Chromosome 1q. Stem Cell Rep. 2024, 19, 1217–1232. [Google Scholar] [CrossRef]
- Keller, A.; Lei, Y.; Krivec, N.; de Deckersberg, E.C.; Dziedzicka, D.; Markouli, C.; Sermon, K.; Geens, M.; Spits, C.; Couvreu de Deckersberg, E.; et al. Gains of 12p13.31 Delay WNT-Mediated Initiation of HPSC Differentiation and Promote Residual Pluripotency in a Cell Cycle Dependent Manner. bioRxiv 2021. [Google Scholar] [CrossRef]
- Avery, S.; Hirst, A.J.; Baker, D.; Lim, C.Y.; Alagaratnam, S.; Skotheim, R.I.; Lothe, R.A.; Pera, M.F.; Colman, A.; Robson, P.; et al. BCL-XL Mediates the Strong Selective Advantage of a 20q11.21 Amplification Commonly Found in Human Embryonic Stem Cell Cultures. Stem Cell Rep. 2013, 1, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.T.; Geens, M.; Mertzanidou, A.; Jacobs, K.; Heirman, C.; Breckpot, K.; Spits, C. Gain of 20q11.21 in Human Embryonic Stem Cells Improves Cell Survival by Increased Expression of Bcl-XL. Mol. Hum. Reprod. 2014, 20, 168–177. [Google Scholar] [CrossRef]
- Markouli, C.; De Deckersberg, E.C.; Regin, M.; Nguyen, H.T.T.; Zambelli, F.; Keller, A.; Dziedzicka, D.; De Kock, J.; Tilleman, L.; Van Nieuwerburgh, F.; et al. Gain of 20q11.21 in Human Pluripotent Stem Cells Impairs TGF-β-Dependent Neuroectodermal Commitment. Stem Cell Rep. 2019, 13, 163–176. [Google Scholar] [CrossRef]
- Jo, H.Y.; Lee, Y.; Ahn, H.; Han, H.J.; Kwon, A.; Kim, B.Y.; Ha, H.Y.; Kim, S.C.; Kim, J.H.; Kim, Y.O.; et al. Functional in Vivo and in Vitro Effects of 20q11.21 Genetic Aberrations on HPSC Differentiation. Sci. Rep. 2020, 10, 18582. [Google Scholar] [CrossRef]
- Price, C.J.; Stavish, D.; Gokhale, P.J.; Stevenson, B.A.; Sargeant, S.; Lacey, J.; Rodriguez, T.A.; Barbaric, I. Genetically Variant Human Pluripotent Stem Cells Selectively Eliminate Wild-Type Counterparts through YAP-Mediated Cell Competition. Dev. Cell 2021, 56, 2455–2470.e10. [Google Scholar] [CrossRef]
- Andrews, P.W.; Ben-David, U.; Benvenisty, N.; Coffey, P.; Eggan, K.; Knowles, B.B.; Nagy, A.; Pera, M.; Reubinoff, B.; Rugg-Gunn, P.J.; et al. Assessing the Safety of Human Pluripotent Stem Cells and Their Derivatives for Clinical Applications. Stem Cell Rep. 2017, 9, 1–4. [Google Scholar] [CrossRef]
- Na, J.; Baker, D.; Zhang, J.; Andrews, P.W.; Barbaric, I. Aneuploidy in Pluripotent Stem Cells and Implications for Cancerous Transformation. Protein Cell 2014, 5, 569–579. [Google Scholar] [CrossRef]
- Xiao, R.; Xu, D.; Zhang, M.; Chen, Z.; Cheng, L.; Du, S.; Lu, M.; Zhou, T.; Li, R.; Bai, F.; et al. Aneuploid Embryonic Stem Cells Drive Teratoma Metastasis. Nat. Commun. 2024, 15, 1087. [Google Scholar] [CrossRef] [PubMed]
- Ben-David, U.; Arad, G.; Weissbein, U.; Mandefro, B.; Maimon, A.; Golan-Lev, T.; Narwani, K.; Clark, A.T.; Andrews, P.W.; Benvenisty, N.; et al. Aneuploidy Induces Profound Changes in Gene Expression, Proliferation and Tumorigenicity of Human Pluripotent Stem Cells. Nat. Commun. 2014, 5, 4825. [Google Scholar] [CrossRef]
- Goldring, C.E.P.; Duffy, P.A.; Benvenisty, N.; Andrews, P.W.; Ben-David, U.; Eakins, R.; French, N.; Hanley, N.A.; Kelly, L.; Kitteringham, N.R.; et al. Assessing the Safety of Stem Cell Therapeutics. Cell Stem Cell 2011, 8, 618–628. [Google Scholar] [CrossRef]
- Gore, A.; Li, Z.; Fung, H.; Young, J.; Agarwal, S.; Canto, I.; Giorgetti, A.; Israel, M.; Lee, J.; Loh, Y.; et al. Somatic Coding Mutations in Human Induced Pluripotent Stem Cells. Nature 2011, 471, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Lezmi, E.; Jung, J.; Benvenisty, N. High Prevalence of Acquired Cancer-Related Mutations in 146 Human Pluripotent Stem Cell Lines and Their Differentiated Derivatives. Nat. Biotechnol. 2024, 42, 896–913. [Google Scholar] [CrossRef] [PubMed]
- Avior, Y.; Lezmi, E.; Eggan, K.; Benvenisty, N. Cancer-Related Mutations Identified in Primed Human Pluripotent Stem Cells. Cell Stem Cell 2021, 28, 10–11. [Google Scholar] [CrossRef]
- Merkle, F.T.; Ghosh, S.; Kamitaki, N.; Mitchell, J.; Avior, Y.; Mello, C.; Kashin, S.; Mekhoubad, S.; Ilic, D.; Charlton, M.; et al. Human Pluripotent Stem Cells Recurrently Acquire and Expand Dominant Negative P53 Mutations. Nature 2017, 545, 229–233. [Google Scholar] [CrossRef]
- Mateizel, I.; Spits, C.; De Rycke, M.; Liebaers, I.; Sermon, K. Derivation, Culture, and Characterization of VUB HESC Lines. Vitr. Cell. Dev. Biol. Anim. 2010, 46, 300–308. [Google Scholar] [CrossRef] [PubMed]
- Geens, M.; Mateizel, I.; Sermon, K.; De Rycke, M.; Spits, C.; Cauffman, G.; Devroey, P.; Tournaye, H.; Liebaers, I.; Van de Velde, H. Human Embryonic Stem Cell Lines Derived from Single Blastomeres of Two 4-Cell Stage Embryos. Hum. Reprod. 2009, 24, 2709–2717. [Google Scholar] [CrossRef] [PubMed]
- Mateizel, I.; De Temmerman, N.; Ullmann, U.; Cauffman, G.; Sermon, K.; Van de Velde, H.; De Rycke, M.; Degreef, E.; Devroey, P.; Liebaers, I.; et al. Derivation of Human Embryonic Stem Cell Lines from Embryos Obtained after IVF and after PGD for Monogenic Disorders. Hum. Reprod. 2006, 21, 503–511. [Google Scholar] [CrossRef]
- Bayindir, B.; Dehaspe, L.; Brison, N.; Brady, P.; Ardui, S.; Kammoun, M.; Van der Veken, L.; Lichtenbelt, K.; Van den Bogaert, K.; Van Houdt, J.; et al. Noninvasive Prenatal Testing Using a Novel Analysis Pipeline to Screen for All Autosomal Fetal Aneuploidies Improves Pregnancy Management. Eur. J. Hum. Genet. 2015, 23, 1286–1293. [Google Scholar] [CrossRef] [PubMed]
- Kazemi-Sefat, G.E.; Keramatipour, M.; Talebi, S.; Kavousi, K.; Sajed, R.; Kazemi-Sefat, N.A.; Mousavizadeh, K. The Importance of CDC27 in Cancer: Molecular Pathology and Clinical Aspects. Cancer Cell Int. 2021, 21, 160. [Google Scholar] [CrossRef]
- Krivec, N.; Couvreu de Deckersberg, E.; Lei, Y.; Al Delbany, D.; Regin, M.; Verhulst, S.; van Grunsven, L.A.; Sermon, K.; Spits, C. Gain of 1q Confers an MDM4-Driven Growth Advantage to Undifferentiated and Differentiating HESC While Altering Their Differentiation Capacity. bioRxiv 2023. [Google Scholar] [CrossRef]
- Stavish, D.; Price, C.J.; Gelezauskaite, G.; Leonhard, K.A.; Taapken, S.M.; McIntire, E.M.; Laing, O.; James, B.M.; Riley, J.J.; Zerbib, J.; et al. Cytogenetic Resource Enables Mechanistic Resolution of Changing Trends in Human Pluripotent Stem Cell Aberrations Linked to Feeder-Free Culture. bioRxiv 2023. [Google Scholar] [CrossRef]
- Halliwell, J.A.; Baker, D.; Judge, K.; Quail, M.A.; Oliver, K.; Betteridge, E.; Skelton, J.; Andrews, P.W.; Barbaric, I. Nanopore Sequencing Indicates That Tandem Amplification of Chromosome 20q11.21 in Human Pluripotent Stem Cells Is Driven by Break-Induced Replication. Stem Cells Dev. 2021, 30, 578–586. [Google Scholar] [CrossRef]
- Spits, C.; Mateizel, I.; Geens, M.; Mertzanidou, A.; Staessen, C.; Vandeskelde, Y.; Van der Elst, J.; Liebaers, I.; Sermon, K. Recurrent Chromosomal Abnormalities in Human Embryonic Stem Cells. Nat. Biotechnol. 2008, 26, 1361–1363. [Google Scholar] [CrossRef]
- Lei, Y.; Al Delbany, D.; Krivec, N.; Regin, M.; Couvreu de Deckersberg, E.; Janssens, C.; Ghosh, M.; Sermon, K.; Spits, C. SALL3 Mediates the Loss of Neuroectodermal Differentiation Potential in Human Embryonic Stem Cells with Chromosome 18q Loss. Stem Cell Rep. 2024, 19, 562–578. [Google Scholar] [CrossRef]
- Amir, H.; Touboul, T.; Sabatini, K.; Chhabra, D.; Garitaonandia, I.; Loring, J.F.; Morey, R.; Laurent, L.C. Spontaneous Single-Copy Loss of TP53 in Human Embryonic Stem Cells Markedly Increases Cell Proliferation and Survival. Stem Cells 2017, 35, 872–885. [Google Scholar] [CrossRef] [PubMed]
- Rouhani, F.J.; Zou, X.; Danecek, P.; Badja, C.; Amarante, T.D.; Koh, G.; Wu, Q.; Memari, Y.; Durbin, R.; Martincorena, I.; et al. Substantial Somatic Genomic Variation and Selection for BCOR Mutations in Human Induced Pluripotent Stem Cells. Nat. Genet. 2022, 54, 1406. [Google Scholar] [CrossRef] [PubMed]
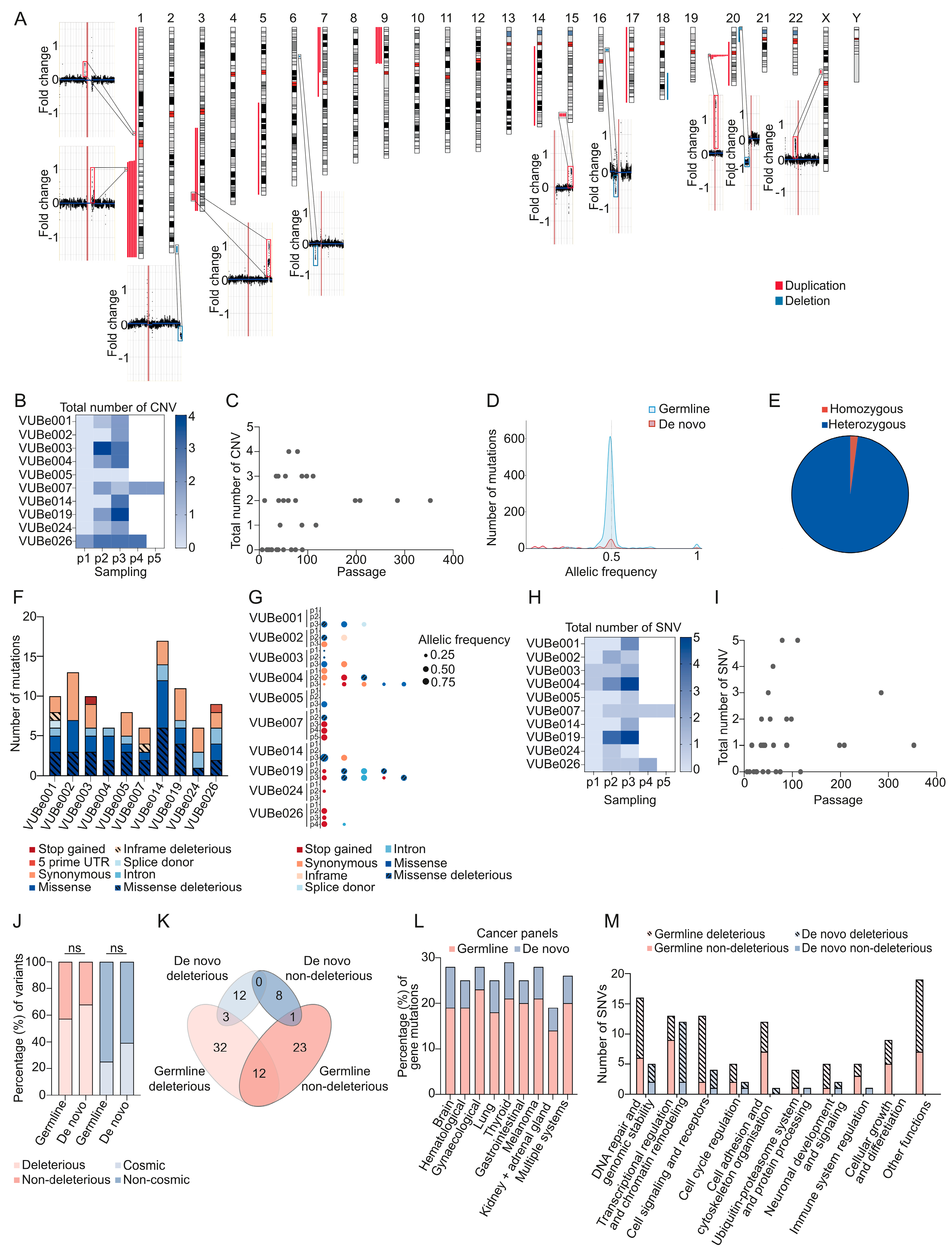
 . Mutations found in duplicated regions of the genome are marked with ▼.
. Mutations found in duplicated regions of the genome are marked with ▼.
. Mutations found in duplicated regions of the genome are marked with ▼.
. Mutations found in duplicated regions of the genome are marked with ▼.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al Delbany, D.; Ghosh, M.S.; Krivec, N.; Huyghebaert, A.; Regin, M.; Duong, M.C.; Lei, Y.; Sermon, K.; Olsen, C.; Spits, C. De Novo Cancer Mutations Frequently Associate with Recurrent Chromosomal Abnormalities during Long-Term Human Pluripotent Stem Cell Culture. Cells 2024, 13, 1395. https://doi.org/10.3390/cells13161395
Al Delbany D, Ghosh MS, Krivec N, Huyghebaert A, Regin M, Duong MC, Lei Y, Sermon K, Olsen C, Spits C. De Novo Cancer Mutations Frequently Associate with Recurrent Chromosomal Abnormalities during Long-Term Human Pluripotent Stem Cell Culture. Cells. 2024; 13(16):1395. https://doi.org/10.3390/cells13161395
Chicago/Turabian StyleAl Delbany, Diana, Manjusha S. Ghosh, Nuša Krivec, Anfien Huyghebaert, Marius Regin, Mai Chi Duong, Yingnan Lei, Karen Sermon, Catharina Olsen, and Claudia Spits. 2024. "De Novo Cancer Mutations Frequently Associate with Recurrent Chromosomal Abnormalities during Long-Term Human Pluripotent Stem Cell Culture" Cells 13, no. 16: 1395. https://doi.org/10.3390/cells13161395