
TRIM44, a Novel Prognostic Marker, Supports the Survival of Proteasome-Resistant Multiple Myeloma Cells
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Antibodies
2.3. Reagents
2.4. Apoptosis Assay
2.5. Confocal Microscopy
2.6. Immunoprecipitation and Immunoblotting
2.7. CHX Chase Assay
2.8. Cell Lysate Fraction
2.9. RNA Extraction and Real-Time PCR
2.10. Patient Dataset Analysis
2.11. Gene Expression and Mutation Status
2.12. scRNA-Seq Data Analysis
2.13. Statistical Analysis
3. Results
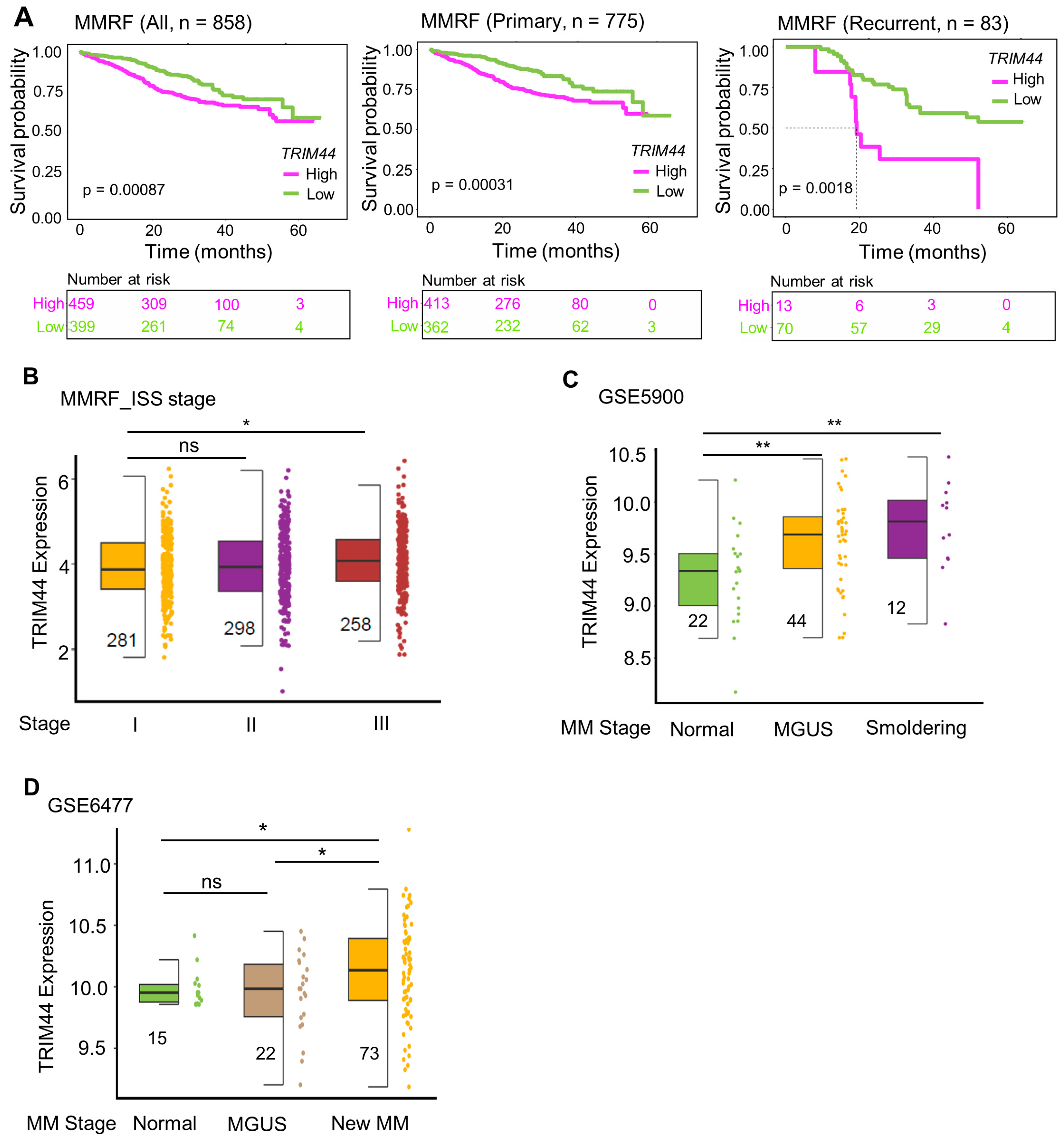
3.1. High TRIM44 Expression Is Associated with Poor Prognosis in the MMRF Cohort
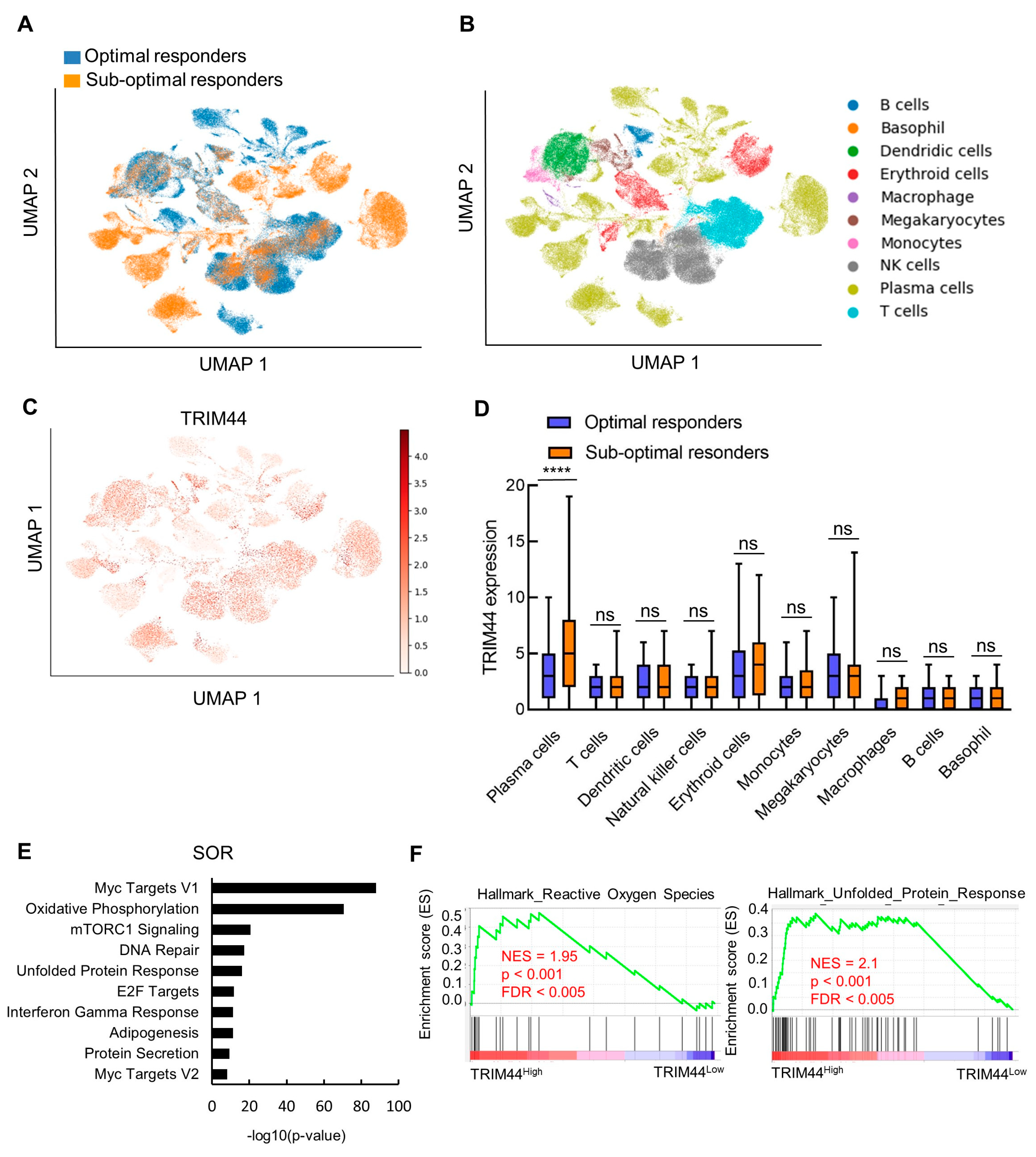
3.2. Profile of TRIM44 Expression at Single-Cell Resolution during MM Progression
3.3. TRIM44 Levels Strongly Correlate with an Increased Unfolded Protein Response in Patients Exhibiting Low Response to Bortezomib
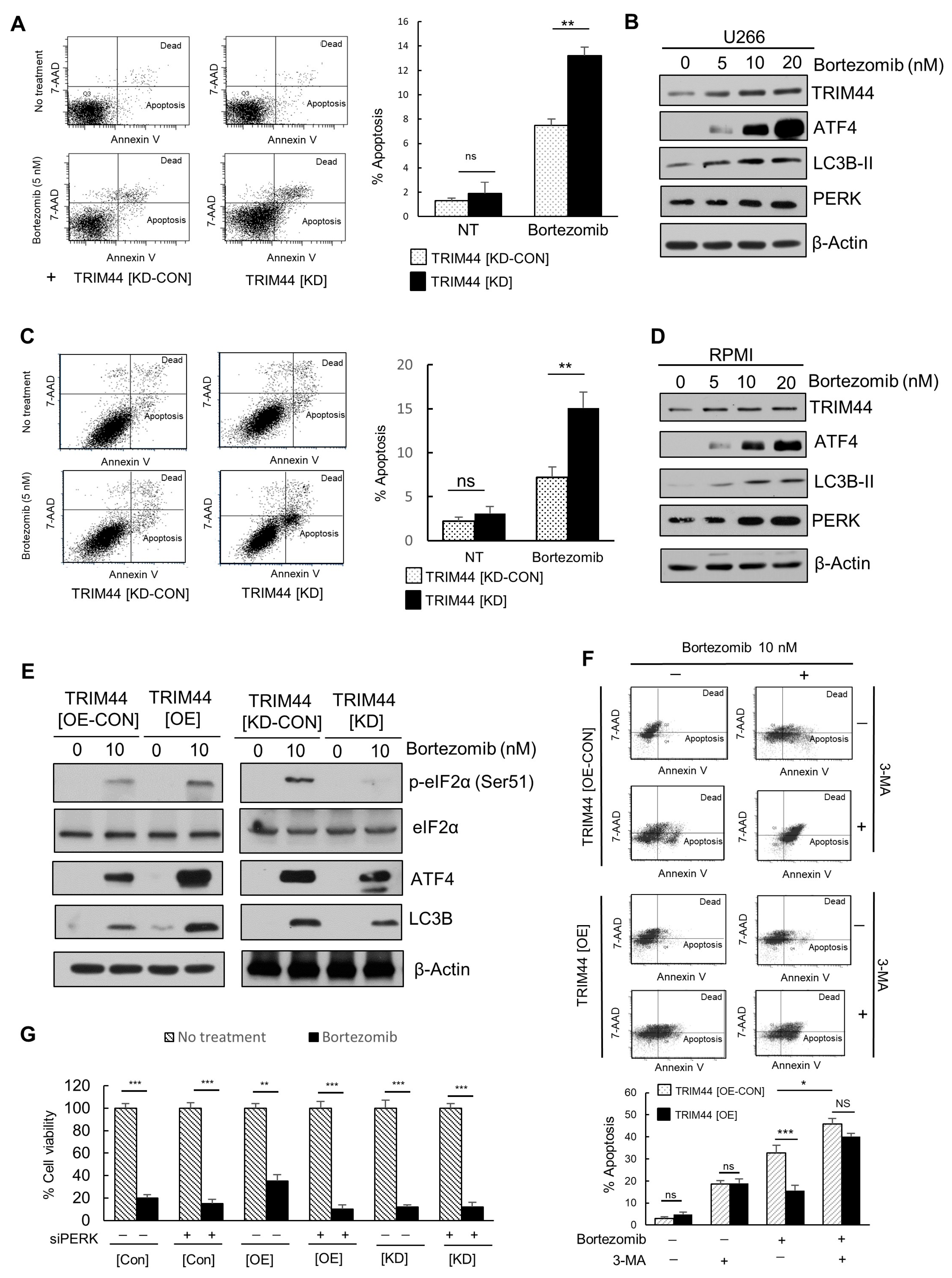
3.4. TRIM44 Is Upregulated and Enhances the Survival of Therapy-Resistant MM Cells Treated with Bortezomib
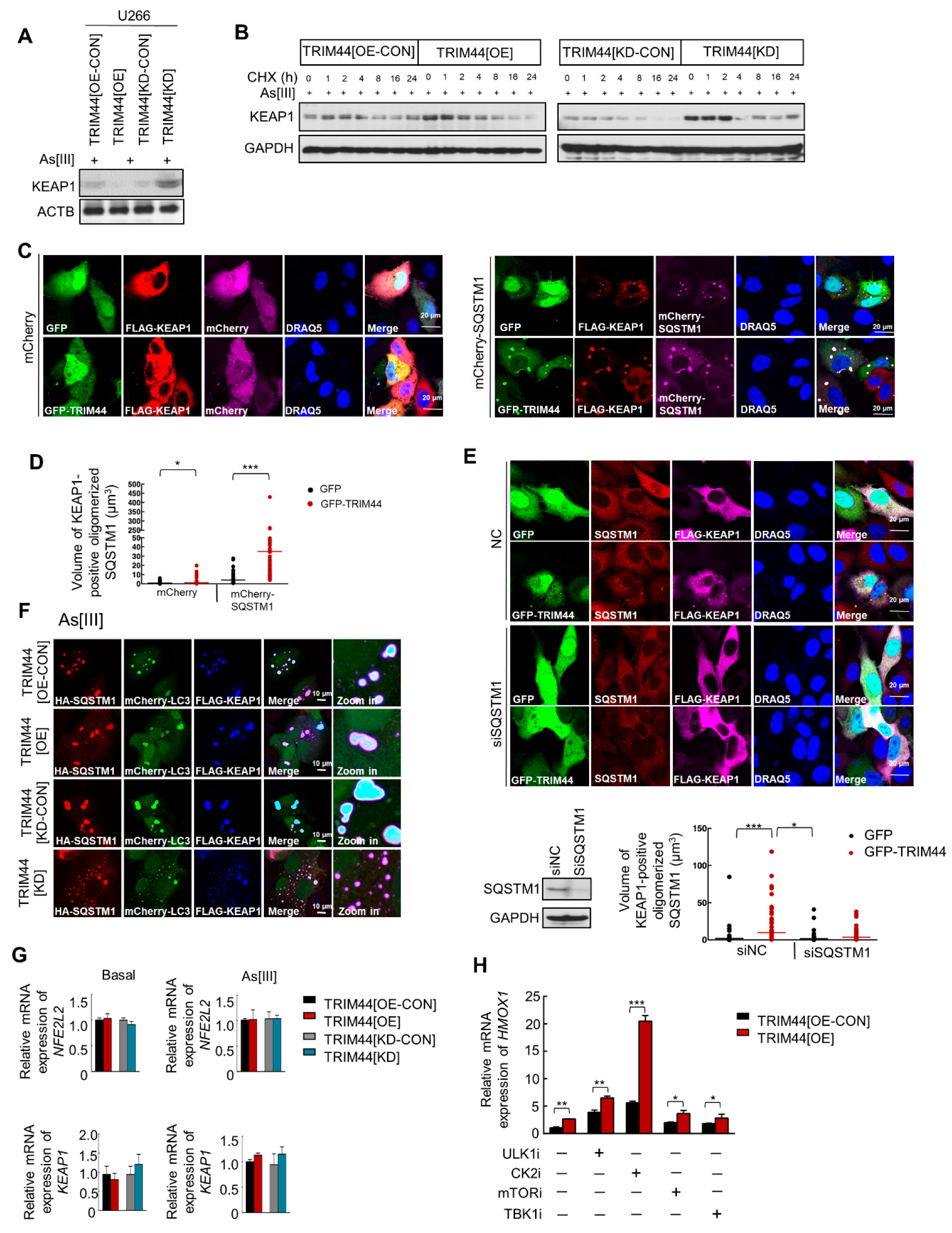
3.5. TRIM44 Promotes the Oligomerization of SQSTM1, Which Is Essential for Its Phosphorylation in Response to Oxidative Stress
3.6. TRIM44-Mediated SQSTM1 Oligomerization Sequesters KEAP1 and Activates NRF2, Thereby Enhancing MM Cell Survival under Oxidative Stress
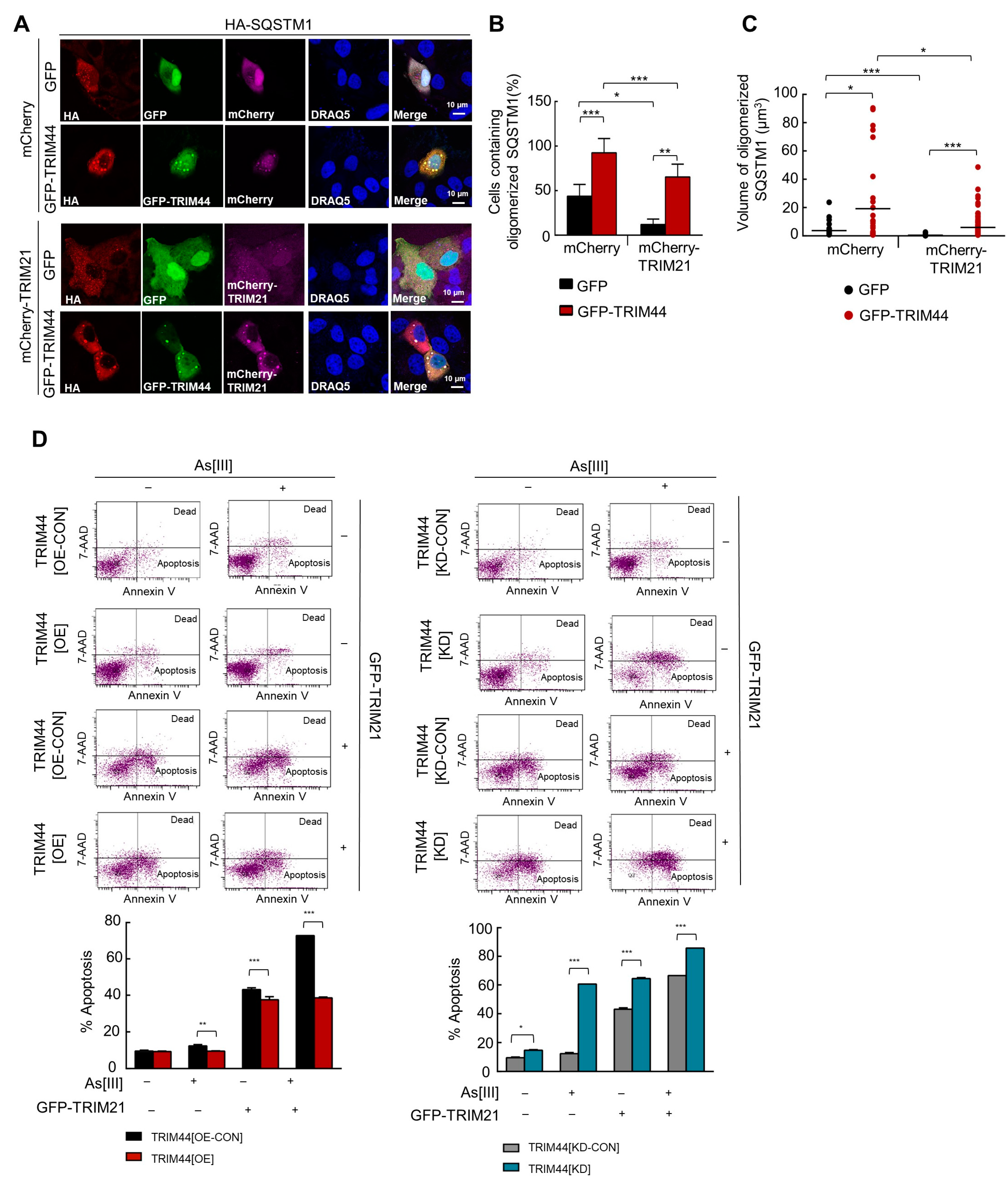
3.7. TRIM44 Counteracts TRIM21-Mediated Suppression of MM Survival under Oxidative Stress
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mateos, M.-V.; Cavo, M.; Blade, J.; Dimopoulos, M.A.; Suzuki, K.; Jakubowiak, A.; Knop, S.; Doyen, C.; Lucio, P.; Nagy, Z.; et al. Overall survival with daratumumab, bortezomib, melphalan, and prednisone in newly diagnosed multiple myeloma (ALCYONE): A randomised, open-label, phase 3 trial. Lancet 2020, 395, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Padala, S.A.; Barsouk, A.; Barsouk, A.; Rawla, P.; Vakiti, A.; Kolhe, R.; Kota, V.; Ajebo, G.H. Epidemiology, Staging, and Management of Multiple Myeloma. Med. Sci. 2021, 9, 3. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, Z.S.; Kim, E.B.; Raje, N. Bone Disease in Multiple Myeloma: Biologic and Clinical Implications. Cells 2022, 11, 2308. [Google Scholar] [CrossRef] [PubMed]
- Lipchick, B.C.; Fink, E.E.; Nikiforov, M.A. Oxidative stress and proteasome inhibitors in multiple myeloma. Pharmacol. Res. 2016, 105, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.; Palombella, V.J.; A Sausville, E.; Johnson, J.; Destree, A.; Lazarus, D.D.; Maas, J.; Pien, C.S.; Prakash, S.; Elliott, P.J. Proteasome inhibitors: A novel class of potent and effective antitumor agents. Cancer Res. 1999, 59, 2615–2622. [Google Scholar]
- Nawrocki, S.T.; Carew, J.S.; Pino, M.S.; Highshaw, R.A.; Dunner, K.; Huang, P.; Abbruzzese, J.L.; McConkey, D.J. Bortezomib sensitizes pancreatic cancer cells to endoplasmic reticulum stress-mediated apoptosis. Cancer Res. 2005, 65, 11658–11666. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Milani, M.; Rzymski, T.; Mellor, H.R.; Pike, L.; Bottini, A.; Generali, D.; Harris, A.L. The role of ATF4 stabilization and autophagy in resistance of breast cancer cells treated with Bortezomib. Cancer Res. 2009, 69, 4415–4423. [Google Scholar] [CrossRef]
- Li, X.; He, S.; Ma, B. Autophagy and autophagy-related proteins in cancer. Mol. Cancer 2020, 19, 12. [Google Scholar] [CrossRef]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 and Autophagy. Methods Mol. Biol. 2008, 445, 77–88. [Google Scholar] [CrossRef]
- Katsuragi, Y.; Ichimura, Y.; Komatsu, M. p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J. 2015, 282, 4672–4678. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef]
- Ichimura, Y.; Waguri, S.; Sou, Y.-S.; Kageyama, S.; Hasegawa, J.; Ishimura, R.; Saito, T.; Yang, Y.; Kouno, T.; Fukutomi, T.; et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol. Cell. 2013, 51, 618–631. [Google Scholar] [CrossRef]
- Carroll, B.; Otten, E.G.; Manni, D.; Stefanatos, R.; Menzies, F.M.; Smith, G.R.; Jurk, D.; Kenneth, N.; Wilkinson, S.; Passos, J.F.; et al. Oxidation of SQSTM1/p62 mediates the link between redox state and protein homeostasis. Nat. Commun. 2018, 9, 256. [Google Scholar] [CrossRef]
- Chen, Z.; Lin, T.-C.; Bi, X.; Lu, G.; Dawson, B.C.; Miranda, R.; Medeiros, L.J.; McNiece, I.; McCarty, N. TRIM44 promotes quiescent multiple myeloma cell occupancy and survival in the osteoblastic niche via HIF-1α stabilization. Leukemia 2019, 33, 469–486. [Google Scholar] [CrossRef] [PubMed]
- Lyu, L.; Chen, Z.; McCarty, N. TRIM44 links the UPS to SQSTM1/p62-dependent aggrephagy and removing misfolded proteins. Autophagy 2021, 18, 783–798. [Google Scholar] [CrossRef] [PubMed]
- Lyu, L.; Lin, T.C.; McCarty, N. TRIM44 mediated p62 deubiquitination enhances DNA damage repair by increasing nuclear FLNA and 53BP1 expression. Oncogene 2021, 40, 5116–5130. [Google Scholar] [CrossRef]
- Settino, M.; Arbitrio, M.; Scionti, F.; Caracciolo, D.; Di Martino, M.T.; Tagliaferri, P.; Tassone, P.; Cannataro, M. MMRF-CoMMpass Data Integration and Analysis for Identifying Prognostic Markers. In International Conference on Computational Science; Springer: Cham, Switzerland, 2020; Volume 12139, pp. 564–571. [Google Scholar] [CrossRef]
- Palumbo, A.; Avet-Loiseau, H.; Oliva, S.; Lokhorst, H.M.; Goldschmidt, H.; Rosinol, L.; Richardson, P.; Caltagirone, S.; Lahuerta, J.J.; Facon, T.; et al. Revised International Staging System for Multiple Myeloma: A Report from International Myeloma Working Group. J. Clin. Oncol. 2015, 33, 2863–2869. [Google Scholar] [CrossRef]
- Kyle, R.A.; Rajkumar, S.V. Multiple myeloma. Blood 2008, 111, 2962–2972. [Google Scholar] [CrossRef]
- Landgren, O. Monoclonal gammopathy of undetermined significance and smoldering multiple myeloma: Biological insights and early treatment strategies. Hematol. Am. Soc. Hematol. Educ. Program. 2013, 2013, 478. [Google Scholar] [CrossRef]
- Rajkumar, S.V. Multiple myeloma: 2011 update on diagnosis, risk-stratification, and management. Am. J. Hematol. 2011, 86, 57–65. [Google Scholar] [CrossRef]
- Kyle, R.A.; Remstein, E.D.; Therneau, T.M.; Dispenzieri, A.; Kurtin, P.J.; Hodnefield, J.M.; Larson, D.R.; Plevak, M.F.; Jelinek, D.F.; Fonseca, R.; et al. Clinical course and prognosis of smoldering (asymptomatic) multiple myeloma. N. Engl. J. Med. 2007, 356, 2582–2590. [Google Scholar] [CrossRef]
- Kyle, R.A.; Larson, D.R.; Therneau, T.M.; Dispenzieri, A.; Kumar, S.; Cerhan, J.R.; Rajkumar, S.V. Long-Term Follow-up of Monoclonal Gammopathy of Undetermined Significance. N. Engl. J. Med. 2018, 378, 241–249. [Google Scholar] [CrossRef]
- Lakshman, A.; Rajkumar, S.V.; Buadi, F.K.; Binder, M.; Gertz, M.A.; Lacy, M.Q.; Dispenzieri, A.; Dingli, D.; Fonder, A.L.; Hayman, S.R.; et al. Risk stratification of smoldering multiple myeloma incorporating revised IMWG diagnostic criteria. Blood Cancer J. 2018, 8, 59. [Google Scholar] [CrossRef]
- Rajkumar, S.V.; Gupta, V.; Fonseca, R.; Dispenzieri, A.; Gonsalves, W.I.; Larson, D.; Ketterling, R.P.; Lust, J.A.; Kyle, R.A.; Kumar, S.K. Impact of primary molecular cytogenetic abnormalities and risk of progression in smoldering multiple myeloma. Leukemia 2013, 27, 1738–1744. [Google Scholar] [CrossRef]
- Boiarsky, R.; Haradhvala, N.J.; Alberge, J.-B.; Sklavenitis-Pistofidis, R.; Mouhieddine, T.H.; Zavidij, O.; Shih, M.-C.; Firer, D.; Miller, M.; El-Khoury, H.; et al. Single cell characterization of myeloma and its precursor conditions reveals transcriptional signatures of early tumorigenesis. Nat. Commun. 2022, 13, 7040. [Google Scholar] [CrossRef] [PubMed]
- Stewart, A.K.; Bergsagel, P.L.; Greipp, P.R.; Dispenzieri, A.; A Gertz, M.; Hayman, S.R.; Kumar, S.; Lacy, M.Q.; A Lust, J.; Russell, S.J.; et al. A practical guide to defining high-risk myeloma for clinical trials, patient counseling and choice of therapy. Leukemia 2007, 21, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Kalff, A.; Spencer, A. The t(4;14) translocation and FGFR3 overexpression in multiple myeloma: Prognostic implications and current clinical strategies. Blood Cancer J. 2012, 2, e89. [Google Scholar] [CrossRef] [PubMed]
- Qiang, Y.-W.; Ye, S.; Huang, Y.; Chen, Y.; Van Rhee, F.; Epstein, J.; Walker, B.A.; Morgan, G.J.; Davies, F.E. MAFb protein confers intrinsic resistance to proteasome inhibitors in multiple myeloma. BMC Cancer 2018, 18, 724. [Google Scholar] [CrossRef]
- Kozalak, G.; Koşar, A. Autophagy-related mechanisms for treatment of multiple myeloma. Cancer Drug Resist. 2023, 6, 838–857. [Google Scholar] [CrossRef]
- Obeng, E.A.; Carlson, L.M.; Gutman, D.M.; Harrington, W.J.; Jr Lee, K.P.; Boise, L.H. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood 2006, 107, 4907–4916. [Google Scholar] [CrossRef]
- Jung, S.-H.; Park, S.-S.; Lim, J.-Y.; Sohn, S.Y.; Kim, N.Y.; Kim, D.; Lee, S.H.; Chung, Y.-J.; Min, C.-K. Single-cell analysis of multiple myelomas refines the molecular features of bortezomib treatment responsiveness. Exp. Mol. Med. 2022, 54, 1967–1978. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Wang, F.; Tao, P. t-Distributed Stochastic Neighbor Embedding Method with the Least Information Loss for Macromolecular Simulations. J. Chem. Theory Comput. 2018, 14, 5499–5510. [Google Scholar] [CrossRef] [PubMed]
- Cohen, Y.C.; Zada, M.; Wang, S.-Y.; Bornstein, C.; David, E.; Moshe, A.; Li, B.; Shlomi-Loubaton, S.; Gatt, M.E.; Gur, C.; et al. Identification of resistance pathways and therapeutic targets in relapsed multiple myeloma patients through single-cell sequencing. Nat. Med. 2021, 27, 491–503. [Google Scholar] [CrossRef]
- Fribley, A.; Zeng, Q.; Wang, C.Y. Proteasome inhibitor PS-341 induces apoptosis through induction of endoplasmic reticulum stress-reactive oxygen species in head and neck squamous cell carcinoma cells. Mol. Cell. Biol. 2004, 24, 9695–9704. [Google Scholar] [CrossRef]
- Zeeshan, H.M.; Lee, G.H.; Kim, H.R.; Chae, H.J. Endoplasmic Reticulum Stress and Associated ROS. Int. J. Mol. Sci. 2016, 17, 327. [Google Scholar] [CrossRef]
- Li, X.; Liang, M.; Jiang, J.; He, R.; Wang, M.; Guo, X.; Shen, M.; Qin, R. Combined inhibition of autophagy and Nrf2 signaling augments bortezomib-induced apoptosis by increasing ROS production and ER stress in pancreatic cancer cells. Int. J. Biol. Sci. 2018, 14, 1291–1305. [Google Scholar] [CrossRef]
- Ichimura, Y.; Kumanomidou, T.; Sou, Y.-S.; Mizushima, T.; Ezaki, J.; Ueno, T.; Kominami, E.; Yamane, T.; Tanaka, K.; Komatsu, M. Structural basis for sorting mechanism of p62 in selective autophagy. J. Biol. Chem. 2008, 283, 22847–22857. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, K.; Fujikawa, N.; Komatsu, M.; Ishii, T.; Unno, M.; Akaike, T.; Motohashi, H.; Yamamoto, M. Keap1 degradation by autophagy for the maintenance of redox homeostasis. Proc. Natl. Acad. Sci. USA 2012, 109, 13561–13566. [Google Scholar] [CrossRef]
- Ichimura, Y.; Komatsu, M. Activation of p62/SQSTM1-Keap1-Nuclear Factor Erythroid 2-Related Factor 2 Pathway in Cancer. Front. Oncol. 2018, 8, 210. [Google Scholar] [CrossRef]
- Hatakeyama, S. TRIM Family Proteins: Roles in Autophagy, Immunity, and Carcinogenesis. Trends Biochem, Sci. 2017, 42, 297–311. [Google Scholar] [CrossRef] [PubMed]
- Cambiaghi, V.; Giuliani, V.; Lombardi, S.; Marinelli, C.; Toffalorio, F.; Pelicci, P.G. TRIM proteins in cancer. Adv. Exp. Med. Biol. 2012, 770, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Tao, S.; Liao, L.; Li, Y.; Li, H.; Li, Z.; Lin, L.; Wan, X.; Yang, X.; Chen, L. TRIM25 promotes the cell survival and growth of hepatocellular carcinoma through targeting Keap1-Nrf2 pathway. Nat. Commun. 2020, 11, 348. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Fang, Y.; Liu, T. TRIM32 Promotes the Growth of Gastric Cancer Cells through Enhancing AKT Activity and Glucose Transportation. Biomed. Res. Int. 2020, 2020, 4027627. [Google Scholar] [CrossRef]
- Jin, Z.; Liu, L.; Yu, Y.; Li, D.; Zhu, X.; Yan, D.; Zhu, Z. TRIM59: A potential diagnostic and prognostic biomarker in human tumors. PLoS ONE 2021, 16, e0257445. [Google Scholar] [CrossRef]
- Hsu, C.Y.; Yanagi, T.; Ujiie, H. TRIM29 in Cutaneous Squamous Cell Carcinoma. Front. Med. 2021, 8, 804166. [Google Scholar] [CrossRef]
- Järvinen, A.; Autio, R.; Kilpinen, S.; Saarela, M.; Leivo, I.; Grénman, R.; Mäkitie, A.A.; Monni, O. High-resolution copy number and gene expression microarray analyses of head and neck squamous cell carcinoma cell lines of tongue and larynx. Genes Chromosomes Cancer 2008, 47, 500–509. [Google Scholar] [CrossRef]
- Luo, Q.; Lin, H.; Ye, X.; Huang, J.; Lu, S.; Xu, L. Trim44 facilitates the migration and invasion of human lung cancer cells via the NF-κB signaling pathway. Int. J. Clin. Oncol. 2015, 20, 508–517. [Google Scholar] [CrossRef]
- Tan, Y.; Yao, H.; Hu, J.; Liu, L. Knockdown of TRIM44 Inhibits the Proliferation and Invasion in Prostate Cancer Cells. Oncol. Res. 2017, 25, 1253–1259. [Google Scholar] [CrossRef]
- Meng, F.; Ding, J.; Xu, W.; Luo, C.; Chen, X.; Zhang, R.; Sui, L.; Hu, Y.; Liu, S.; Shi, G.; et al. Knockdown of TRIM44 inhibits the progression of ovarian cancer and is related to the FOXM1-EZH2 signaling pathway. Transl. Cancer Res. 2022, 11, 414–425. [Google Scholar] [CrossRef]
- Zhu, X.; Wu, Y.; Miao, X.; Li, C.; Yin, H.; Yang, S.; Lu, X.; Liu, Y.; Chen, Y.; Shen, R.; et al. High expression of TRIM44 is associated with enhanced cell proliferation, migration, invasion, and resistance to doxorubicin in hepatocellular carcinoma. Tumour Biol. 2016, 37, 14615–14628. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Takayama, K.; Fujimura, T.; Ashikari, D.; Obinata, D.; Takahashi, S.; Ikeda, K.; Kakutani, S.; Urano, T.; Fukuhara, H.; et al. A novel prognostic factor TRIM44 promotes cell proliferation and migration, and inhibits apoptosis in testicular germ cell tumor. Cancer Sci. 2017, 108, 32–41. [Google Scholar] [CrossRef]
- Kawaguchi, T.; Komatsu, S.; Ichikawa, D.; Hirajima, S.; Nishimura, Y.; Konishi, H.; Shiozaki, A.; Fujiwara, H.; Okamoto, K.; Tsuda, H.; et al. Overexpression of TRIM44 is related to invasive potential and malignant outcomes in esophageal squamous cell carcinoma. Tumour Biol. 2017, 39, 1010428317700409. [Google Scholar] [CrossRef] [PubMed]
- Kashimoto, K.; Komatsu, S.; Ichikawa, D.; Arita, T.; Konishi, H.; Nagata, H.; Takeshita, H.; Nishimura, Y.; Hirajima, S.; Kawaguchi, T.; et al. Overexpression of TRIM44 contributes to malignant outcome in gastric carcinoma. Cancer Sci. 2012, 103, 2021–2026. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, H.; Azuma, K.; Ikeda, K.; Sugitani, I.; Kinowaki, K.; Fujii, T.; Osaki, A.; Saeki, T.; Horie-Inoue, K.; Inoue, S. TRIM44 Is a Poor Prognostic Factor for Breast Cancer Patients as a Modulator of NF-κB Signaling. Int. J. Mol. Sci. 2017, 18, 1931. [Google Scholar] [CrossRef]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef]
- Shintani, T.; Klionsky, D.J. Autophagy in health and disease: A double-edged sword. Science 2004, 306, 990–995. [Google Scholar] [CrossRef]
- Al-Odat, O.S.; Guirguis, D.A.; Schmalbach, N.K.; Yao, G.; Budak-Alpdogan, T.; Jonnalagadda, S.C.; Pandey, M.K. Autophagy and Apoptosis: Current Challenges of Treatment and Drug Resistance in Multiple Myeloma. Int. J. Mol. Sci. 2022, 24, 644. [Google Scholar] [CrossRef]
- Chang, H.; Zou, Z. Targeting autophagy to overcome drug resistance: Further developments. J. Hematol. Oncol. 2020, 13, 159. [Google Scholar] [CrossRef]
- Mandell, M.A.; Jain, A.; Arko-Mensah, J.; Chauhan, S.; Kimura, T.; Dinkins, C.; Silvestri, G.; Münch, J.; Kirchhoff, F.; Simonsen, A.; et al. TRIM proteins regulate autophagy and can target autophagic substrates by direct recognition. Dev. Cell. 2014, 30, 394–409. [Google Scholar] [CrossRef]
- Yorimitsu, T.; Klionsky, D.J. Endoplasmic reticulum stress: A new pathway to induce autophagy. Autophagy 2007, 3, 160–162. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.I.; Gill, D.J.; Perisic, O.; Quinn, M.T.; Williams, R.L. PB1 domain-mediated heterodimerization in NADPH oxidase and signaling complexes of atypical protein kinase C with Par6 and p62. Mol. Cell. 2003, 12, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Bjørkøy, G.; Lamark, T.; Brech, A.; Outzen, H.; Perander, M.; Øvervatn, A.; Stenmark, H.; Johansen, T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005, 171, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Park, J.S.; Lee, Y.S.; Han, J.; Lee, D.-K.; Kwon, S.W.; Han, D.H.; Lee, Y.-H.; Bae, S.H. SQSTM1/p62 activates NFE2L2/NRF2 via ULK1-mediated autophagic KEAP1 degradation and protects mouse liver from lipotoxicity. Autophagy 2020, 16, 1949–1973. [Google Scholar] [CrossRef] [PubMed]
- Rotariu, D.; Babes, E.E.; Tit, D.M.; Moisi, M.; Bustea, C.; Stoicescu, M.; Radu, A.-F.; Vesa, C.M.; Behl, T.; Bungau, A.F.; et al. Oxidative stress—Complex pathological issues concerning the hallmark of cardiovascular and metabolic disorders. Biomed. Pharmacother. 2022, 152, 113238. [Google Scholar] [CrossRef]
- Pérez-Galán, P.; Roué, G.; Villamor, N.; Montserrat, E.; Campo, E.; Colomer, D. The proteasome inhibitor bortezomib induces apoptosis in mantle-cell lymphoma through generation of ROS and Noxa activation independent of p53 status. Blood 2006, 107, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.-Y.; Farooqi, A.A.; Ou-Yang, F.; Hou, M.-F.; Huang, H.-W.; Wang, H.-R.; Li, K.-T.; Fayyaz, S.; Shu, C.-W.; Chang, H.-W. Oxidative stress-modulating drugs have preferential anticancer effects—Involving the regulation of apoptosis, DNA damage, endoplasmic reticulum stress, autophagy, metabolism, and migration. Semin. Cancer Biol. 2019, 58, 109–117. [Google Scholar] [CrossRef]
- Shimizu, Y.; Hendershot, L.M. Oxidative folding: Cellular strategies for dealing with the resultant equimolar production of reactive oxygen species. Antioxid. Redox Signal. 2009, 11, 2317–2331. [Google Scholar] [CrossRef]
- Weniger, M.A.; Rizzatti, E.G.; Pérez-Galán, P.; Liu, D.; Wang, Q.; Munson, P.J.; Raghavachari, N.; White, T.; Tweito, M.M.; Dunleavy, K.; et al. Treatment-induced oxidative stress and cellular antioxidant capacity determine response to bortezomib in mantle cell lymphoma. Clin. Cancer Res. 2011, 17, 5101–5112. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vu, T.; Wang, Y.; Fowler, A.; Simieou, A.; McCarty, N. TRIM44, a Novel Prognostic Marker, Supports the Survival of Proteasome-Resistant Multiple Myeloma Cells. Cells 2024, 13, 1431. https://doi.org/10.3390/cells13171431
Vu T, Wang Y, Fowler A, Simieou A, McCarty N. TRIM44, a Novel Prognostic Marker, Supports the Survival of Proteasome-Resistant Multiple Myeloma Cells. Cells. 2024; 13(17):1431. https://doi.org/10.3390/cells13171431
Chicago/Turabian StyleVu, Trung, Yuqin Wang, Annaliese Fowler, Anton Simieou, and Nami McCarty. 2024. "TRIM44, a Novel Prognostic Marker, Supports the Survival of Proteasome-Resistant Multiple Myeloma Cells" Cells 13, no. 17: 1431. https://doi.org/10.3390/cells13171431