
The Complement System Is Essential for Arteriogenesis by Enhancing Sterile Inflammation as a Relevant Step in Collateral Artery Growth

 , ,
, , 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Protocol and Treatments
2.2. Experimental Procedures and Tissue Harvesting
2.3. Histology and Immunohistology
2.4. Flow Cytometry Analyses of Whole Blood
2.5. qRT-PCR Analyses
2.6. Statistical Analyses
3. Results
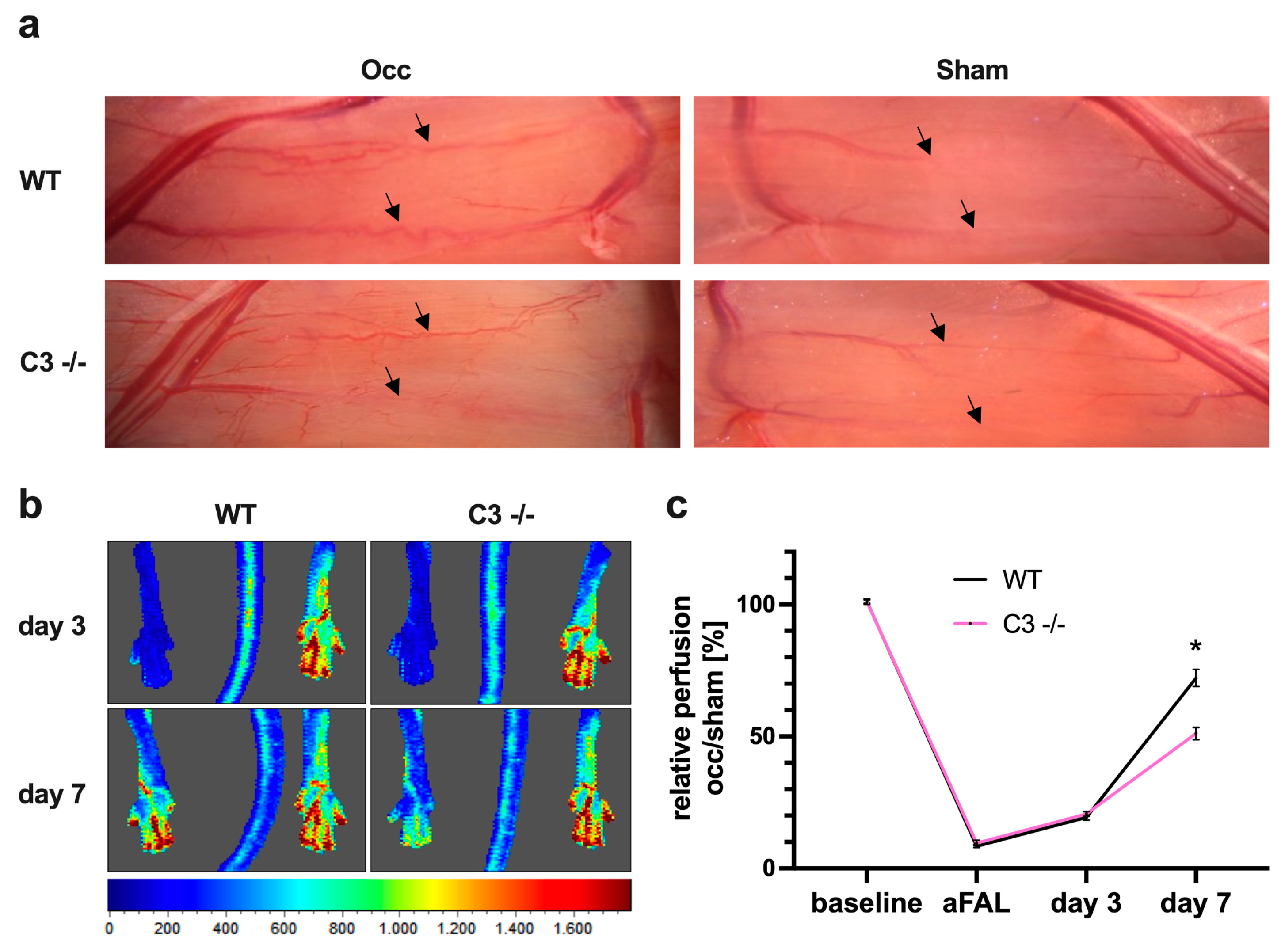
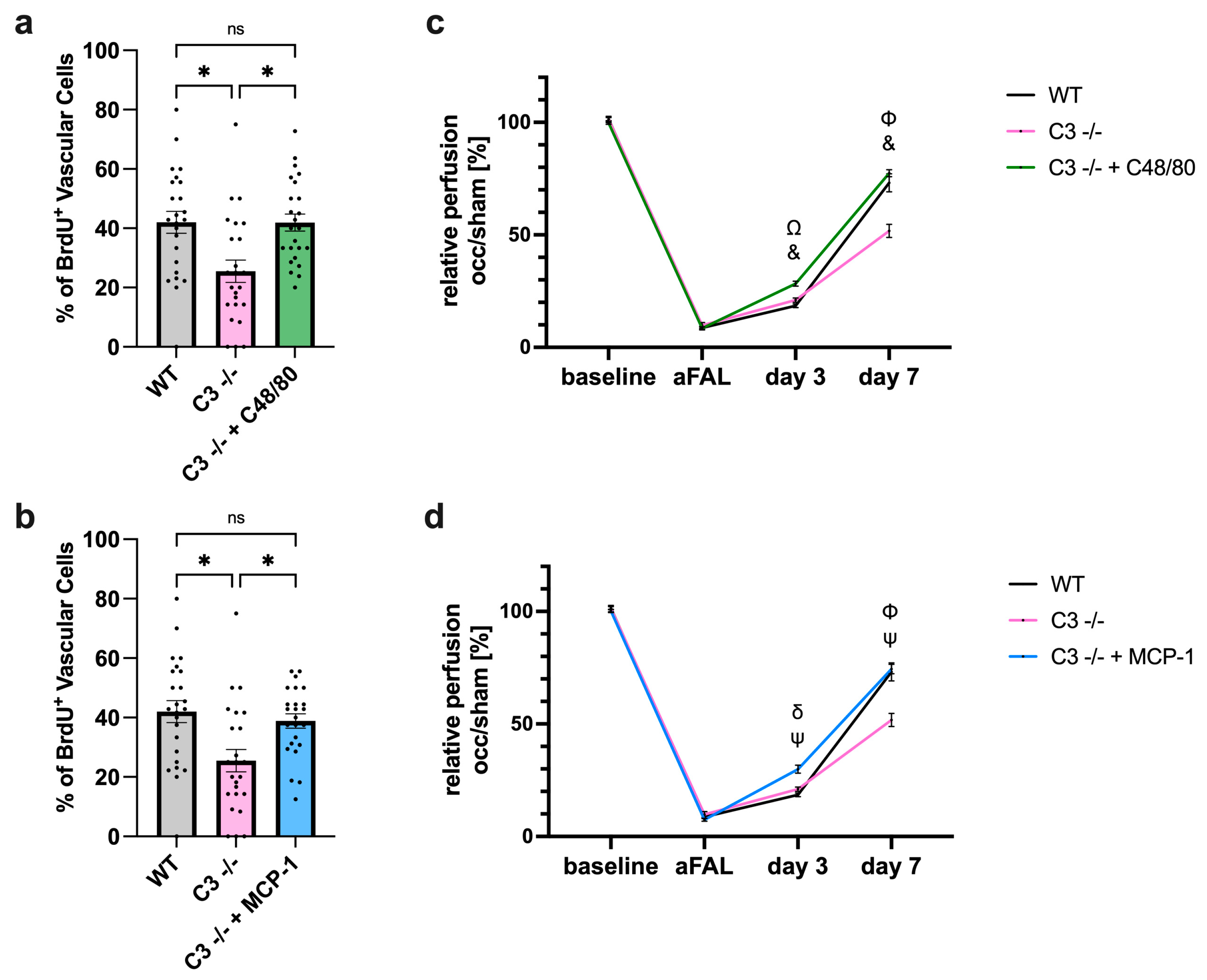
3.1. The Absence of C3 in Mice Reduces Perfusion Recovery in Arteriogenesis
3.1.1. Impact of C3 Deficiency on Perfusion Recovery
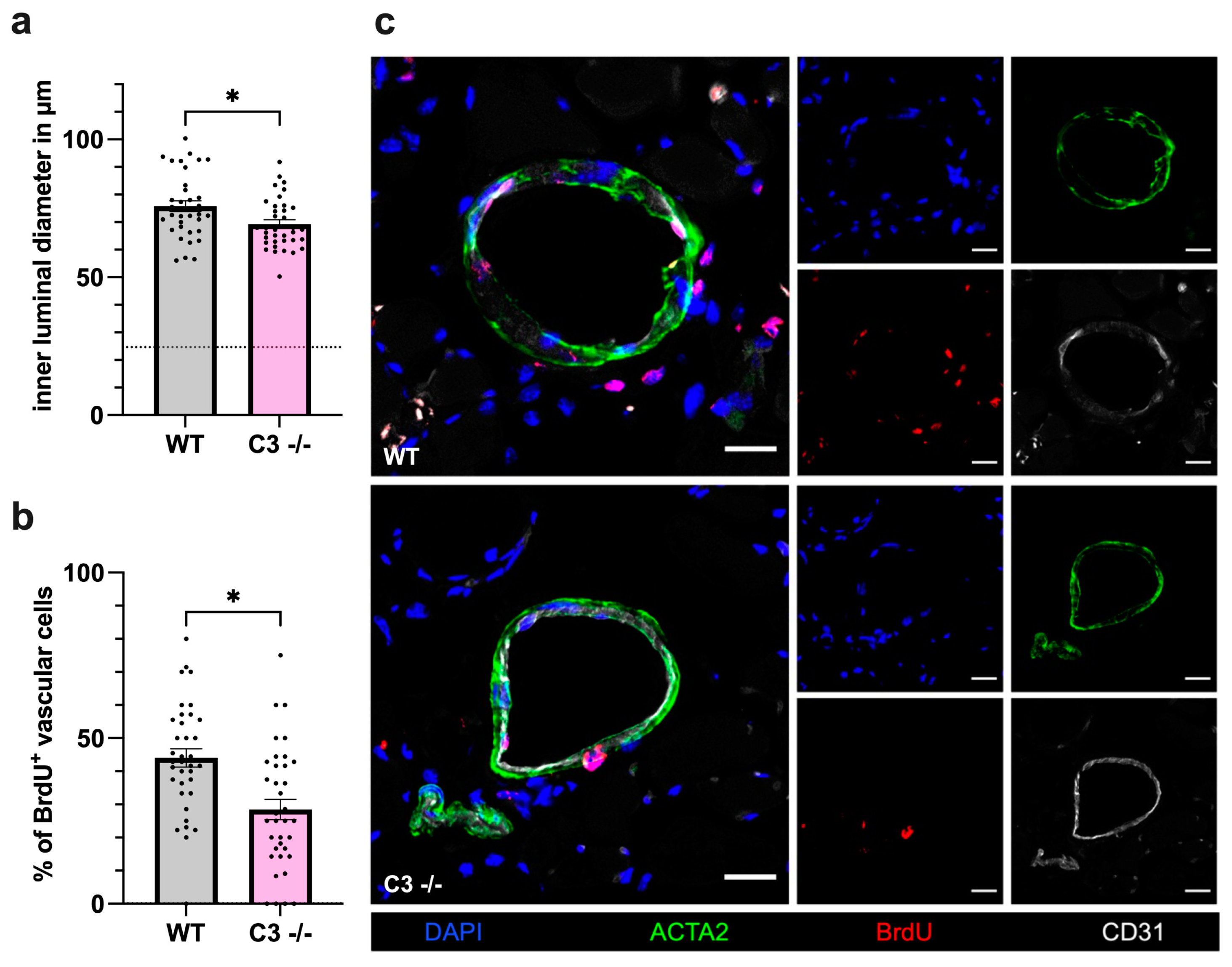
3.1.2. Impact of C3 Deficiency on Vascular Cell Proliferation
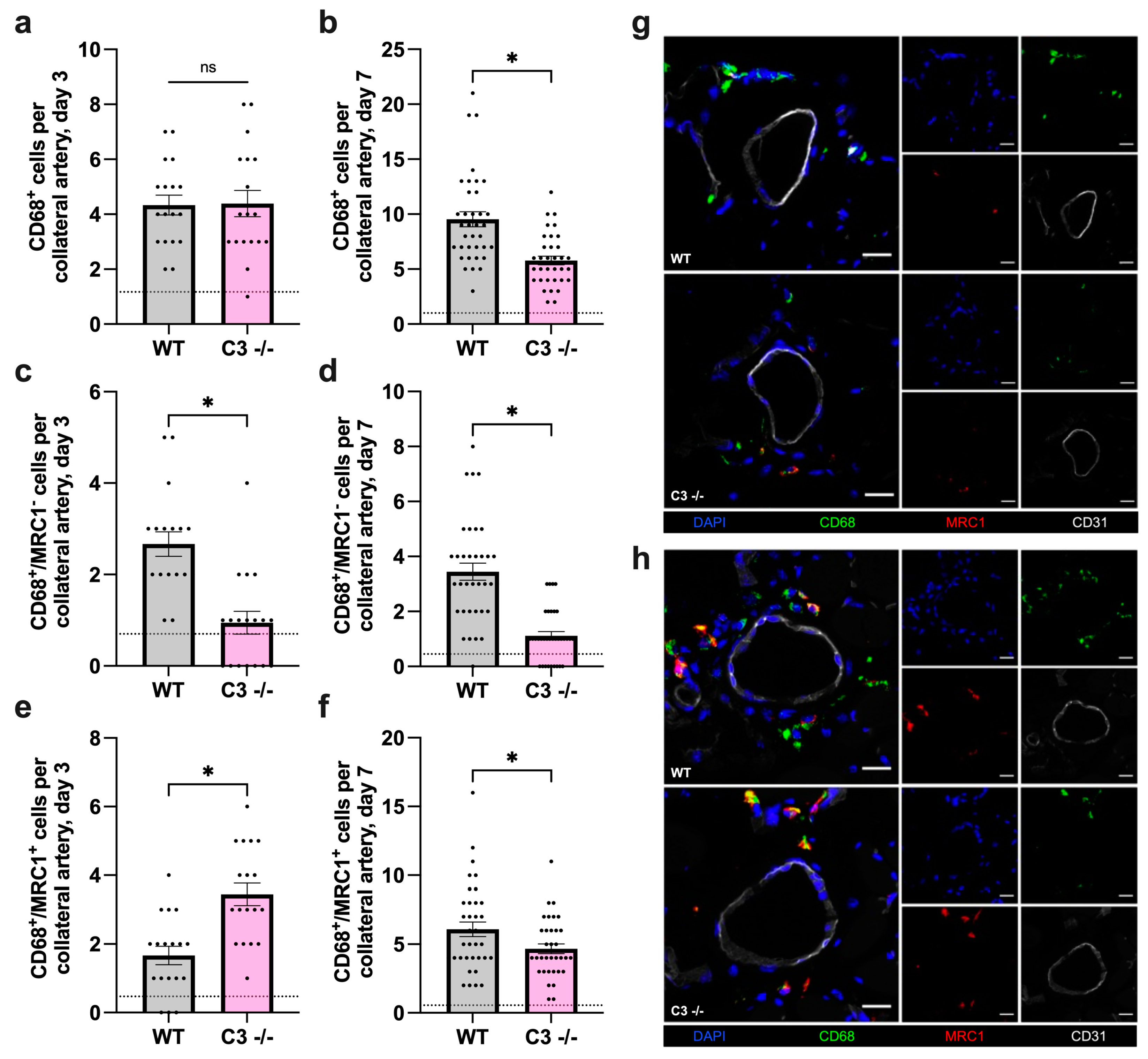
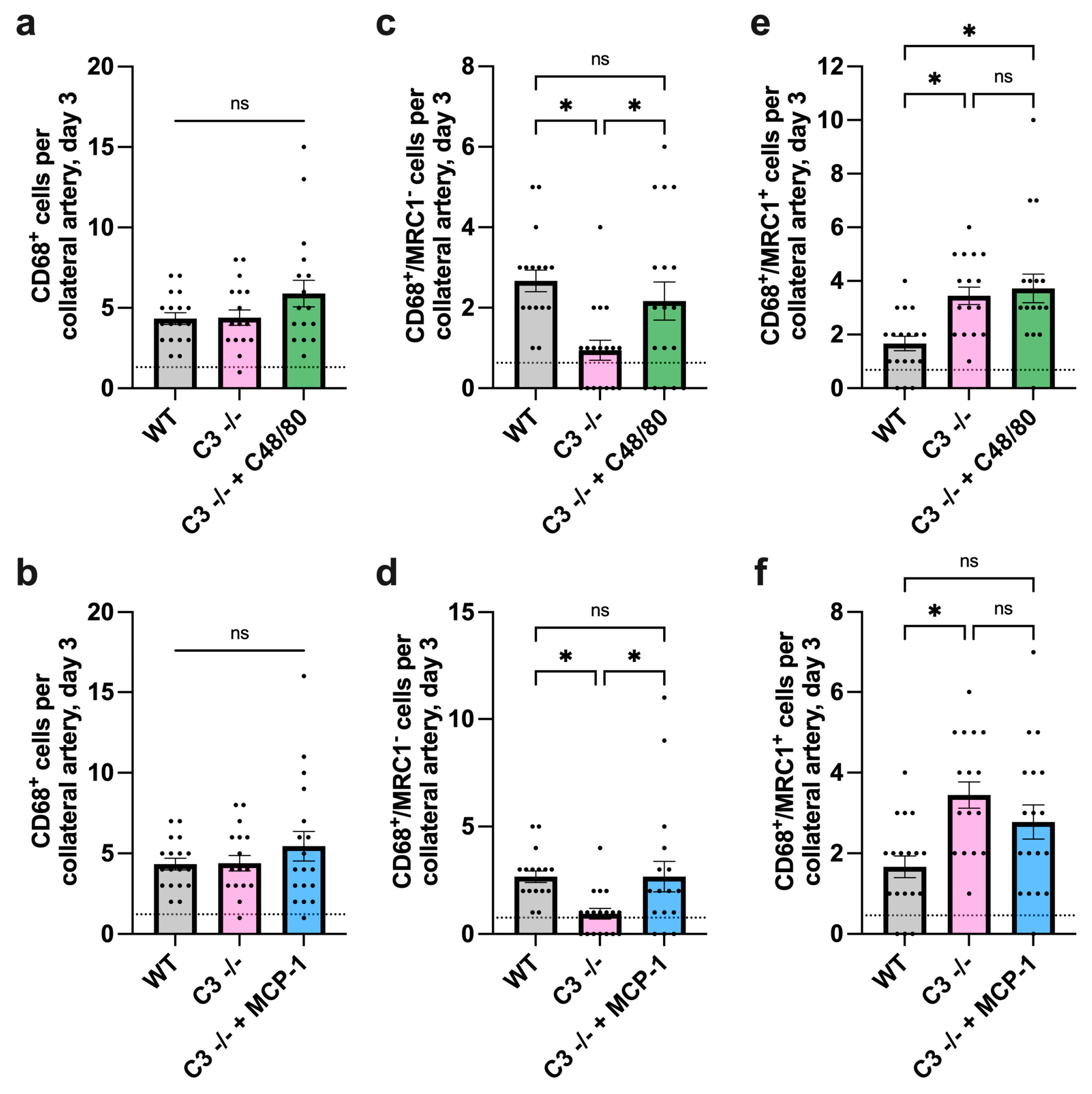
3.2. The Absence of C3 Leads to Diminished Macrophage Recruitment and Alters Macrophage Polarization during Arteriogenesis in Mice
3.2.1. Impact of C3 Deficiency on Macrophage Recruitment and Polarization 3 Days after FAL
3.2.2. Impact of C3 Deficiency on Macrophage Recruitment and Polarization 7 Days after FAL
3.3. The Absence of C3 Reveals No Impact on Platelet–Leukocyte Aggregate Formation and Perivascular Mast Cell Activation in Arteriogenesis in Mice
3.3.1. Impact of C3 Deficiency on Platelet–Leukocyte Aggregate Formation
3.3.2. Impact of C3 Deficiency on Mast Cell Recruitment and Activation
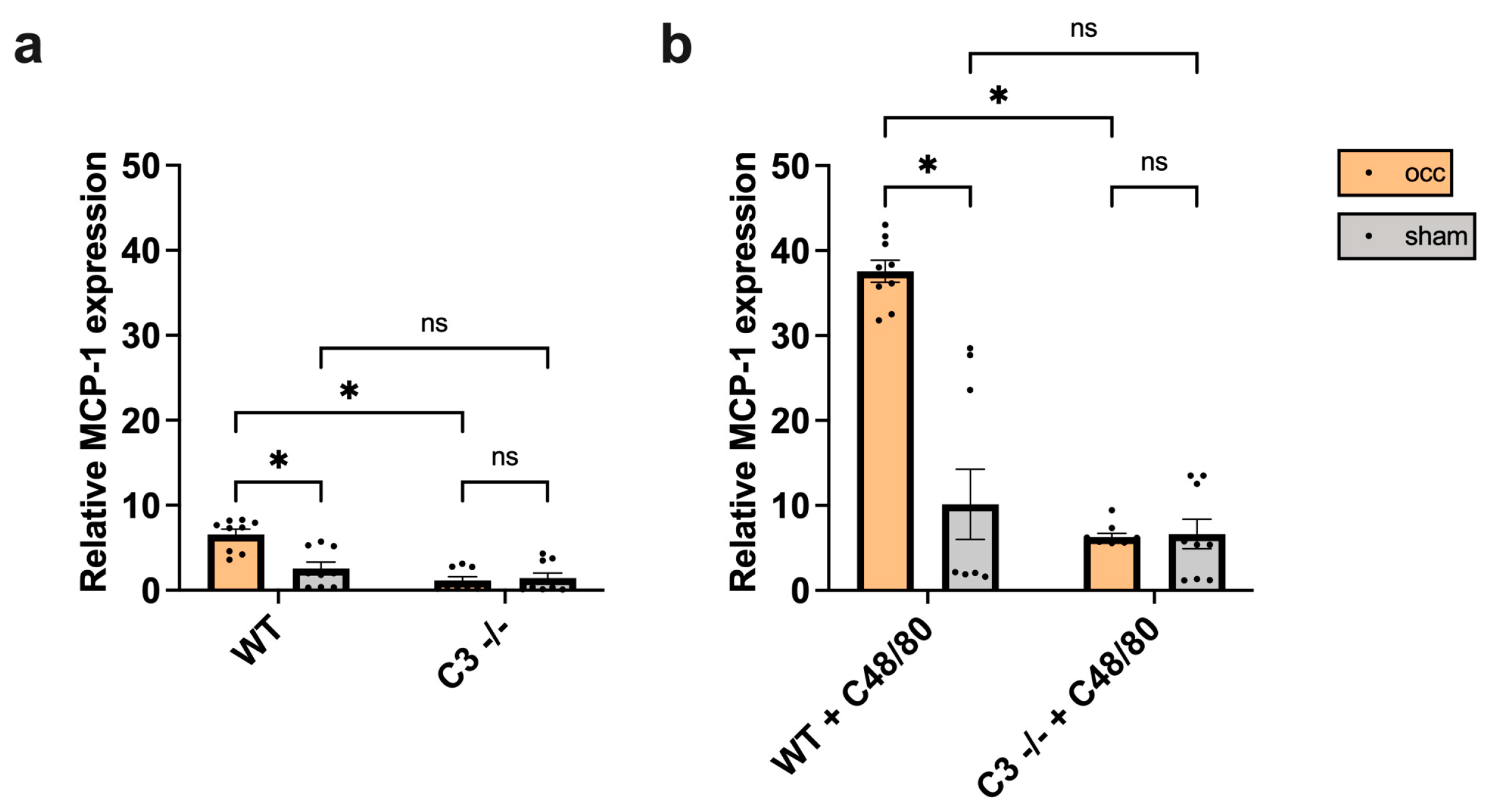
3.4. The Absence of C3 Interferes with MCP-1 Expression in Growing Collateral Arteries in Mice
3.4.1. Impact of C3 Deficiency on MCP-1 Expression
3.4.2. Impact of C48/80 on MCP-1 Expression in C3 −/− Mice
3.5. Forced Mast Cell Activation Rescues Perfusion Recovery in C3 −/− Mice via Promotion of MCP-1 Expression and Subsequent M1-Like Macrophage Recruitment
3.5.1. Impact of C48/80 or MCP-1 on Macrophage Recruitment and Polarization in C3 −/− Mice
3.5.2. Impact of C48/80 or MCP-1 on Vascular Cell Proliferation and Perfusion Recovery in C3 −/− Mice
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Faber, J.E.; Chilian, W.M.; Deindl, E.; van Royen, N.; Simons, M. A brief etymology of the collateral circulation. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1854–1859. [Google Scholar] [CrossRef]
- Schaper, W. Collateral circulation: Past and present. Basic. Res. Cardiol. 2009, 104, 5–21. [Google Scholar] [CrossRef] [PubMed]
- Tzima, E.; Irani-Tehrani, M.; Kiosses, W.B.; Dejana, E.; Schultz, D.A.; Engelhardt, B.; Cao, G.; DeLisser, H.; Schwartz, M.A. A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature 2005, 437, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Hoefer, I.E.; van Royen, N.; Rectenwald, J.E.; Deindl, E.; Hua, J.; Jost, M.; Grundmann, S.; Voskuil, M.; Ozaki, C.K.; Piek, J.J.; et al. Arteriogenesis proceeds via ICAM-1/Mac-1- mediated mechanisms. Circ. Res. 2004, 94, 1179–1185. [Google Scholar] [CrossRef] [PubMed]
- Lasch, M.; Kleinert, E.C.; Meister, S.; Kumaraswami, K.; Buchheim, J.I.; Grantzow, T.; Lautz, T.; Salpisti, S.; Fischer, S.; Troidl, K.; et al. Extracellular RNA released due to shear stress controls natural bypass growth by mediating mechanotransduction in mice. Blood 2019, 134, 1469–1479. [Google Scholar] [CrossRef] [PubMed]
- Chandraratne, S.; von Bruehl, M.L.; Pagel, J.I.; Stark, K.; Kleinert, E.; Konrad, I.; Farschtschi, S.; Coletti, R.; Gärtner, F.; Chillo, O.; et al. Critical role of platelet glycoprotein ibα in arterial remodeling. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 589–597. [Google Scholar] [CrossRef]
- Yang, J.; Furie, B.C.; Furie, B. The biology of P-selectin glycoprotein ligand-1: Its role as a selectin counterreceptor in leukocyte-endothelial and leukocyte-platelet interaction. Thromb. Haemost. 1999, 81, 1–7. [Google Scholar]
- Chillo, O.; Kleinert, E.C.; Lautz, T.; Lasch, M.; Pagel, J.I.; Heun, Y.; Troidl, K.; Fischer, S.; Caballero-Martinez, A.; Mauer, A.; et al. Perivascular mast cells govern shear stress-induced arteriogenesis by orchestrating leukocyte function. Cell. Rep. 2016, 16, 2197–2207. [Google Scholar] [CrossRef]
- Deshmane, S.L.; Kremlev, S.; Amini, S.; Sawaya, B.E. Monocyte chemoattractant protein-1 (MCP-1): An overview. J. Interferon Cytokine Res. 2009, 29, 313–326. [Google Scholar] [CrossRef]
- Arras, M.; Ito, W.D.; Scholz, D.; Winkler, B.; Schaper, J.; Schaper, W. Monocyte activation in angiogenesis and collateral growth in the rabbit hindlimb. J. Clin. Invest. 1998, 101, 40–50. [Google Scholar] [CrossRef]
- Deindl, E.; Hoefer, I.E.; Fernandez, B.; Barancik, M.; Heil, M.; Strniskova, M.; Schaper, W. Involvement of the fibroblast growth factor system in adaptive and chemokine-induced arteriogenesis. Circ. Res. 2003, 92, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Heil, M.; Ziegelhoeffer, T.; Pipp, F.; Kostin, S.; Martin, S.; Clauss, M.; Schaper, W. Blood monocyte concentration is critical for enhancement of collateral artery growth. Am. J. Physiol. Heart. Circ. Physiol. 2002, 283, H2411–H2419. [Google Scholar] [CrossRef]
- Hoefer, I.E.; van Royen, N.; Rectenwald, J.E.; Bray, E.J.; Abouhamze, Z.; Moldawer, L.L.; Voskuil, M.; Piek, J.J.; Buschmann, I.R.; Ozaki, C.K. Direct evidence for tumor necrosis factor-alpha signaling in arteriogenesis. Circulation 2002, 105, 1639–1641. [Google Scholar] [CrossRef] [PubMed]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef] [PubMed]
- Gratchev, A.; Guillot, P.; Hakiy, N.; Politz, O.; Orfanos, C.E.; Schledzewski, K.; Goerdt, S. Alternatively activated macrophages differentially express fibronectin and its splice variants and the extracellular matrix protein betaIG-H. Scand. J. Immunol. 2001, 53, 386–392. [Google Scholar] [CrossRef]
- Troidl, C.; Jung, G.; Troidl, K.; Hoffmann, J.; Mollmann, H.; Nef, H.; Schaper, W.; Hamm, C.W.; Schmitz-Rixen, T. The temporal and spatial distribution of macrophage subpopulations during arteriogenesis. Curr. Vasc. Pharmacol. 2013, 11, 5–12. [Google Scholar] [CrossRef]
- Merle, N.S.; Church, S.E.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement system part I–molecular mechanisms of activation and regulation. Front. Immunol. 2015, 6, 262. [Google Scholar] [CrossRef] [PubMed]
- Howard, M.C.; Nauser, C.L.; Farrar, C.A.; Sacks, S.H. Complement in ischaemia-reperfusion injury and transplantation. Semin. Immunopathol. 2021, 43, 789–797. [Google Scholar] [CrossRef]
- Coss, S.L.; Zhou, D.; Chua, G.T.; Aziz, R.A.; Hoffman, R.P.; Wu, Y.L.; Ardoin, S.P.; Atkinson, J.P.; Yu, C.Y. The complement system and human autoimmune diseases. J. Autoimmun. 2023, 137, 102979. [Google Scholar] [CrossRef]
- Petr, V.; Thurman, J.M. The role of complement in kidney disease. Nat. Rev. Nephrol. 2023, 19, 771–787. [Google Scholar] [CrossRef]
- Mollnes, T.E.; Huber-Lang, M. Complement in sepsis-when science meets clinics. FEBS. Lett. 2020, 594, 2621–2632. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Arguinzonis, M.; Diaz-Riera, E.; Peña, E.; Escate, R.; Juan-Babot, O.; Mata, P.; Badimon, L.; Padro, T. Alternative C3 complement system: Lipids and atherosclerosis. Int. J. Mol. Sci. 2021, 22, 5122. [Google Scholar] [CrossRef] [PubMed]
- Gnanaguru, G.; Tabor, S.J.; Bonilla, G.M.; Sadreyev, R.; Yuda, K.; Köhl, J.; Connor, K.M. Microglia refine developing retinal astrocytic and vascular networks through the complement C3/C3aR axis. Development 2023, 150. [Google Scholar] [CrossRef]
- Long, Q.; Cao, X.; Bian, A.; Li, Y. C3a increases VEGF and decreases PEDF mRNA levels in human retinal pigment epithelial cells. Biomed. Res. Int. 2016, 2016, 6958752. [Google Scholar] [CrossRef] [PubMed]
- Gotz, P.; Braumandl, A.; Kubler, M.; Kumaraswami, K.; Ishikawa-Ankerhold, H.; Lasch, M.; Deindl, E. C3 deficiency leads to increased angiogenesis and elevated pro-angiogenic leukocyte recruitment in ischemic muscle tissue. Int. J. Mol. Sci. 2021, 22, 5800. [Google Scholar] [CrossRef]
- Markiewski, M.M.; Daugherity, E.; Reese, B.; Karbowniczek, M. The role of complement in angiogenesis. Antibodies 2020, 9, 67. [Google Scholar] [CrossRef]
- Götz, P.; Azubuike-Osu, S.O.; Braumandl, A.; Arnholdt, C.; Kübler, M.; Richter, L.; Lasch, M.; Bobrowski, L.; Preissner, K.T.; Deindl, E. Cobra venom factor boosts arteriogenesis in mice. Int. J. Mol. Sci. 2022, 23, 8454. [Google Scholar] [CrossRef]
- Scholz, D.; Ziegelhoeffer, T.; Helisch, A.; Wagner, S.; Friedrich, C.; Podzuweit, T.; Schaper, W. Contribution of arteriogenesis and angiogenesis to postocclusive hindlimb perfusion in mice. J. Mol. Cell. Cardiol. 2002, 34, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Chomczynski, P.; Sacchi, N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 1987, 162, 156–159. [Google Scholar] [CrossRef]
- Xu, J.M.; Shi, G.P. Emerging role of mast cells and macrophages in cardiovascular and metabolic diseases. Endocr. Rev. 2012, 33, 71–108. [Google Scholar] [CrossRef]
- Michelson, A.D.; Barnard, M.R.; Krueger, L.A.; Valeri, C.R.; Furman, M.I. Circulating monocyte-platelet aggregates are a more sensitive marker of in vivo platelet activation than platelet surface P-selectin: Studies in baboons, human coronary intervention, and human acute myocardial infarction. Circulation 2001, 104, 1533–1537. [Google Scholar] [CrossRef] [PubMed]
- Ito, W.D.; Arras, M.; Winkler, B.; Scholz, D.; Schaper, J.; Schaper, W. Monocyte chemotactic protein-1 increases collateral and peripheral conductance after femoral artery occlusion. Circ. Res. 1997, 80, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Gschwandtner, M.; Derler, R.; Midwood, K.S. More than just attractive: How CCL2 influences myeloid cell behavior beyond chemotaxis. Front. Immunol. 2019, 10, 2759. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, K.; Liang, X.; Li, Y.; Zhang, Y.; Zhang, C.; Wei, H.; Luo, R.; Ge, S.; Xu, G. Complement C3 produced by macrophages promotes renal fibrosis via IL-17A secretion. Front. Immunol. 2018, 9, 2385. [Google Scholar] [CrossRef] [PubMed]
- Zhong, F.; Hu, Z.; Jiang, K.; Lei, B.; Wu, Z.; Yuan, G.; Luo, H.; Dong, C.; Tang, B.; Zheng, C.; et al. Complement C3 activation regulates the production of tRNA-derived fragments Gly-tRFs and promotes alcohol-induced liver injury and steatosis. Cell Res. 2019, 29, 548–561. [Google Scholar] [CrossRef]
- Pei, Y.; Zhang, J.; Qu, J.; Rao, Y.; Li, D.; Gai, X.; Chen, Y.; Liang, Y.; Sun, Y. Complement component 3 protects human bronchial epithelial cells from cigarette smoke-induced oxidative stress and prevents incessant apoptosis. Front. Immunol. 2022, 13, 1035930. [Google Scholar] [CrossRef]
- Huber-Lang, M.; Sarma, J.V.; Zetoune, F.S.; Rittirsch, D.; Neff, T.A.; McGuire, S.R.; Lambris, J.D.; Warner, R.L.; Flierl, M.A.; Hoesel, L.M.; et al. Generation of C5a in the absence of C3: A new complement activation pathway. Nat. Med. 2006, 12, 682–687. [Google Scholar] [CrossRef]
- Roumenina, L.T. Terminal complement without C5 convertase? Blood 2021, 137, 431–432. [Google Scholar] [CrossRef]
- Ramos, T.N.; Darley, M.M.; Weckbach, S.; Stahel, P.F.; Tomlinson, S.; Barnum, S.R. The C5 convertase is not required for activation of the terminal complement pathway in murine experimental cerebral malaria. J. Biol. Chem. 2012, 287, 24734–24738. [Google Scholar] [CrossRef]
- Ruan, C.C.; Ge, Q.; Li, Y.; Li, X.D.; Chen, D.R.; Ji, K.D.; Wu, Y.J.; Sheng, L.J.; Yan, C.; Zhu, D.L.; et al. Complement-mediated macrophage polarization in perivascular adipose tissue contributes to vascular injury in deoxycorticosterone acetate-salt mice. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 598–606. [Google Scholar] [CrossRef]
- Bohlson, S.S.; O’Conner, S.D.; Hulsebus, H.J.; Ho, M.M.; Fraser, D.A. Complement, c1q, and c1q-related molecules regulate macrophage polarization. Front. Immunol. 2014, 5, 402. [Google Scholar] [CrossRef] [PubMed]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Imhof, B.A.; Aurrand-Lions, M. Adhesion mechanisms regulating the migration of monocytes. Nat. Rev. Immunol. 2004, 4, 432–444. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Lisi, L.; Ciotti, G.M.; Braun, D.; Kalinin, S.; Currò, D.; Dello Russo, C.; Coli, A.; Mangiola, A.; Anile, C.; Feinstein, D.L.; et al. Expression of iNOS, CD163 and ARG-1 taken as M1 and M2 markers of microglial polarization in human glioblastoma and the surrounding normal parenchyma. Neurosci. Lett. 2017, 645, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Italiani, P.; Boraschi, D. From monocytes to M1/M2 macrophages: Phenotypical vs. functional differentiation. Front. Immunol. 2014, 5, 514. [Google Scholar] [CrossRef]
- Schultz, K.; Murthy, V.; Tatro, J.B.; Beasley, D. Endogenous interleukin-1 alpha promotes a proliferative and proinflammatory phenotype in human vascular smooth muscle cells. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H2927–H2934. [Google Scholar] [CrossRef]
- Lee, G.L.; Wu, J.Y.; Tsai, C.S.; Lin, C.Y.; Tsai, Y.T.; Lin, C.S.; Wang, Y.F.; Yet, S.F.; Hsu, Y.J.; Kuo, C.C. TLR4-Activated MAPK-IL-6 axis regulates vascular smooth muscle cell function. Int. J. Mol. Sci. 2016, 17, 1394. [Google Scholar] [CrossRef]
- Bonin, P.D.; Fici, G.J.; Singh, J.P. Interleukin-1 promotes proliferation of vascular smooth muscle cells in coordination with PDGF or a monocyte derived growth factor. Exp. Cell Res. 1989, 181, 475–482. [Google Scholar] [CrossRef]
- Rajasekaran, M.; Sul, O.J.; Choi, E.K.; Kim, J.E.; Suh, J.H.; Choi, H.S. MCP-1 deficiency enhances browning of adipose tissue via increased M2 polarization. J. Endocrinol. 2019, 242, 91–101. [Google Scholar] [CrossRef]
- Nio, Y.; Yamauchi, T.; Iwabu, M.; Okada-Iwabu, M.; Funata, M.; Yamaguchi, M.; Ueki, K.; Kadowaki, T. Monocyte chemoattractant protein-1 (MCP-1) deficiency enhances alternatively activated M2 macrophages and ameliorates insulin resistance and fatty liver in lipoatrophic diabetic A-ZIP transgenic mice. Diabetologia 2012, 55, 3350–3358. [Google Scholar] [CrossRef]
- Venkatesha, R.T.; Berla Thangam, E.; Zaidi, A.K.; Ali, H. Distinct regulation of C3a-induced MCP-1/CCL2 and RANTES/CCL5 production in human mast cells by extracellular signal regulated kinase and PI3 kinase. Mol. Immunol. 2005, 42, 581–587. [Google Scholar] [CrossRef]
- Yang, Y.H.; Tsai, I.J.; Chang, C.J.; Chuang, Y.H.; Hsu, H.Y.; Chiang, B.L. The interaction between circulating complement proteins and cutaneous microvascular endothelial cells in the development of childhood Henoch-Schönlein Purpura. PLoS ONE 2015, 10, e0120411. [Google Scholar] [CrossRef]
- Laudes, I.J.; Chu, J.C.; Huber-Lang, M.; Guo, R.F.; Riedemann, N.C.; Sarma, J.V.; Mahdi, F.; Murphy, H.S.; Speyer, C.; Lu, K.T.; et al. Expression and function of C5a receptor in mouse microvascular endothelial cells. J. Immunol. 2002, 169, 5962–5970. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Zhang, X.; Miwa, T.; Song, W.C. Complement promotes the development of inflammatory T-helper 17 cells through synergistic interaction with Toll-like receptor signaling and interleukin-6 production. Blood 2009, 114, 1005–1015. [Google Scholar] [CrossRef]
- Huang, X.; Li, Z.; Shen, X.; Nie, N.; Shen, Y. IL-17 upregulates MCP-1 expression via Act1 / TRAF6 / TAK1 in experimental autoimmune myocarditis. Cytokine 2022, 152, 155823. [Google Scholar] [CrossRef] [PubMed]
- Hata, T.; Takahashi, M.; Hida, S.; Kawaguchi, M.; Kashima, Y.; Usui, F.; Morimoto, H.; Nishiyama, A.; Izawa, A.; Koyama, J.; et al. Critical role of Th17 cells in inflammation and neovascularization after ischaemia. Cardiovasc. Res. 2011, 90, 364–372. [Google Scholar] [CrossRef] [PubMed]
- De Martinis, M.; Sirufo, M.M.; Suppa, M.; Ginaldi, L. IL-33/IL-31 Axis in Osteoporosis. Int. J. Mol. Sci. 2020, 21, 1239. [Google Scholar] [CrossRef]
- Murdaca, G.; Greco, M.; Tonacci, A.; Negrini, S.; Borro, M.; Puppo, F.; Gangemi, S. IL-33/IL-31 Axis in immune-mediated and allergic diseases. Int. J. Mol. Sci. 2019, 20, 5856. [Google Scholar] [CrossRef]
- Pinto, S.M.; Subbannayya, Y.; Rex, D.A.B.; Raju, R.; Chatterjee, O.; Advani, J.; Radhakrishnan, A.; Keshava Prasad, T.S.; Wani, M.R.; Pandey, A. A network map of IL-33 signaling pathway. J. Cell Commun. Signal 2018, 12, 615–624. [Google Scholar] [CrossRef]
- Singh, S.; Anshita, D.; Ravichandiran, V. MCP-1: Function, regulation, and involvement in disease. Int. Immunopharmacol. 2021, 101, 107598. [Google Scholar] [CrossRef] [PubMed]
- Owsiany, K.M.; Deaton, R.A.; Soohoo, K.G.; Tram Nguyen, A.; Owens, G.K. Dichotomous roles of smooth muscle cell-derived MCP1 (Monocyte Chemoattractant Protein 1) in development of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2022, 42, 942–956. [Google Scholar] [CrossRef]
- Al Samarraie, A.; Pichette, M.; Rousseau, G. Role of the gut microbiome in the development of atherosclerotic cardiovascular disease. Int. J. Mol. Sci. 2023, 24, 5420. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.; Wang, Z.; Levison, B.S.; Koeth, R.A.; Britt, E.B.; Fu, X.; Wu, Y.; Hazen, S.L. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N. Engl. J. Med. 2013, 368, 1575–1584. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, S.H.; Warrier, M. Trimethylamine N-Oxide, the microbiome, and heart and kidney disease. Annu. Rev. Nutr. 2017, 37, 157–181. [Google Scholar] [CrossRef]
- Seldin, M.M.; Meng, Y.; Qi, H.; Zhu, W.; Wang, Z.; Hazen, S.L.; Lusis, A.J.; Shih, D.M. Trimethylamine N-Oxide promotes vascular inflammation through signaling of mitogen-activated protein kinase and nuclear factor-κB. J. Am. Heart Assoc. 2016, 5. [Google Scholar] [CrossRef]
- Wu, M.; Zheng, W.; Song, X.; Bao, B.; Wang, Y.; Ramanan, D.; Yang, D.; Liu, R.; Macbeth, J.C.; Do, E.A.; et al. Gut complement induced by the microbiota combats pathogens and spares commensals. Cell 2024, 187, 897–913. [Google Scholar] [CrossRef]
- Li, H.; Xie, X.; Bai, G.; Qiang, D.; Zhang, L.; Liu, H.; He, Y.; Tang, Y.; Li, L. Vitamin D deficiency leads to the abnormal activation of the complement system. Immunol. Res. 2023, 71, 29–38. [Google Scholar] [CrossRef]
- Ao, T.; Kikuta, J.; Ishii, M. The effects of vitamin D on immune system and inflammatory diseases. Biomolecules 2021, 11, 1624. [Google Scholar] [CrossRef]
- Khanolkar, S.; Hirani, S.; Mishra, A.; Vardhan, S.; Hirani, S.; Prasad, R.; Wanjari, M. Exploring the role of vitamin D in atherosclerosis and its impact on cardiovascular events: A comprehensive review. Cureus 2023, 15, e42470. [Google Scholar] [CrossRef]
- Ruytinx, P.; Proost, P.; Van Damme, J.; Struyf, S. Chemokine-induced macrophage polarization in inflammatory conditions. Front. Immunol. 2018, 9, 1930. [Google Scholar] [CrossRef] [PubMed]
- Cochain, C.; Zernecke, A. Macrophages and immune cells in atherosclerosis: Recent advances and novel concepts. Basic Res. Cardiol. 2015, 110, 34. [Google Scholar] [CrossRef] [PubMed]
- Georgakis, M.K.; van der Laan, S.W.; Asare, Y.; Mekke, J.M.; Haitjema, S.; Schoneveld, A.H.; de Jager, S.C.A.; Nurmohamed, N.S.; Kroon, J.; Stroes, E.S.G.; et al. Monocyte-chemoattractant protein-1 levels in human atherosclerotic lesions associate with plaque vulnerability. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 2038–2048. [Google Scholar] [CrossRef] [PubMed]
- Hong, K.H.; Ryu, J.; Han, K.H. Monocyte chemoattractant protein-1-induced angiogenesis is mediated by vascular endothelial growth factor-A. Blood 2005, 105, 1405–1407. [Google Scholar] [CrossRef] [PubMed]
- Adams, R.H.; Alitalo, K. Molecular regulation of angiogenesis and lymphangiogenesis. Nat. Rev. Mol. Cell Biol. 2007, 8, 464–478. [Google Scholar] [CrossRef]
- Wang, J. Neutrophils in tissue injury and repair. Cell Tissue Res. 2018, 371, 531–539. [Google Scholar] [CrossRef]
- Du Cheyne, C.; Tay, H.; De Spiegelaere, W. The complex TIE between macrophages and angiogenesis. Anat. Histol. Embryol. 2020, 49, 585–596. [Google Scholar] [CrossRef]
- Gurevich, D.B.; Severn, C.E.; Twomey, C.; Greenhough, A.; Cash, J.; Toye, A.M.; Mellor, H.; Martin, P. Live imaging of wound angiogenesis reveals macrophage orchestrated vessel sprouting and regression. Embo. J. 2018, 37. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Geml, A.; Baur, C.; Götz, P.; Elbs, K.; Lasch, M.; Faro, A.; Preissner, K.T.; Deindl, E. The Complement System Is Essential for Arteriogenesis by Enhancing Sterile Inflammation as a Relevant Step in Collateral Artery Growth. Cells 2024, 13, 1405. https://doi.org/10.3390/cells13171405
Geml A, Baur C, Götz P, Elbs K, Lasch M, Faro A, Preissner KT, Deindl E. The Complement System Is Essential for Arteriogenesis by Enhancing Sterile Inflammation as a Relevant Step in Collateral Artery Growth. Cells. 2024; 13(17):1405. https://doi.org/10.3390/cells13171405
Chicago/Turabian StyleGeml, Amanda, Carolin Baur, Philipp Götz, Katharina Elbs, Manuel Lasch, Anna Faro, Klaus T. Preissner, and Elisabeth Deindl. 2024. "The Complement System Is Essential for Arteriogenesis by Enhancing Sterile Inflammation as a Relevant Step in Collateral Artery Growth" Cells 13, no. 17: 1405. https://doi.org/10.3390/cells13171405
APA StyleGeml, A., Baur, C., Götz, P., Elbs, K., Lasch, M., Faro, A., Preissner, K. T., & Deindl, E. (2024). The Complement System Is Essential for Arteriogenesis by Enhancing Sterile Inflammation as a Relevant Step in Collateral Artery Growth. Cells, 13(17), 1405. https://doi.org/10.3390/cells13171405