
Focal Cerebral Ischemia Induces Expression of Glutaminyl Cyclase along with Downstream Molecular and Cellular Inflammatory Responses
 , ,
, ,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Characterization of KO Mice
2.2. Genotyping
2.3. Experimental Stroke
2.4. Evaluation of Functional Outcome
Overall Physical Condition
2.5. Mouse Plasma and Brain Tissue Preparation
2.6. Calculation of Infarct Volumes
2.7. Immunohistochemistry
2.7.1. Single Labeling of NeuN and HuC/D, QC, isoQC and CCL2 in Mouse Brain Sections
2.7.2. Double and Triple Immunofluorescent Labeling in the Mouse Brain
2.8. Microscopy
2.8.1. Light Microscopy
2.8.2. Fluorescence Microscopy
2.8.3. Image Analysis
2.9. Quantification of Chemokines in Brain Tissue and Plasma
2.9.1. Cerebrum Preparation
2.9.2. BCA Protein Assay
2.9.3. Bio Plex Analysis
2.10. Statistical Analysis
3. Results
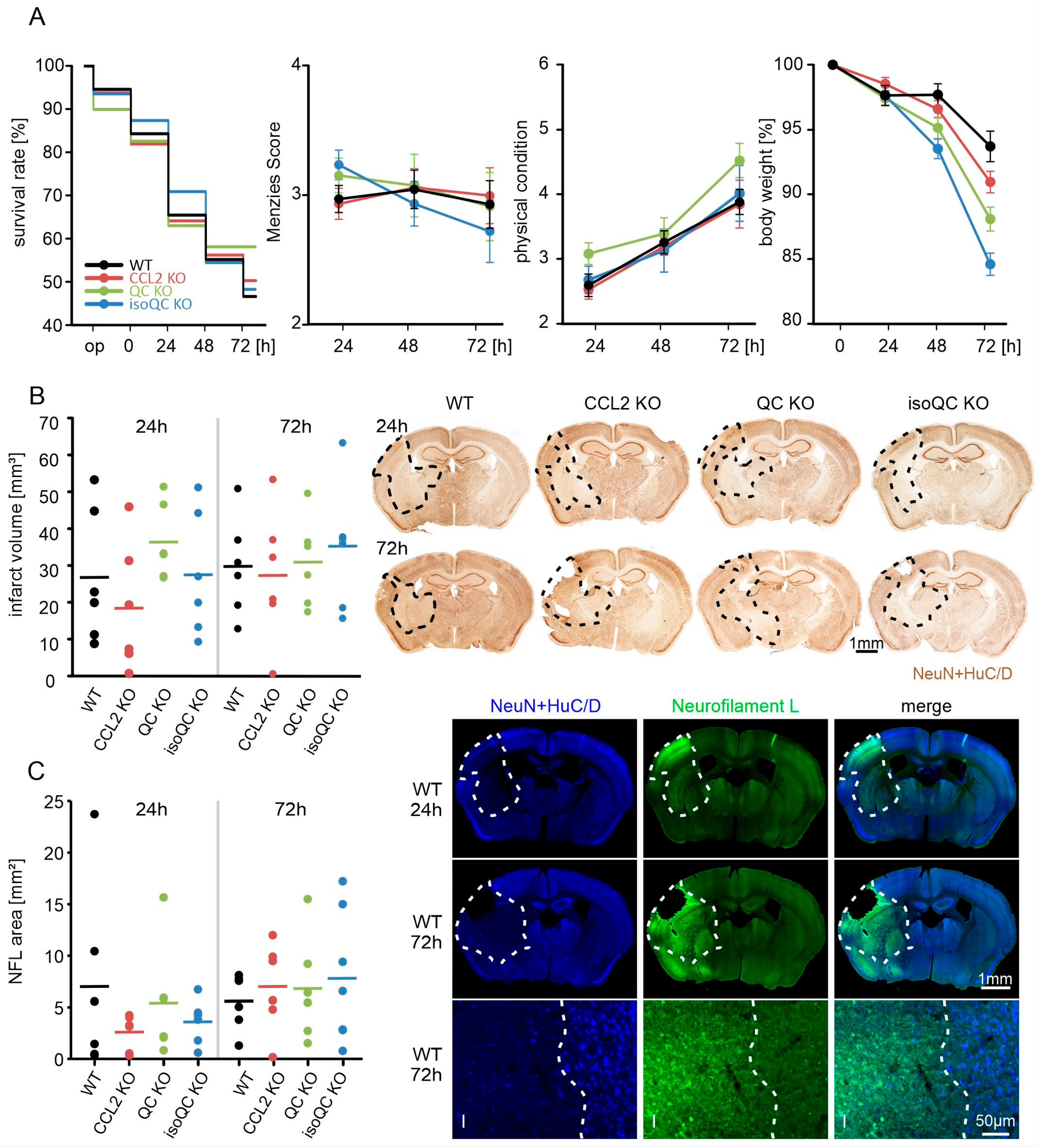
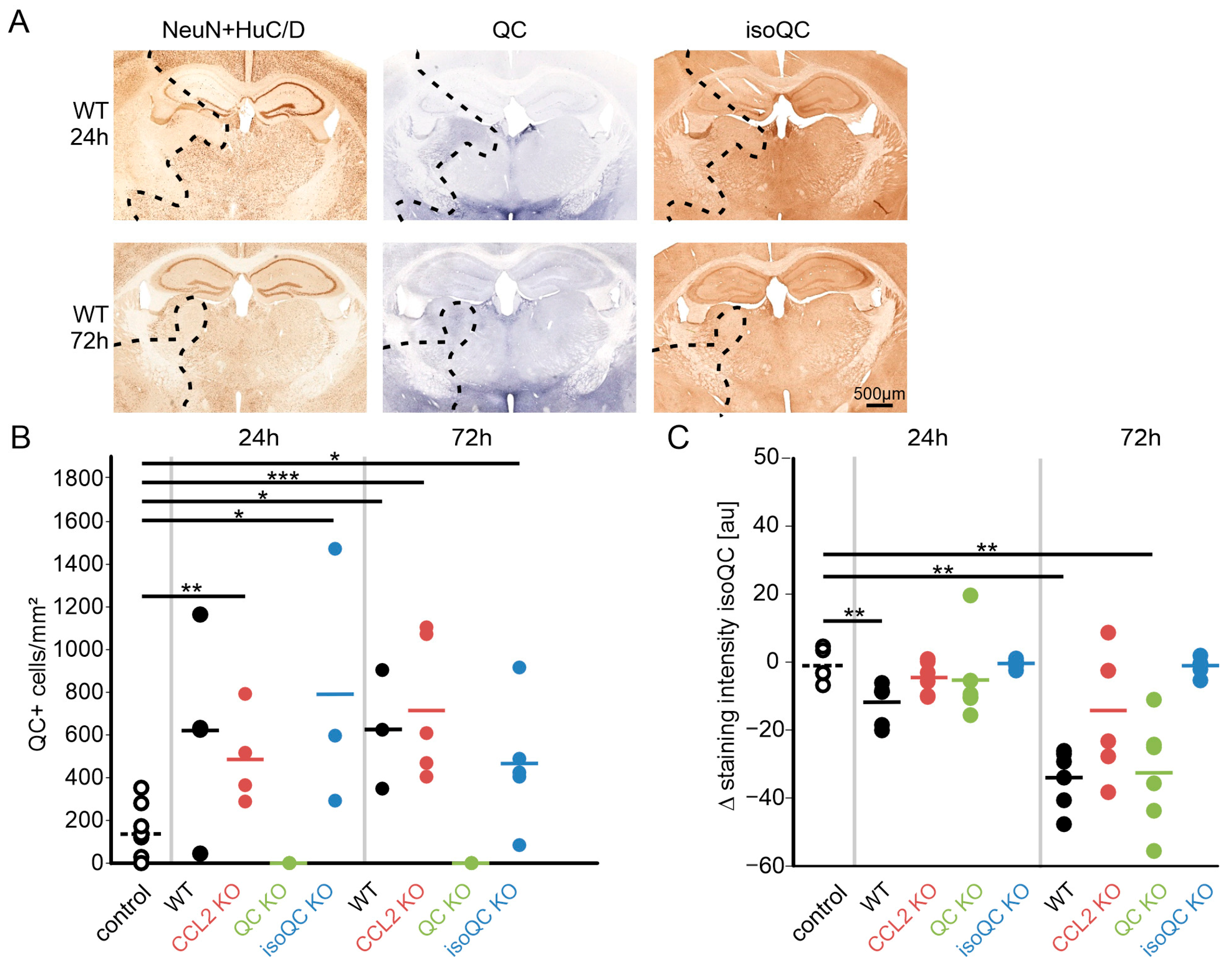
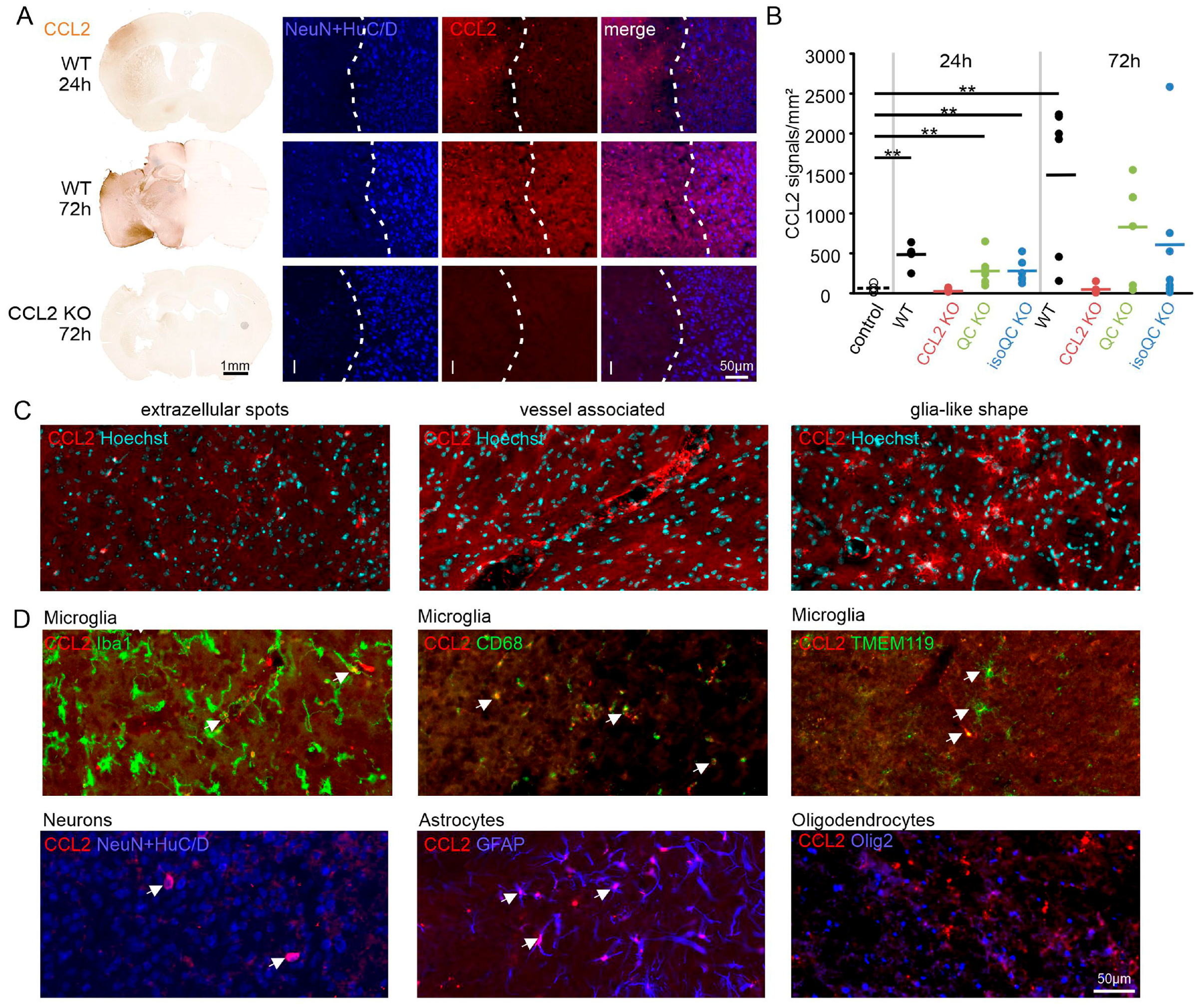
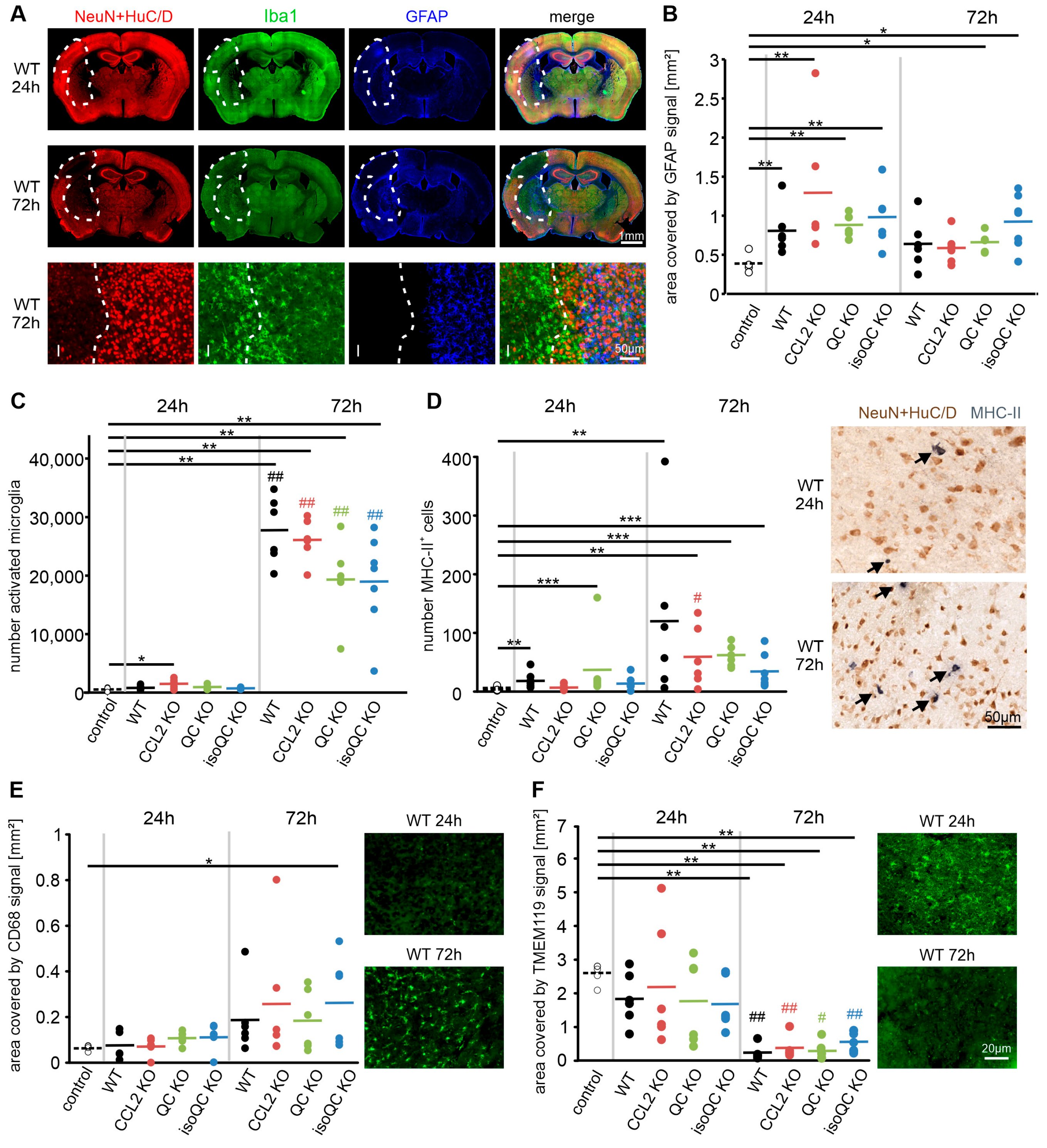
3.1. Consequences of Focal Cerebral Ischemia in Mice with Lacking Genes for QC, isoQC, and CCL2
3.2. Consequences of Increased QC and CCL2 Expression in Ischemic Brain Tissue
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Busby, W.H.; Quackenbush, G.E.; Humm, J.; Youngblood, W.W.; Kizer, J.S. An enzyme(s) that converts glutaminyl-peptides into pyroglutamyl-peptides. J. Biol. Chem. 1987, 262, 8532–8536. [Google Scholar] [CrossRef] [PubMed]
- Fischer, W.H.; Spiess, J. Identification of a mammalian glutaminyl cyclase converting glutaminyl into pyroglutamyl peptides. Proc. Natl. Acad. Sci. USA 1987, 84, 3628–3632. [Google Scholar] [CrossRef] [PubMed]
- Böckers, T.M.; Kreutz, M.R.; Pohl, T. Glutaminyl-cyclase expression in the bovine/porcine hypothalamus and pituitary. J. Neuroendocrinol. 1995, 7, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Hartlage-Rübsamen, M.; Staffa, K.; Waniek, A.; Wermann, M.; Hoffmann, T.; Cynis, H.; Schilling, S.; Demuth, H.U.; Roßner, S. Developmental expression and subcellular localization of glutaminyl cyclase in mouse brain. Int. J. Dev. Neurosci. 2009, 27, 825–835. [Google Scholar] [CrossRef]
- Becker, A.; Eichentopf, R.; Sedlmeier, R.; Waniek, A.; Cynis, H.; Koch, B.; Stephan, A.; Bäuscher, C.; Kohlmann, S.; Hoffmann, T.; et al. IsoQC (QPCTL) knock-out mice suggest differential substrate conversion by glutaminyl cyclase isoenzymes. Biol. Chem. 2016, 397, 45–55. [Google Scholar] [CrossRef]
- Morawski, M.; Hartlage-Rübsamen, M.; Jäger, C.; Waniek, A.; Schilling, S.; Schwab, C.; McGeer, P.L.; Arendt, T.; Demuth, H.U.; Roßner, S. Distinct glutaminyl cyclase expression in Edinger-Westphal nucleus, locus coeruleus and nucleus basalis Meynert contributes to pGlu-Abeta pathology in Alzheimer’s disease. Acta Neuropathol. 2010, 120, 195–207. [Google Scholar] [CrossRef]
- Hartlage-Rübsamen, M.; Morawski, M.; Waniek, A.; Jäger, C.; Zeitschel, U.; Koch, B.; Cynis, H.; Schilling, S.; Schliebs, R.; Demuth, H.U.; et al. Glutaminyl cyclase contributes to the formation of focal and diffuse pyroglutamate (pGlu)-Aβ deposits in hippocampus via distinct cellular mechanisms. Acta Neuropathol. 2011, 121, 705–719. [Google Scholar] [CrossRef]
- Hartlage-Rübsamen, M.; Bluhm, A.; Moceri, S.; Machner, L.; Köppen, J.; Schenk, M.; Hilbrich, I.; Holzer, M.; Weidenfeller, M.; Richter, F.; et al. A glutaminyl cyclase-catalyzed α-synuclein modification identified in human synucleinopathies. Acta Neuropathol. 2021, 142, 399–421. [Google Scholar] [CrossRef]
- Schilling, S.; Zeitschel, U.; Hoffmann, T.; Heiser, U.; Francke, M.; Kehlen, A.; Holzer, M.; Hutter-Paier, B.; Prokesch, M.; Windisch, M.; et al. Glutaminyl cyclase inhibition attenuates pyroglutamate Abeta and Alzheimer’s disease-like pathology. Nat. Med. 2008, 14, 1106–1111. [Google Scholar] [CrossRef]
- Jimenez-Sanchez, M.; Lam, W.; Hannus, M.; Sönnichsen, B.; Imarisio, S.; Fleming, A.; Tarditi, A.; Menzies, F.; Dami, T.E.; Xu, C.; et al. siRNA screen identifies QPCT as a druggable target for Huntington’s disease. Nat. Chem. Biol. 2015, 11, 347–354. [Google Scholar] [CrossRef]
- Alexandru, A.; Jagla, W.; Graubner, S.; Becker, A.; Bäuscher, C.; Kohlmann, S.; Sedlmeier, R.; Raber, K.A.; Cynis, H.; Rönicke, R.; et al. Selective hippocampal neurodegeneration in transgenic mice expressing small amounts of truncated Aβ is induced by pyroglutamate-Aβ formation. J. Neurosci. 2011, 31, 12790–12801. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, T.; Meyer, A.; Heiser, U.; Kurat, S.; Böhme, L.; Kleinschmidt, M.; Bühring, K.U.; Hutter-Paier, B.; Farcher, M.; Demuth, H.U.; et al. Glutaminyl cyclase inhibitor PQ912 improves cognition in mouse models of Alzheimer’s disease-studies on relation to effective target occupancy. J. Pharmacol. Exp. Ther. 2017, 362, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Jawhar, S.; Wirths, O.; Schilling, S.; Graubner, S.; Demuth, H.U.; Bayer, T.A. Overexpression of glutaminyl cyclase, the enzyme responsible for pyroglutamate A{beta} formation, induces behavioral deficits, and glutaminyl cyclase knock-out rescues the behavioral phenotype in 5XFAD mice. J. Biol. Chem. 2011, 286, 4454–4460. [Google Scholar] [CrossRef] [PubMed]
- Lues, I.; Weber, F.; Meyer, A.; Bühring, U.; Hoffmann, T.; Kühn-Wache, K.; Manhart, S.; Heiser, U.; Pokorny, R.; Chiesa, J.; et al. A phase 1 study to evaluate the safety and pharmacokinetics of PQ912, a glutaminyl cyclase inhibitor, in healthy subjects. Alzheimers Dement. 2015, 1, 182–195. [Google Scholar] [CrossRef]
- Scheltens, P.; Hallikainen, M.; Grimmer, T.; Duning, T.; Gouw, A.A.; Teunissen, C.E.; Wink, A.M.; Maruff, P.; Harrison, J.; van Baal, C.M.; et al. Safety, tolerability and efficacy of the glutaminyl cyclase inhibitor PQ912 in Alzheimer’s disease: Results of a randomized, double-blind, placebo-controlled phase 2a study. Alzheimers Res. Ther. 2018, 10, 107. [Google Scholar] [CrossRef]
- Vijverberg, E.G.B.; Axelsen, T.M.; Bihlet, A.R.; Henriksen, K.; Weber, F.; Fuchs, K.; Harrison, J.E.; Kühn-Wache, K.; Alexandersen, P.; Prins, N.D.; et al. Rationale and study design of a randomized, placebo-controlled, double-blind phase 2b trial to evaluate efficacy, safety, and tolerability of an oral glutaminyl cyclase inhibitor varoglutamstat (PQ912) in study participants with MCI and mild AD-VIVIAD. Alzheimers Res. Ther. 2021, 13, 142. [Google Scholar] [CrossRef]
- Höfling, C.; Indrischek, H.; Höpcke, T.; Waniek, A.; Cynis, H.; Koch, B.; Schilling, S.; Morawski, M.; Demuth, H.U.; Roßner, S.; et al. Mouse strain and brain region-specific expression of the glutaminyl cyclases QC and isoQC. Int. J. Dev. Neurosci. 2014, 36, 64–73. [Google Scholar] [CrossRef]
- Hartlage-Rübsamen, M.; Waniek, A.; Meissner, J.; Morawski, M.; Schilling, S.; Jäger, C.; Kleinschmidt, M.; Cynis, H.; Kehlen, A.; Arendt, T.; et al. Isoglutaminyl cyclase contributes to CCL2-driven neuroinflammation in Alzheimer’s disease. Acta Neuropathol. 2015, 129, 565–583. [Google Scholar] [CrossRef]
- Cynis, H.; Rahfeld, J.U.; Stephan, A.; Kehlen, A.; Koch, B.; Wermann, M.; Demuth, H.U.; Schilling, S. Isolation of an isoenzyme of human glutaminyl cyclase: Retention in the Golgi complex suggests involvement in the protein maturation machinery. J. Mol. Biol. 2008, 379, 966–980. [Google Scholar] [CrossRef]
- Cynis, H.; Hoffmann, T.; Friedrich, D.; Kehlen, A.; Gans, K.; Kleinschmidt, M.; Rahfeld, J.U.; Wolf, R.; Wermann, M.; Stephan, A.; et al. The isoenzyme of glutaminyl cyclase is an important regulator of monocyte infiltration under inflammatory conditions. EMBO Mol. Med. 2011, 3, 545–558. [Google Scholar] [CrossRef]
- Kehlen, A.; Haegele, M.; Böhme, L.; Cynis, H.; Hoffmann, T.; Demuth, H.U. N-terminal pyroglutamate formation in CX3CL1 is essential for its full biologic activity. Biosci. Rep. 2017, 37, BSR20170712. [Google Scholar] [CrossRef]
- Cynis, H.; Kehlen, A.; Haegele, M.; Hoffmann, T.; Heiser, U.; Fujii, M.; Shibazaki, Y.; Yoneyama, H.; Schilling, S.; Demuth, H.U. Inhibition of glutaminyl cyclases alleviates CCL2-mediated inflammation of non-alcoholic fatty liver disease in mice. Int. J. Exp. Pathol. 2013, 94, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Hellvard, A.; Maresz, K.; Schilling, S.; Graubner, S.; Heiser, U.; Jonsson, R.; Cynis, H.; Demuth, H.U.; Potempa, J.; Mydel, P. Glutaminyl cyclases as novel targets for the treatment of septic arthritis. J. Infect. Dis. 2013, 207, 768–777. [Google Scholar] [CrossRef] [PubMed]
- Gyoneva, S.; Ransohoff, R.M. Inflammatory reaction after traumatic brain injury: Therapeutic potential of targeting cell-cell communication by chemokines. Trends Pharmacol. Sci. 2015, 36, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Arisi, G.M.; Foresti, M.L.; Katki, K.; Shapiro, L.A. Increased CCL2, CCL3, CCL5, and IL-1β cytokine concentration in piriform cortex, hippocampus, and neocortex after pilocarpine-induced seizures. J. Neuroinflammation 2015, 12, 129. [Google Scholar] [CrossRef] [PubMed]
- Leung, S.Y.; Wong, M.P.; Chung, L.P.; Chan, A.S.; Yuen, S.T. Monocyte chemoattractant protein-1 expression and macrophage infiltration in gliomas. Acta Neuropathol. 1997, 93, 518–527. [Google Scholar] [CrossRef]
- Lindemann, C.; Marschall, V.; Weigert, A.; Klingebiel, T.; Fulda, S. Smac mimetic-induced upregulation of CCL2/MCP-1 triggers migration and invasion of glioblastoma cells and influences the tumor microenvironment in a paracrine manner. Neoplasia 2015, 17, 481–489. [Google Scholar] [CrossRef]
- Sokolova, A.; Hill, M.D.; Rahimi, F.; Warden, L.A.; Halliday, G.M.; Shepherd, C.E. Monocyte chemoattractant protein-1 plays a dominant role in the chronic inflammation observed in Alzheimer’s disease. Brain Pathol. 2009, 19, 392–398. [Google Scholar] [CrossRef]
- Westin, K.; Buchhave, P.; Nielsen, H.; Minthon, L.; Janciauskiene, S.; Hansson, O. CCL2 is associated with a faster rate of cognitive decline during early stages of Alzheimer’s disease. PLoS ONE 2012, 7, e30525. [Google Scholar] [CrossRef]
- Worthmann, H.; Tryc, A.B.; Goldbecker, A.; Ma, Y.T.; Tountopoulou, A.; Hahn, A.; Dengler, R.; Lichtinghagen, R.; Weissenborn, K. The temporal profile of inflammatory markers and mediators in blood after acute ischemic stroke differs depending on stroke outcome. Cerebrovasc. Dis. 2010, 30, 85–92. [Google Scholar] [CrossRef]
- Kuriyama, N.; Mizuno, T.; Kita, M.; Nagakane, Y.; Hosomi, A.; Harada, S.; Takeda, K.; Ozasa, K.; Yamada, K.; Tokuda, T.; et al. Predictive markers of blood cytokine and chemokine in recurrent brain infarction. J. Interferon Cytokine Res. 2009, 29, 729–734. [Google Scholar] [CrossRef] [PubMed]
- Schilling, S.; Kohlmann, S.; Bäuscher, C.; Sedlmeier, R.; Koch, B.; Eichentopf, R.; Becker, A.; Cynis, H.; Hoffmann, T.; Berg, S.; et al. Glutaminyl cyclase knock-out mice exhibit slight hypothyroidism but no hypogonadism: Implications for enzyme function and drug development. J. Biol. Chem. 2011, 286, 14199–14208. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Rutledge, B.J.; Gu, L.; Fiorillo, J.; Lukacs, N.W.; Kunkel, S.L.; North, R.; Gerard, C.; Rollins, B.J. Abnormalities in monocyte recruitment and cytokine expression in monocyte chemoattractant protein 1-deficient mice. J. Exp. Med. 1998, 187, 601–608. [Google Scholar] [CrossRef]
- Barendrecht, S.; Schreurs, A.; Geissler, S.; Sabanov, V.; Ilse, V.; Rieckmann, V.; Eichentopf, R.; Künemund, A.; Hietel, B.; Wussow, S.; et al. A novel human tau knock-in mouse model reveals interaction of Abeta and human tau under progressing cerebral amyloidosis in 5xFAD mice. Alzheimers Res. Ther. 2023, 15, 16. [Google Scholar] [CrossRef]
- Longa, E.Z.; Weinstein, P.R.; Carlson, S.; Cummins, R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 1989, 20, 84–91. [Google Scholar] [CrossRef]
- Strecker, J.K.; Minnerup, J.; Gess, B.; Ringelstein, E.B.; Schäbitz, W.R.; Schilling, M. Monocyte chemoattractant protein-1-deficiency impairs the expression of IL-6, IL-1β and G-CSF after transient focal ischemia in mice. PLoS ONE 2011, 6, e25863. [Google Scholar] [CrossRef]
- Menzies, S.A.; Hoff, J.T.; Betz, A.L. Middle cerebral artery occlusion in rats: A neurological and pathological evaluation of a reproducible model. Neurosurgery 1992, 31, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Strecker, J.K.; Minnerup, J.; Schütte-Nütgen, K.; Gess, B.; Schäbitz, W.R.; Schilling, M. Monocyte chemoattractant protein-1-deficiency results in altered blood-brain barrier breakdown after experimental stroke. Stroke 2013, 44, 2536–2544. [Google Scholar] [CrossRef]
- Schuette-Nuetgen, K.; Strecker, J.K.; Minnerup, J.; Ringelstein, E.B.; Schilling, M. MCP-1/CCR-2-double-deficiency severely impairs the migration of hematogenous inflammatory cells following transient cerebral ischemia in mice. Exp. Neurol. 2012, 233, 849–858. [Google Scholar] [CrossRef]
- Gliem, M.; Krammes, K.; Liaw, L.; van Rooijen, N.; Hartung, H.P.; Jander, S. Macrophage-derived osteopontin induces reactive astrocyte polarization and promotes re-establishment of the blood brain barrier after ischemic stroke. Glia 2015, 63, 2198–2207. [Google Scholar] [CrossRef]
- Gliem, M.; Schwaninger, M.; Jander, S. Protective features of peripheral monocytes/macrophages in stroke. Biochim. Biophys. Acta 2016, 1862, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Wattananit, S.; Tornero, D.; Graubardt, N.; Memanishvili, T.; Monni, E.; Tatarishvili, J.; Miskinyte, G.; Ge, R.; Ahlenius, H.; Lindvall, O.; et al. Monocyte-derived macrophages contribute to spontaneous long-term functional recovery after stroke in mice. J. Neurosci. 2016, 36, 4182–4195. [Google Scholar] [CrossRef] [PubMed]
- Ling, Z.; Li, W.; Hu, J.; Li, Y.; Deng, M.; Zhang, S.; Ren, X.; Wu, T.; Xia, J.; Cheng, B.; et al. Targeting CCL2-CCR4 axis suppress cell migration of head and neck squamous cell carcinoma. Cell Death Dis. 2022, 13, 158. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Somasundaram, R.; Berencsi, K.; Caputo, L.; Gimotty, P.; Rani, P.; Guerry, D.; Swoboda, R.; Herlyn, D. Migration of cytotoxic T lymphocytes toward melanoma cells in three-dimensional organotypic culture is dependent on CCL2 and CCR4. Eur. J. Immunol. 2006, 36, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Lieberam, I.; Förster, I. The murine beta-chemokine TARC is expressed by subsets of dendritic cells and attracts primed CD4+ T cells. Eur. J. Immunol. 1999, 29, 2684–2694. [Google Scholar] [CrossRef]
- Hiroyama, T.; Iwama, A.; Nakamura, Y.; Nakauchi, H. Fractalkine shares signal sequence with TARC: Gene structures and expression profiles of two chemokine genes. Genomics 2001, 75, 3–5. [Google Scholar] [CrossRef]
- Lier, J.; Streit, W.J.; Bechmann, I. Beyond activation: Characterizing microglial functional phenotypes. Cells 2021, 10, 2236. [Google Scholar] [CrossRef]
- Spiteri, A.G.; Wishart, C.L.; King, N.J.C. Immovable object meets unstoppable force? Dialogue between resident and peripheral myeloid cells in the inflamed brain. Front. Immunol. 2020, 11, 600822. [Google Scholar] [CrossRef]
- Dirnagl, U.; Iadecola, C.; Moskowitz, M.A. Pathobiology of ischaemic stroke: An integrated view. Trends Neurosci. 1999, 22, 391–397. [Google Scholar] [CrossRef]
- Kehlen, A.; Haegele, M.; Menge, K.; Gans, K.; Immel, U.D.; Hoang-Vu, C.; Klonisch, T.; Demuth, H.U. Role of glutaminyl cyclases in thyroid carcinomas. Endocr. Relat. Cancer 2013, 20, 79–90. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Primer | Sequence 5‘ → 3‘ | Product [bp] | |
|---|---|---|---|---|
| QC KO | Pbd2-1 | Common | GTC CGG TAA GGT GAG GAG AA | |
| Pbd2-2 | KO | TGA TGT GTG CGT TTC AGA GA | 525 (KO) | |
| Pbd2-WT1 | WT | CCA GAG ACA TCC TGG TAA AAC A | 733 (WT) | |
| isoQC KO | 7b | Common | TGT CCA CCC TCC TAC CTC TC | 212 + 106 (KO) (AvrII Digestion) |
| 8b | Common | TCA CCT GCT GCT TGA TTC TG | 318 (WT) (AvrII Digestion) | |
| CCL2 KO | oIMR7415 | KO | GCC AGA GGC CAC TTG TGT AG | 179 (KO) |
| oIMR9219_opt | WT | TGA CAG TCC CCA GAG TCA CAT AG | 287 (WT) | |
| oIMR9220_opt | Common | TCA TTG GGA TCA TCT TGC TGG TGA | ||
| Primary Antibody | Dilution | Host | Company | Cat. No. | Detection | |
|---|---|---|---|---|---|---|
| single labeling | ||||||
| MHC-II | 1:500 | rat | Invitrogen | M5/114.15.2 | Ni-DAB | |
| NeuN | 1:1000 | mouse | Millipore | MAB 377 | DAB | |
| HuC/D | 1:1000 | mouse | Invitrogen | A-21271 | DAB | |
| QC | 1:500 | goat | IZI | 77101 | DAB/Ni-DAB | |
| isoQC | 1:200 | rabbit | IZI | 3285 | DAB | |
| double labeling | ||||||
| NeuN | 1:1000 | mouse | Millipore | MAB 377 | donkey anti-mouse Cy5 | |
| HuC/D + | 1:1000 | mouse | Invitrogen | A-21271 | donkey anti-mouse Cy5 | |
| NFL | 1:200 | rabbit | Synaptic Systems | 171002 | donkey anti-rabbit Cy2 | |
| CCL2 | + | 1:200 | goat | AF479 | R&D | donkey anti-goat Cy3 |
| NeuN | 1:1000 | mouse | Millipore | MAB 377 | donkey anti-mouse Cy5 | |
| HuC/D | 1:1000 | mouse | Invitrogen | A-21271 | donkey anti-mouse Cy5 | |
| Hoechst | 1:10,000 | - | Invitrogen | H3570 | - | |
| Iba1 | 1:500 | rabbit | Wako | 019-19741 | donkey anti-rabbit Cy2 | |
| CD68 | 1:100 | rat | Biorad | MCA1957 | donkey anti-rat Cy2 | |
| TMEM119 | 1:100 | rabbit | abcam | ab209064 | donkey anti-rabbit Cy2 | |
| GFAP | 1:200 | guinea pig | Synaptic Systems | 173001 | donkey anti-guinea pig Cy5 | |
| Olig2 | 1:100 | rabbit | abcam | ab81093 | donkey anti-rabbit Cy5 | |
| triple labeling | ||||||
| NeuN | 1:1000 | mouse | Millipore | MAB 377 | donkey anti-mouse Cy3 | |
| HuC/D + | 1:1000 | mouse | Invitrogen | A-21271 | donkey anti-mouse Cy3 | |
| Iba1 + | 1:500 | rabbit | Wako | 019-19741 | donkey anti-rabbit Cy2 | |
| GFAP | 1:200 | guinea pig | Synaptic Systems | 173001 | donkey anti-guinea pig Cy5 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Höfling, C.; Ulrich, L.; Burghardt, S.; Donkersloot, P.; Opitz, M.; Geissler, S.; Schilling, S.; Cynis, H.; Michalski, D.; Roßner, S. Focal Cerebral Ischemia Induces Expression of Glutaminyl Cyclase along with Downstream Molecular and Cellular Inflammatory Responses. Cells 2024, 13, 1412. https://doi.org/10.3390/cells13171412
Höfling C, Ulrich L, Burghardt S, Donkersloot P, Opitz M, Geissler S, Schilling S, Cynis H, Michalski D, Roßner S. Focal Cerebral Ischemia Induces Expression of Glutaminyl Cyclase along with Downstream Molecular and Cellular Inflammatory Responses. Cells. 2024; 13(17):1412. https://doi.org/10.3390/cells13171412
Chicago/Turabian StyleHöfling, Corinna, Luise Ulrich, Sina Burghardt, Philippa Donkersloot, Michael Opitz, Stefanie Geissler, Stephan Schilling, Holger Cynis, Dominik Michalski, and Steffen Roßner. 2024. "Focal Cerebral Ischemia Induces Expression of Glutaminyl Cyclase along with Downstream Molecular and Cellular Inflammatory Responses" Cells 13, no. 17: 1412. https://doi.org/10.3390/cells13171412