
Metformin Alleviates Inflammation and Induces Mitophagy in Human Retinal Pigment Epithelium Cells Suffering from Mitochondrial Damage
 , and
, and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Cell Treatments
2.3. Cell Viability Assays
2.4. Enzyme-Linked Immunosorbent Assay (ELISA)
2.5. Western Blot
2.6. Real-Time Polymerase Chain Reaction (PCR)
2.7. Immunofluorescence
2.8. MitoTracker Red Staining
2.9. Mitochondrial and Total Cellular ROS Levels
2.10. Statistical Analysis
3. Results
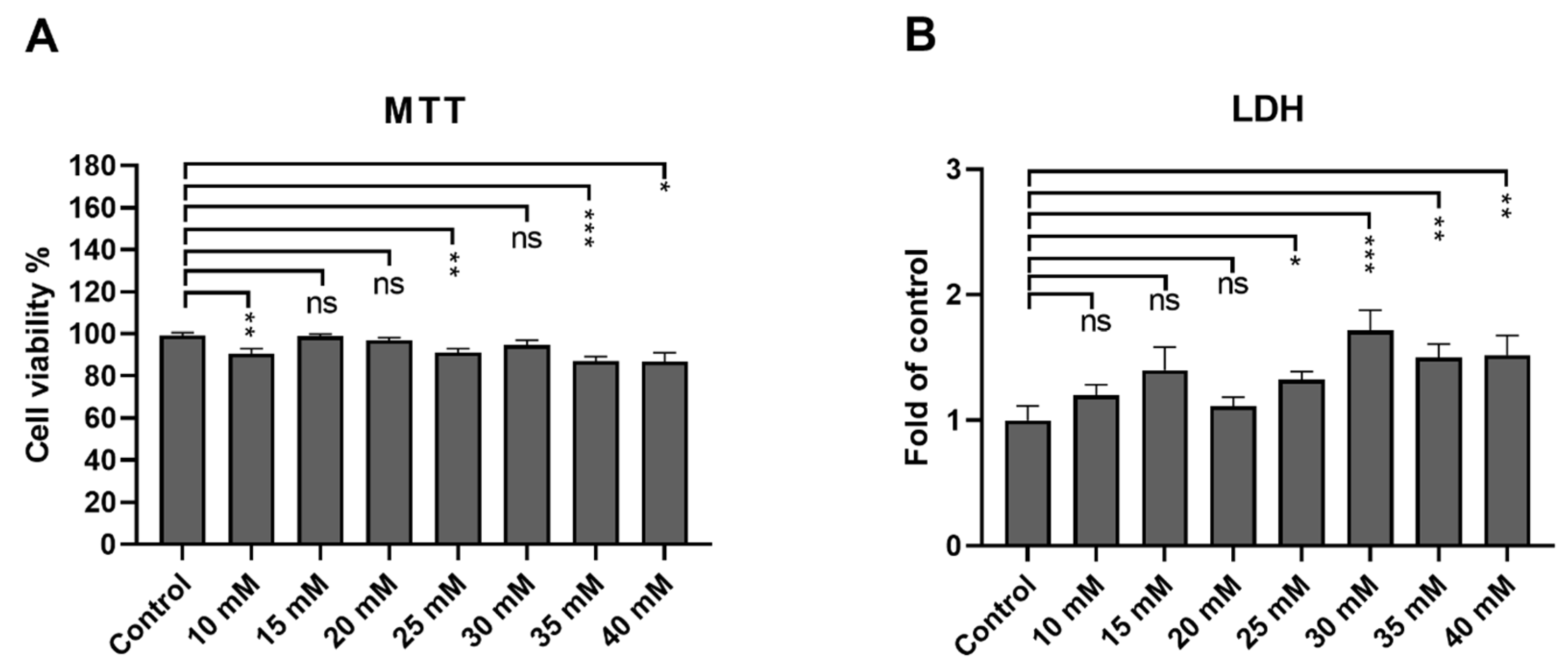
3.1. ARPE-19 Cells Remain Viable after an Exposure up to 40 mM Metformin
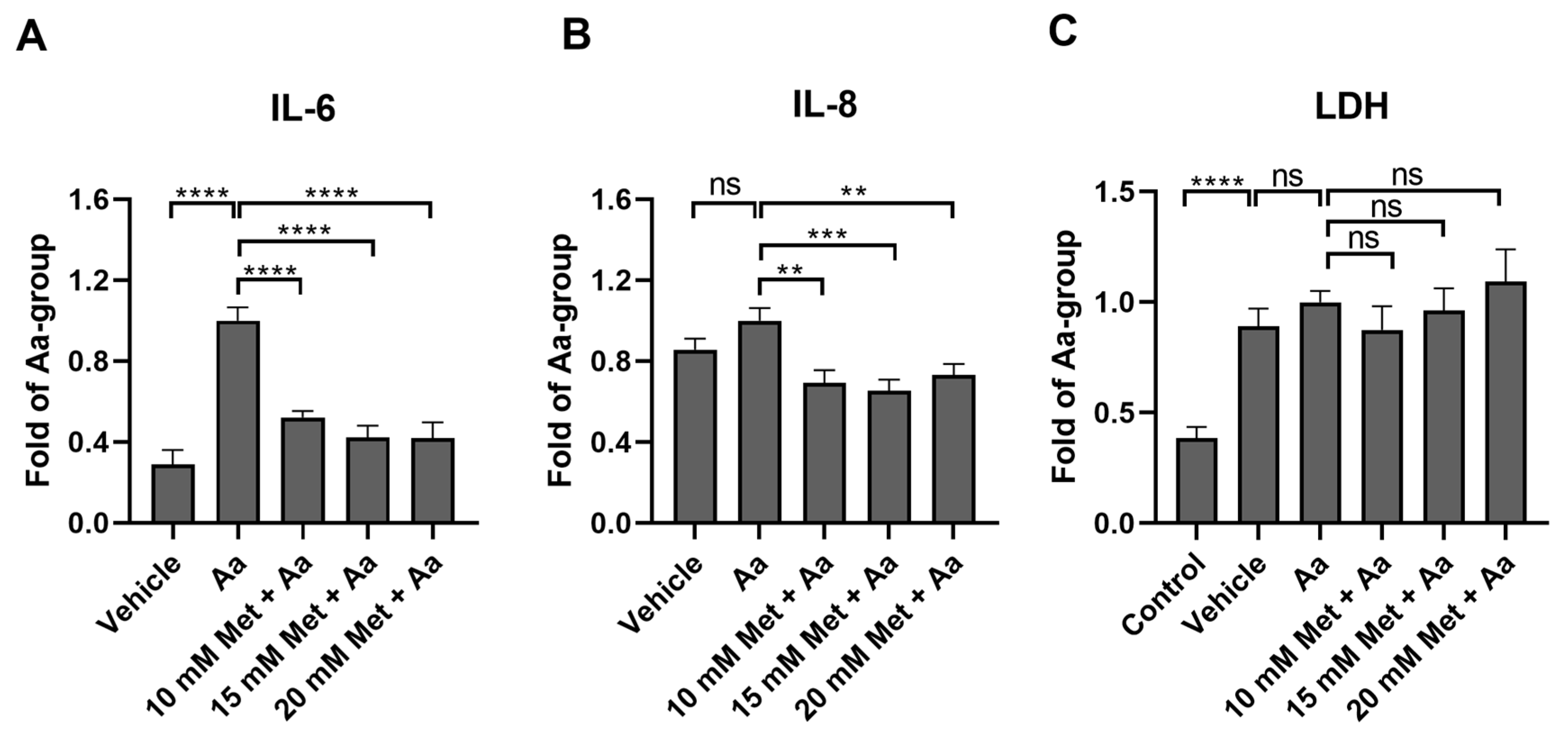
3.2. Metformin Reduces the Levels of Proinflammatory Cytokines IL-6 and IL-8 in ARPE-19 Cells with Induced Mitochondrial Damage
3.3. Anti-Inflammatory Effects of Metformin Are Not Mediated by the Regulation of NF-κB or MAP Kinases
3.4. Metformin Pre-Treatment Prevents Antimycin A-Induced Loss of the Mitochondrial Membrane Potential in ARPE-19 Cells
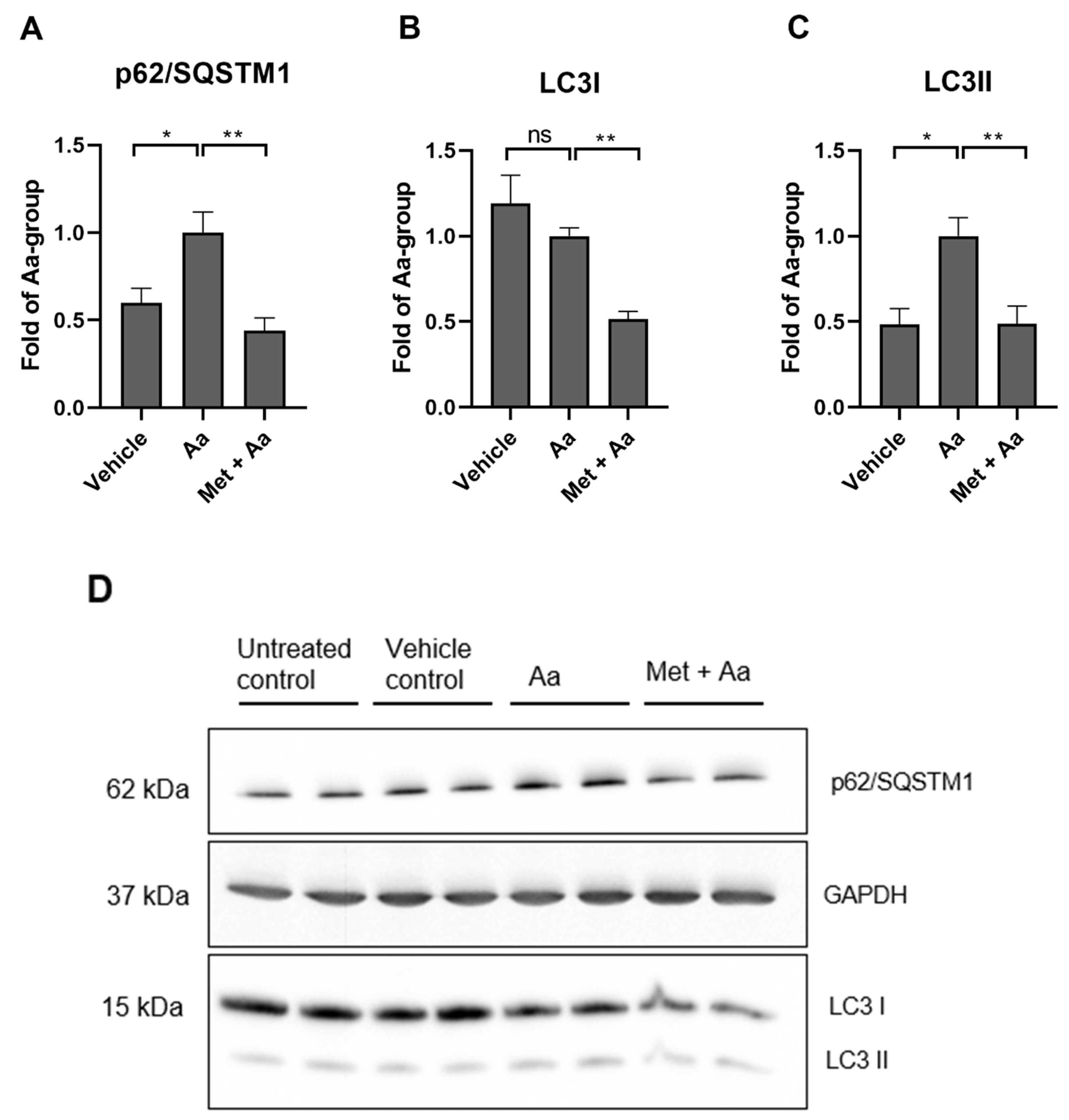
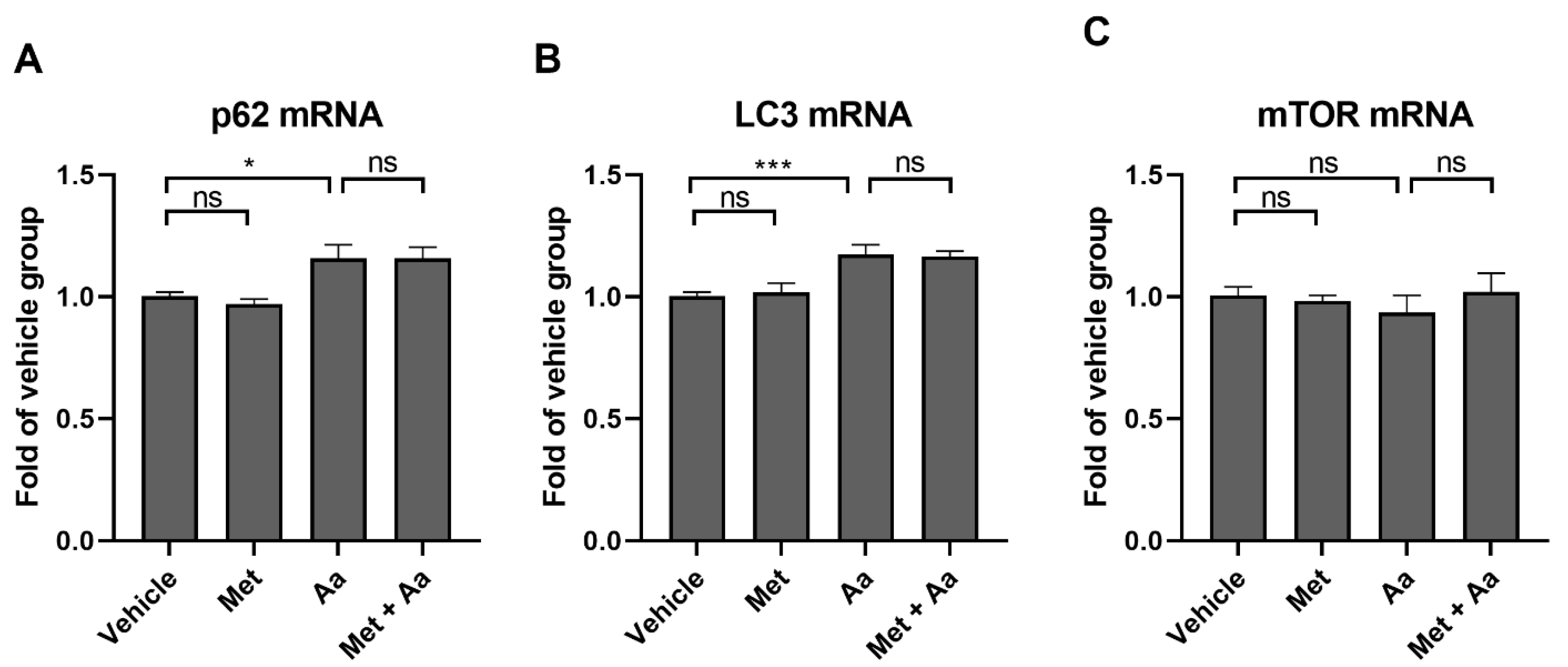
3.5. Metformin Affects the Levels of Autophagy Marker Proteins in ARPE-19 Cells
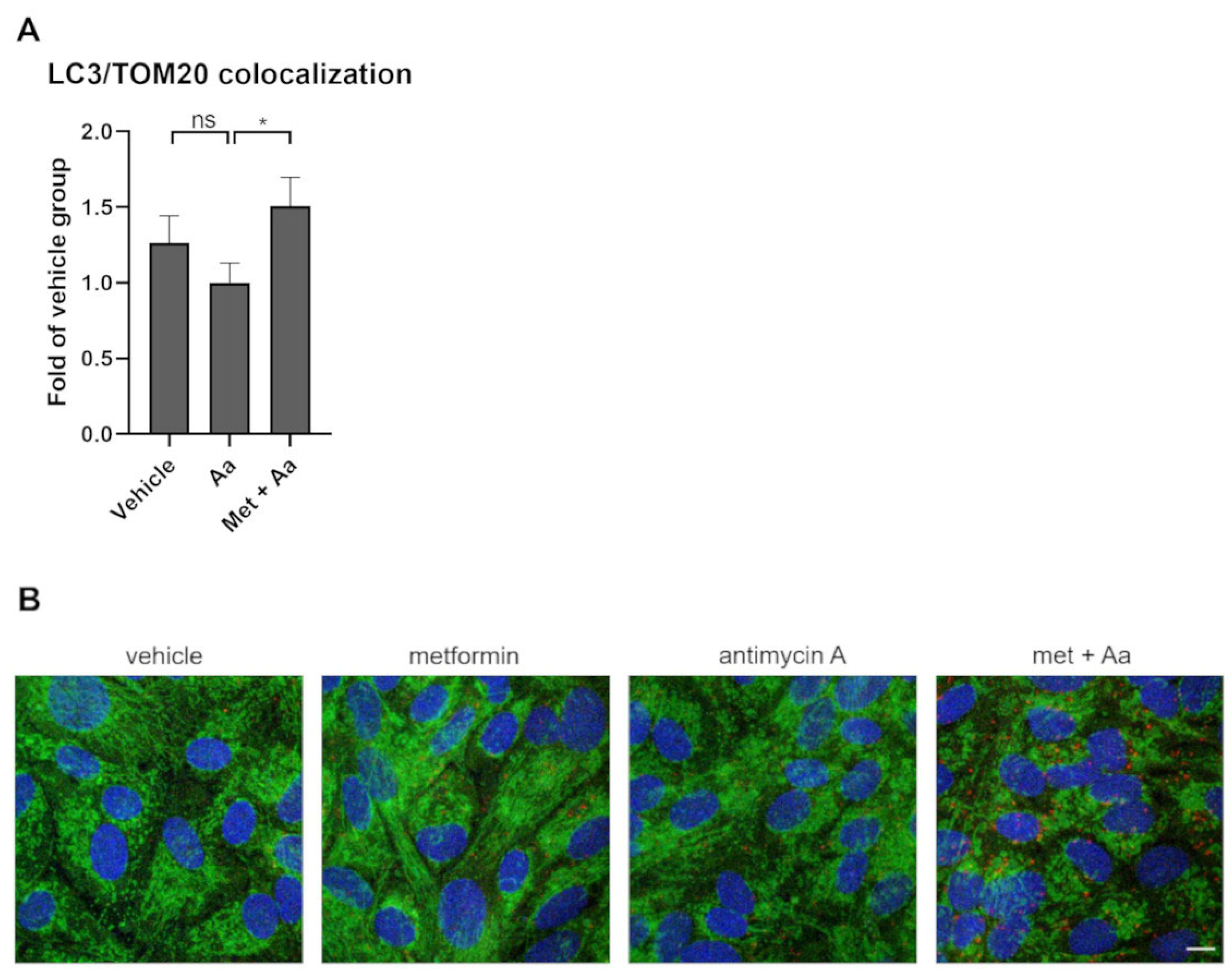
3.6. Metformin Induced Mitophagy in ARPE-19 Cells
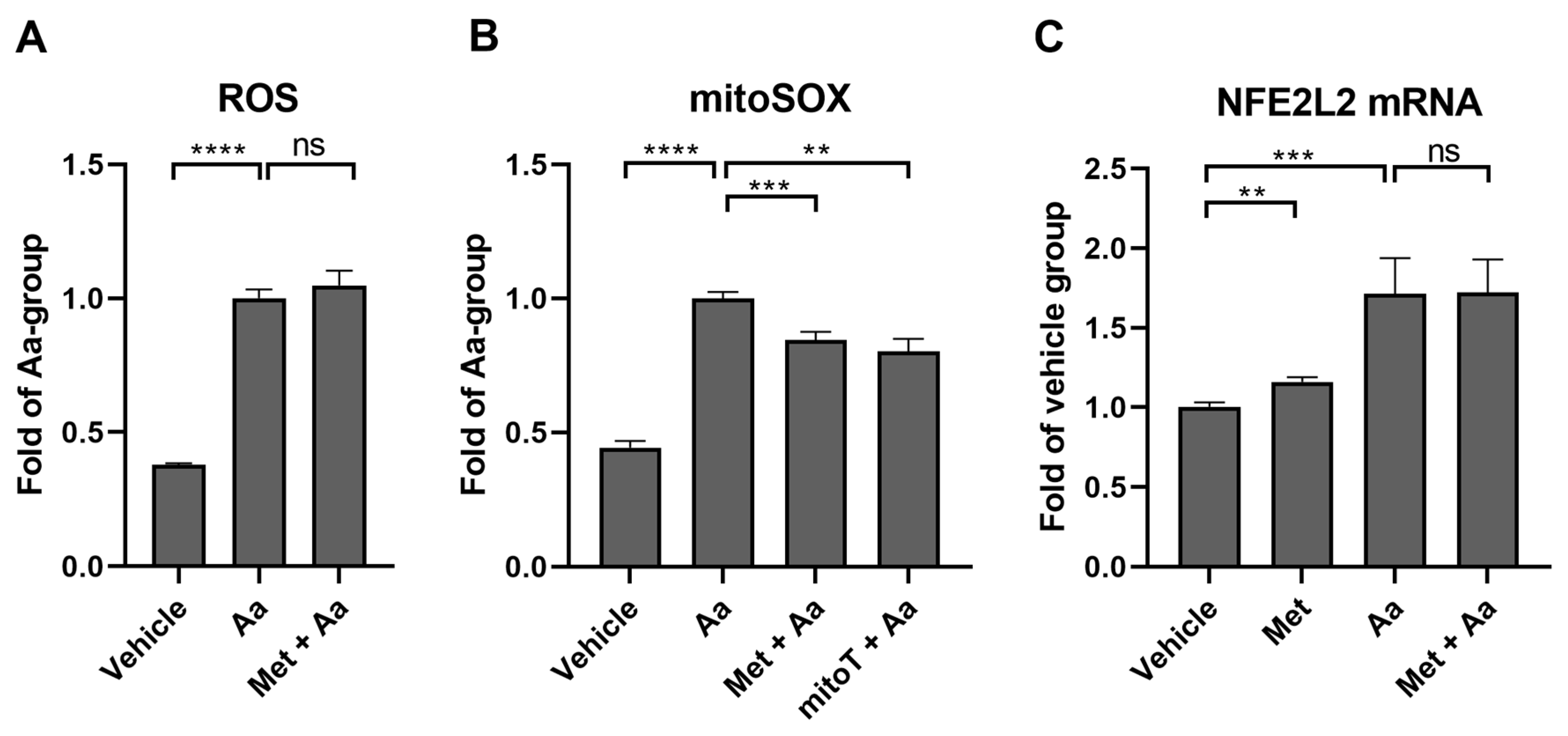
3.7. Metformin Selectively Reduces Antimycin A-Induced Mitochondrial ROS Production in ARPE-19 Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bailey, C.; Day, C. Metformin: Its Botanical Background. Pract. Diabetes Int. 2004, 21, 115–117. [Google Scholar] [CrossRef]
- Raju, T.N. The Nobel Chronicles. Lancet 2000, 355, 1022. [Google Scholar] [CrossRef]
- Johnson, N.P. Metformin Use in Women with Polycystic Ovary Syndrome. Ann. Transl. Med. 2014, 2, 56. [Google Scholar] [CrossRef]
- Triggle, C.R.; Mohammed, I.; Bshesh, K.; Marei, I.; Ye, K.; Ding, H.; MacDonald, R.; Hollenberg, M.D.; Hill, M.A. Metformin: Is It a Drug for All Reasons and Diseases? Metabolism 2022, 133, 155223. [Google Scholar] [CrossRef]
- Foretz, M.; Guigas, B.; Bertrand, L.; Pollak, M.; Viollet, B. Metformin: From Mechanisms of Action to Therapies. Cell Metab. 2014, 20, 953–966. [Google Scholar] [CrossRef]
- Yerevanian, A.; Soukas, A.A. Metformin: Mechanisms in Human Obesity and Weight Loss. Curr. Obes. Rep. 2019, 8, 156–164. [Google Scholar] [CrossRef]
- Brown, E.E.; Ball, J.D.; Chen, Z.; Khurshid, G.S.; Prosperi, M.; Ash, J.D. The Common Antidiabetic Drug Metformin Reduces Odds of Developing Age-Related Macular Degeneration. Investig. Ophthalmol. Vis. Sci. 2019, 60, 1470. [Google Scholar] [CrossRef]
- Chen, Y.-Y.; Shen, Y.-C.; Lai, Y.-J.; Wang, C.-Y.; Lin, K.-H.; Feng, S.-C.; Liang, C.-Y.; Wei, L.-C.; Chou, P. Association between Metformin and a Lower Risk of Age-Related Macular Degeneration in Patients with Type 2 Diabetes. J. Ophthalmol. 2019, 2019, 1649156. [Google Scholar] [CrossRef]
- Blitzer, A.L.; Ham, S.A.; Colby, K.A.; Skondra, D. Association of Metformin Use with Age-Related Macular Degeneration: A Case-Control Study. JAMA Ophthalmol. 2021, 139, 302. [Google Scholar] [CrossRef]
- Khanna, S.; Shaw, L.; Hyman, M.J.; Zhang, J.; Hariprasad, S.; Soo, J.; Flores, A.; Skondra, D. Association of Metformin Use with Risk of Newly Onset Neovascular Age-Related Macular Degeneration Development. Retina 2024, 44, 205–213. [Google Scholar] [CrossRef]
- Aggarwal, S.; Moir, J.; Hyman, M.J.; Kaufmann, G.T.; Flores, A.; Hariprasad, S.M.; Skondra, D. Metformin Use and Age-Related Macular Degeneration in Patients Without Diabetes. JAMA Ophthalmol. 2024, 142, 53–57. [Google Scholar] [CrossRef]
- Wong, W.L.; Su, X.; Li, X.; Cheung, C.M.G.; Klein, R.; Cheng, C.-Y.; Wong, T.Y. Global Prevalence of Age-Related Macular Degeneration and Disease Burden Projection for 2020 and 2040: A Systematic Review and Meta-Analysis. Lancet Glob. Health 2014, 2, e106–e116. [Google Scholar] [CrossRef]
- Deng, Y.; Qiao, L.; Du, M.; Qu, C.; Wan, L.; Li, J.; Huang, L. Age-Related Macular Degeneration: Epidemiology, Genetics, Pathophysiology, Diagnosis, and Targeted Therapy. Genes. Dis. 2022, 9, 62–79. [Google Scholar] [CrossRef]
- Hyttinen, J.M.T.; Viiri, J.; Kaarniranta, K.; Błasiak, J. Mitochondrial Quality Control in AMD: Does Mitophagy Play a Pivotal Role? Cell. Mol. Life Sci. 2018, 75, 2991–3008. [Google Scholar] [CrossRef]
- Kaarniranta, K.; Uusitalo, H.; Blasiak, J.; Felszeghy, S.; Kannan, R.; Kauppinen, A.; Salminen, A.; Sinha, D.; Ferrington, D. Mechanisms of Mitochondrial Dysfunction and Their Impact on Age-Related Macular Degeneration. Prog. Retin. Eye Res. 2020, 79, 100858. [Google Scholar] [CrossRef]
- De Marañón, A.M.; Díaz-Pozo, P.; Canet, F.; Díaz-Morales, N.; Abad-Jiménez, Z.; López-Domènech, S.; Vezza, T.; Apostolova, N.; Morillas, C.; Rocha, M.; et al. Metformin Modulates Mitochondrial Function and Mitophagy in Peripheral Blood Mononuclear Cells from Type 2 Diabetic Patients. Redox Biol. 2022, 53, 102342. [Google Scholar] [CrossRef]
- Liu, H.; Hu, F.-Y.; Xu, P.; Wu, J.-H. Regulation of Mitophagy by Metformin Improves the Structure and Function of Retinal Ganglion Cells Following Excitotoxicity-Induced Retinal Injury. Exp. Eye Res. 2022, 217, 108979. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the Use and Interpretation of Assays for Monitoring Autophagy (4th Edition)1. Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef]
- Bradford, M.M. A Rapid and Sensitive Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Kauppinen, A.; Niskanen, H.; Suuronen, T.; Kinnunen, K.; Salminen, A.; Kaarniranta, K. Oxidative stress activates NLRP3 inflammasomes in ARPE-19 cells—Implications for age-related macular degeneration (AMD). Immunol. Lett. 2012, 147, 29–33. [Google Scholar] [CrossRef]
- Winer, J.; Jung, C.K.S.; Shackel, I.; Williams, P.M. Development and Validation of Real-Time Quantitative Reverse Transcriptase–Polymerase Chain Reaction for Monitoring Gene Expression in Cardiac Myocytes In Vitro. Anal. Biochem. 1999, 270, 41–49. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Manders, E.M.M.; Verbeek, F.J.; Aten, J.A. Measurement of Co-Localization of Objects in Dual-Colour Confocal Images. J. Microsc. 1993, 169, 375–382. [Google Scholar] [CrossRef]
- ISO 10993-5:2009; Biological Evaluation of Medical Devices—Part 5: Tests for In Vitro Cytotoxicity. International Organization for Standardization: Geneva, Switzerland, 2009.
- Hytti, M.; Piippo, N.; Korhonen, E.; Honkakoski, P.; Kaarniranta, K.; Kauppinen, A. Fisetin and Luteolin Protect Human Retinal Pigment Epithelial Cells from Oxidative Stress-Induced Cell Death and Regulate Inflammation. Sci. Rep. 2015, 5, 17645. [Google Scholar] [CrossRef]
- Hsu, M.-L.; Huang, W.-C.; Zhou, Y.-R.; Hu, S.; Huang, C.; Wu, S.-J. Oleuropein Protects Human Retinal Pigment Epithelium Cells from IL-1β–Induced Inflammation by Blocking MAPK/NF-κB Signaling Pathways. Inflammation 2022, 45, 297–307. [Google Scholar] [CrossRef]
- Lee, S.; Lee, E.J.; Lee, G.M.; Yun, J.-H.; Yoo, W. Inhibitory Effect of Fucoidan on TNF-α-Induced Inflammation in Human Retinal Pigment Epithelium Cells. Front. Nutr. 2023, 10, 1162934. [Google Scholar] [CrossRef]
- Hytti, M.; Korhonen, E.; Hyttinen, J.M.T.; Roehrich, H.; Kaarniranta, K.; Ferrington, D.A.; Kauppinen, A. Antimycin A-Induced Mitochondrial Damage Causes Human RPE Cell Death despite Activation of Autophagy. Oxid. Med. Cell. Longev. 2019, 2019, 1583656. [Google Scholar] [CrossRef]
- Vomund, S.; Schäfer, A.; Parnham, M.; Brüne, B.; Von Knethen, A. Nrf2, the Master Regulator of Anti-Oxidative Responses. Int. J. Mol. Sci. 2017, 18, 2772. [Google Scholar] [CrossRef]
- Bharath, L.P.; Nikolajczyk, B.S. The Intersection of Metformin and Inflammation. Am. J. Physiol.—Cell Physiol. 2021, 320, C873–C879. [Google Scholar] [CrossRef]
- Lin, H.; Ao, H.; Guo, G.; Liu, M. The Role and Mechanism of Metformin in Inflammatory Diseases. JIR 2023, 16, 5545–5564. [Google Scholar] [CrossRef]
- Takemura, Y.; Osuga, Y.; Yoshino, O.; Hasegawa, A.; Hirata, T.; Hirota, Y.; Nose, E.; Morimoto, C.; Harada, M.; Koga, K.; et al. Metformin Suppresses Interleukin (IL)-1β-Induced IL-8 Production, Aromatase Activation, and Proliferation of Endometriotic Stromal Cells. J. Clin. Endocrinol. Metab. 2007, 92, 3213–3218. [Google Scholar] [CrossRef]
- Feng, Q.; Ruan, X.; Lu, M.; Bu, S.; Zhang, Y. Metformin Protects Retinal Pigment Epithelium Cells against H2O2-Induced Oxidative Stress and Inflammation via the Nrf2 Signaling Cascade. Graefe’s Arch. Clin. Exp. Ophthalmol. 2023, 262, 1519–1530. [Google Scholar] [CrossRef]
- Petrasca, A.; Hambly, R.; Kearney, N.; Smith, C.M.; Pender, E.K.; Mac Mahon, J.; O’Rourke, A.M.; Ismaiel, M.; Boland, P.A.; Almeida, J.P.; et al. Metformin Has Anti-Inflammatory Effects and Induces Immunometabolic Reprogramming via Multiple Mechanisms in Hidradenitis Suppurativa. Br. J. Dermatol. 2023, 189, 730–740. [Google Scholar] [CrossRef]
- Yang, J.; Liu, H.-C.; Zhang, J.-Q.; Zou, J.-Y.; Zhang, X.; Chen, W.-M.; Gu, Y.; Hong, H. The Effect of Metformin on Senescence of T Lymphocytes. Immun. Ageing 2023, 20, 73. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, M.; Lin, M.; Wei, P. The Immunomodulatory Effects of Metformin in LPS-Induced Macrophages: An In Vitro Study. Inflamm. Res. 2024, 73, 175–181. [Google Scholar] [CrossRef]
- de Araújo, A.A.; Pereira, A.d.S.B.F.; de Medeiros, C.A.C.X.; Brito, G.A.d.C.; Leitão, R.F.d.C.; Araújo, L.d.S.; Guedes, P.M.M.; Hiyari, S.; Pirih, F.Q.; Júnior, R.F.d.A. Effects of Metformin on Inflammation, Oxidative Stress, and Bone Loss in a Rat Model of Periodontitis. PLoS ONE 2017, 12, e0183506. [Google Scholar] [CrossRef]
- Schexnayder, C.; Broussard, K.; Onuaguluchi, D.; Poché, A.; Ismail, M.; McAtee, L.; Llopis, S.; Keizerweerd, A.; McFerrin, H.; Williams, C. Metformin Inhibits Migration and Invasion by Suppressing ROS Production and COX2 Expression in MDA-MB-231 Breast Cancer Cells. Int. J. Mol. Sci. 2018, 19, 3692. [Google Scholar] [CrossRef]
- Picone, P.; Nuzzo, D.; Caruana, L.; Messina, E.; Barera, A.; Vasto, S.; Di Carlo, M. Metformin Increases APP Expression and Processing via Oxidative Stress, Mitochondrial Dysfunction and NF-κB Activation: Use of Insulin to Attenuate Metformin’s Effect. Biochim. Biophys. Acta 2015, 1853, 1046–1059. [Google Scholar] [CrossRef]
- Zhao, X.; Liu, L.; Jiang, Y.; Silva, M.; Zhen, X.; Zheng, W. Protective Effect of Metformin against Hydrogen Peroxide-Induced Oxidative Damage in Human Retinal Pigment Epithelial (RPE) Cells by Enhancing Autophagy through Activation of AMPK Pathway. Oxid. Med. Cell. Longev. 2020, 2020, 2524174. [Google Scholar] [CrossRef]
- Shu, C.-W.; Tsen, C.-L.; Li, M.-S.; Bee, Y.-S.; Lin, S.-H.; Sheu, S.-J. Metformin and Rapamycin Protect Cells from Vital Dye–Induced Damage in Retinal Pigment Epithelial Cells and In Vivo. Graefe’s Arch. Clin. Exp. Ophthalmol. 2020, 258, 557–564. [Google Scholar] [CrossRef]
- Viollet, B.; Guigas, B.; Garcia, N.S.; Leclerc, J.; Foretz, M.; Andreelli, F. Cellular and Molecular Mechanisms of Metformin: An Overview. Clin. Sci. 2012, 122, 253–270. [Google Scholar] [CrossRef]
- Lu, G.; Wu, Z.; Shang, J.; Xie, Z.; Chen, C.; Zhang, C. The Effects of Metformin on Autophagy. Biomed. Pharmacother. 2021, 137, 111286. [Google Scholar] [CrossRef] [PubMed]
- Kauppinen, A. Mitochondria-Associated Inflammasome Activation and Its Impact on Aging and Age-Related Diseases. In Handbook of Immunosenescence; Fulop, T., Franceschi, C., Hirokawa, K., Pawelec, G., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 1–20. ISBN 978-3-319-64597-1. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward | Reverse |
|---|---|---|
| SQSTM1 | 5′-GGA GCA GAT GAG GAA GAT CG-3′ | 3′-CTT CGG ATT CTG GCA TCT GT-5′ |
| MAP1LC3B | 5′-GCA GCA TCC AAC CAA AAT CC-3′ | 3′-CAT TGA GCT GTA AGC GCC TTC T-5′ |
| MTOR | 5′-AGC ATC GGA TGC TTA GGA GTG G-3′ | 3′-CAG CCA GTC ATC TTT GGA GAC C-5′ |
| NFE2L2 | 5′-AAA TTG AGA TTG ATG GAA CAG CGA A-3′ | 3′-TAT GGC CTG GCT TAC ACA TTC A-5′ |
| GAPDH | 5′-GAT CAT CAG CAA TGC CTC CT-3′ | 3′-GGC CAT CCA CAG TCT TCT G-5′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toppila, M.; Ranta-aho, S.; Kaarniranta, K.; Hytti, M.; Kauppinen, A. Metformin Alleviates Inflammation and Induces Mitophagy in Human Retinal Pigment Epithelium Cells Suffering from Mitochondrial Damage. Cells 2024, 13, 1433. https://doi.org/10.3390/cells13171433
Toppila M, Ranta-aho S, Kaarniranta K, Hytti M, Kauppinen A. Metformin Alleviates Inflammation and Induces Mitophagy in Human Retinal Pigment Epithelium Cells Suffering from Mitochondrial Damage. Cells. 2024; 13(17):1433. https://doi.org/10.3390/cells13171433
Chicago/Turabian StyleToppila, Maija, Sofia Ranta-aho, Kai Kaarniranta, Maria Hytti, and Anu Kauppinen. 2024. "Metformin Alleviates Inflammation and Induces Mitophagy in Human Retinal Pigment Epithelium Cells Suffering from Mitochondrial Damage" Cells 13, no. 17: 1433. https://doi.org/10.3390/cells13171433