
Identification and Characterization of Multiple Paneth Cell Types in the Mouse Small Intestine

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
2.2. PC Sort
2.3. RNA Isolation on Bulk PCs
2.4. PC Bulk RNA-Seq
2.5. PC scRNA-Seq
2.6. Bulk and scRNA-Seq Comparison
2.7. Visualizations
2.8. Tissue Preparation Molecular Cartography
2.9. Molecular Cartography Probe Design
2.10. Molecular CartographyTM
2.11. Defa21 ELISA and Lysozyme Activity Assay
3. Results
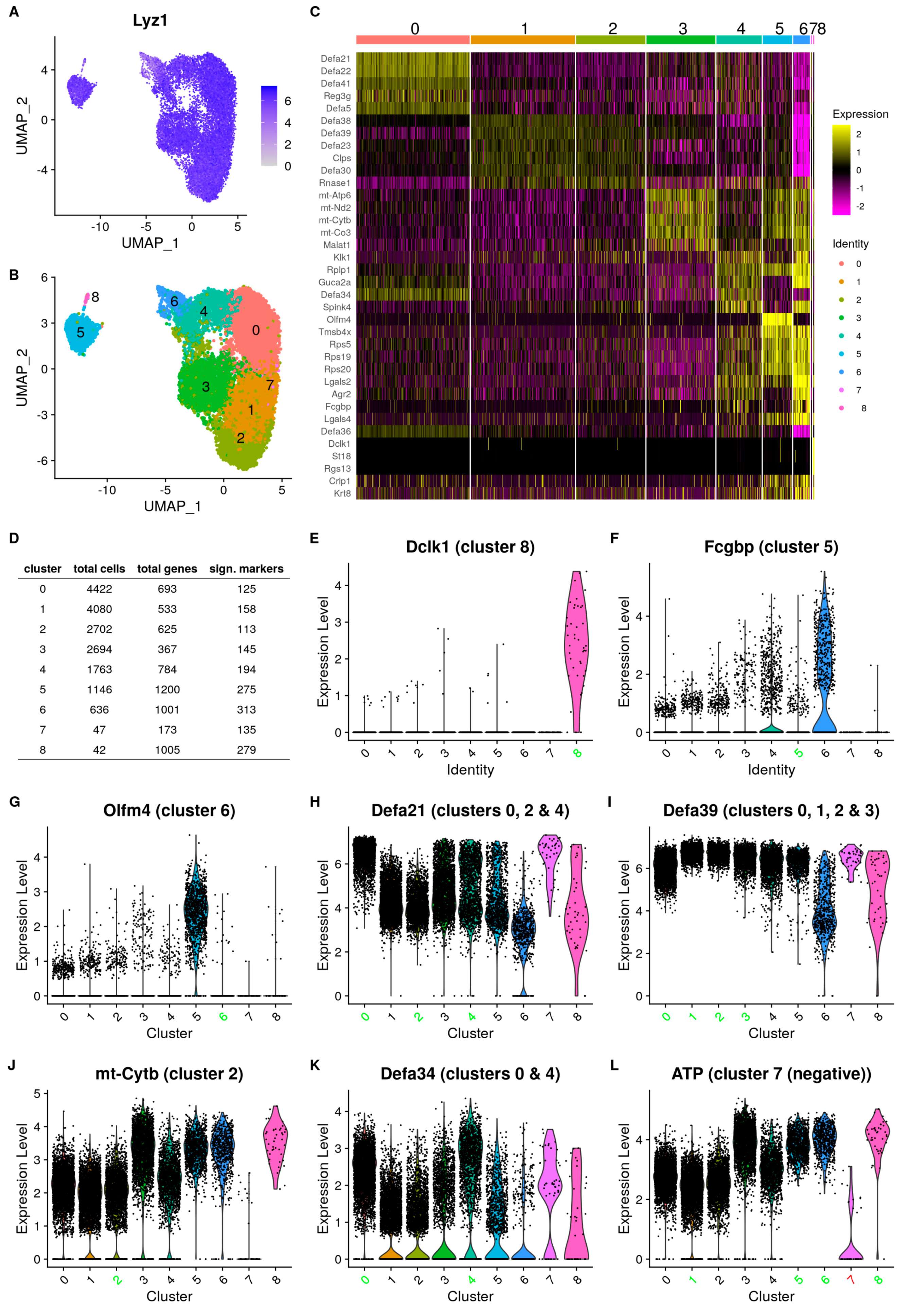
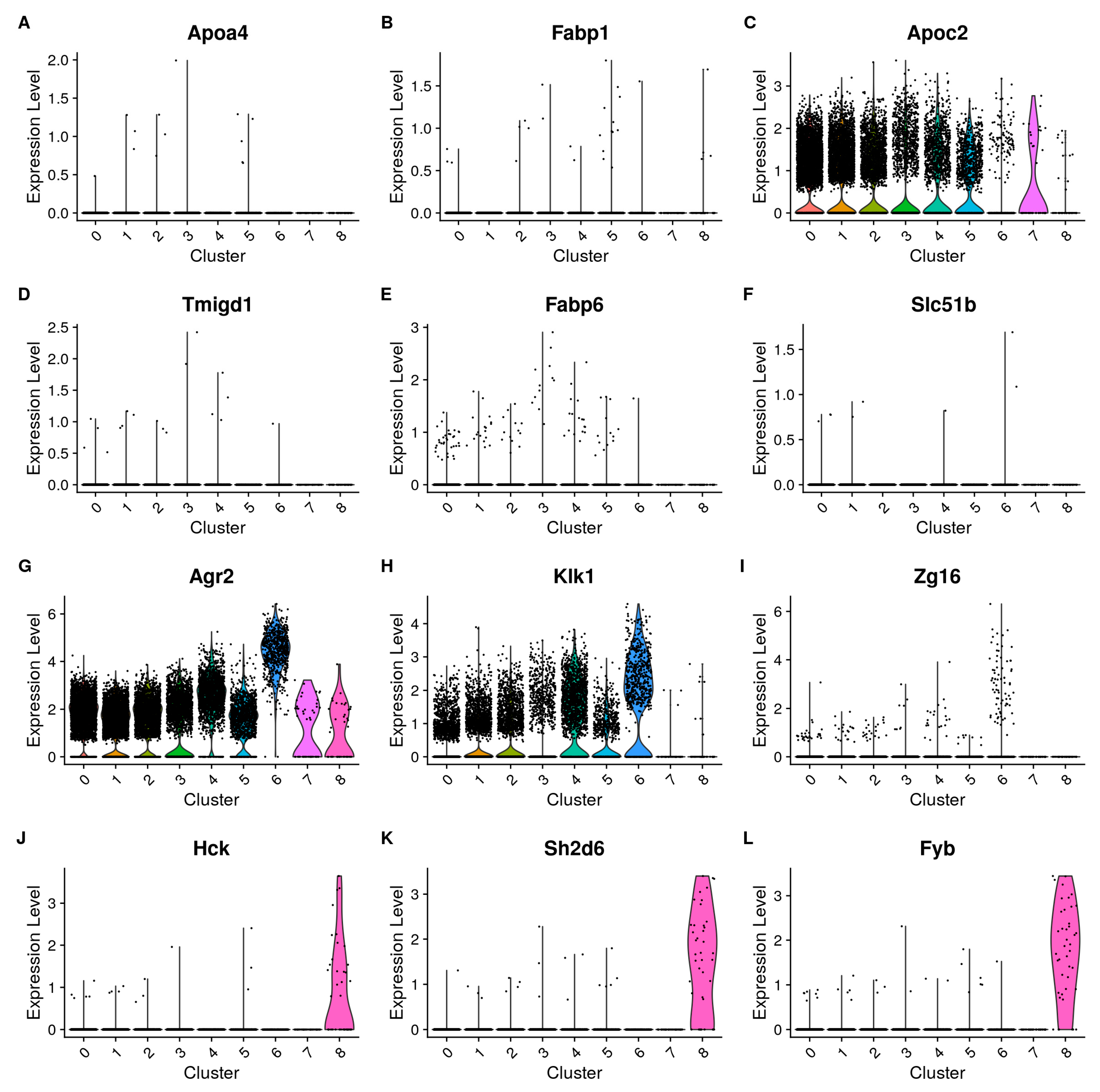
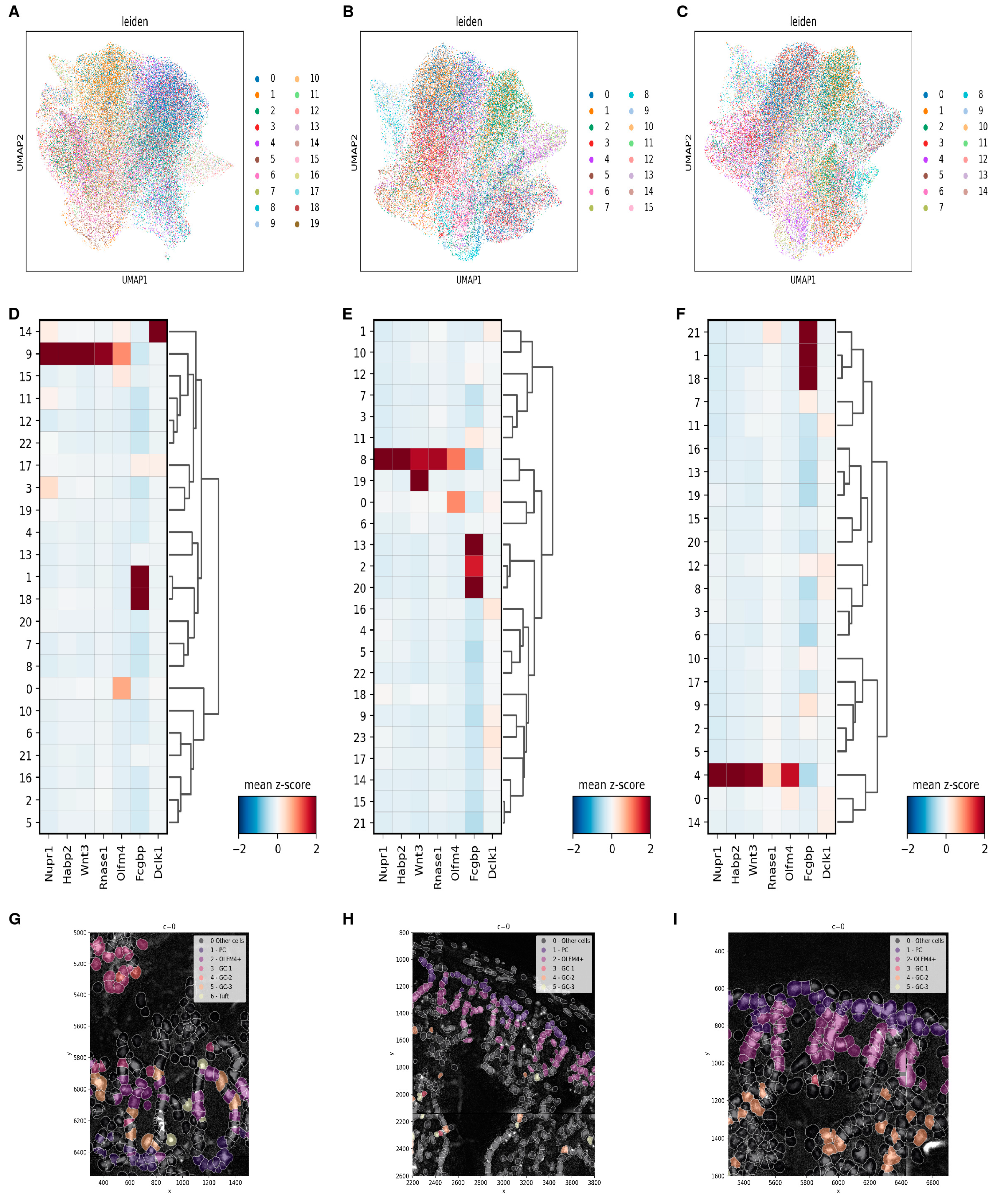
3.1. Single-Cell Analysis Unveils Diverse PC Subpopulations
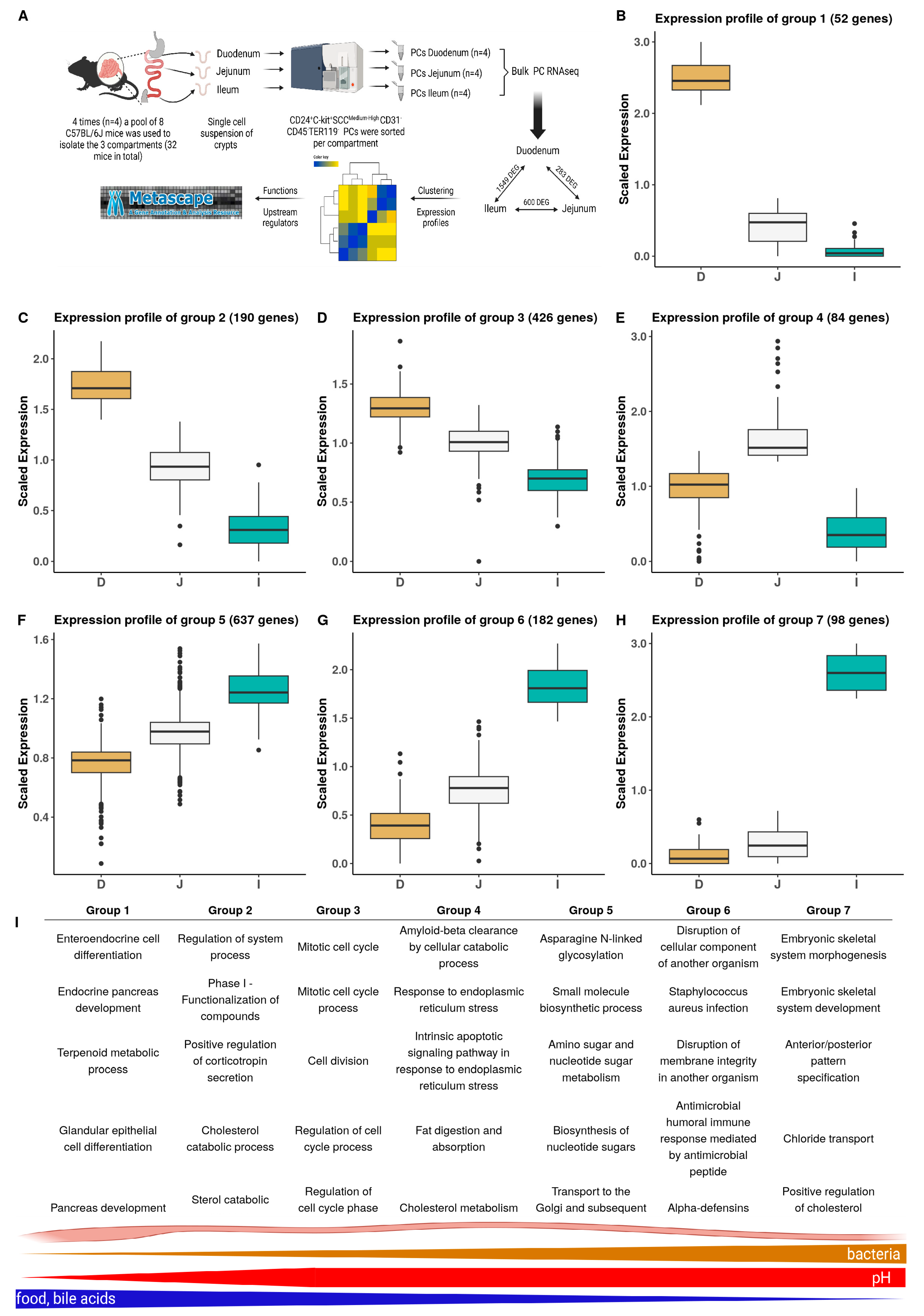
3.2. Regional Variations in PC Transcriptomes along the Small Intestine
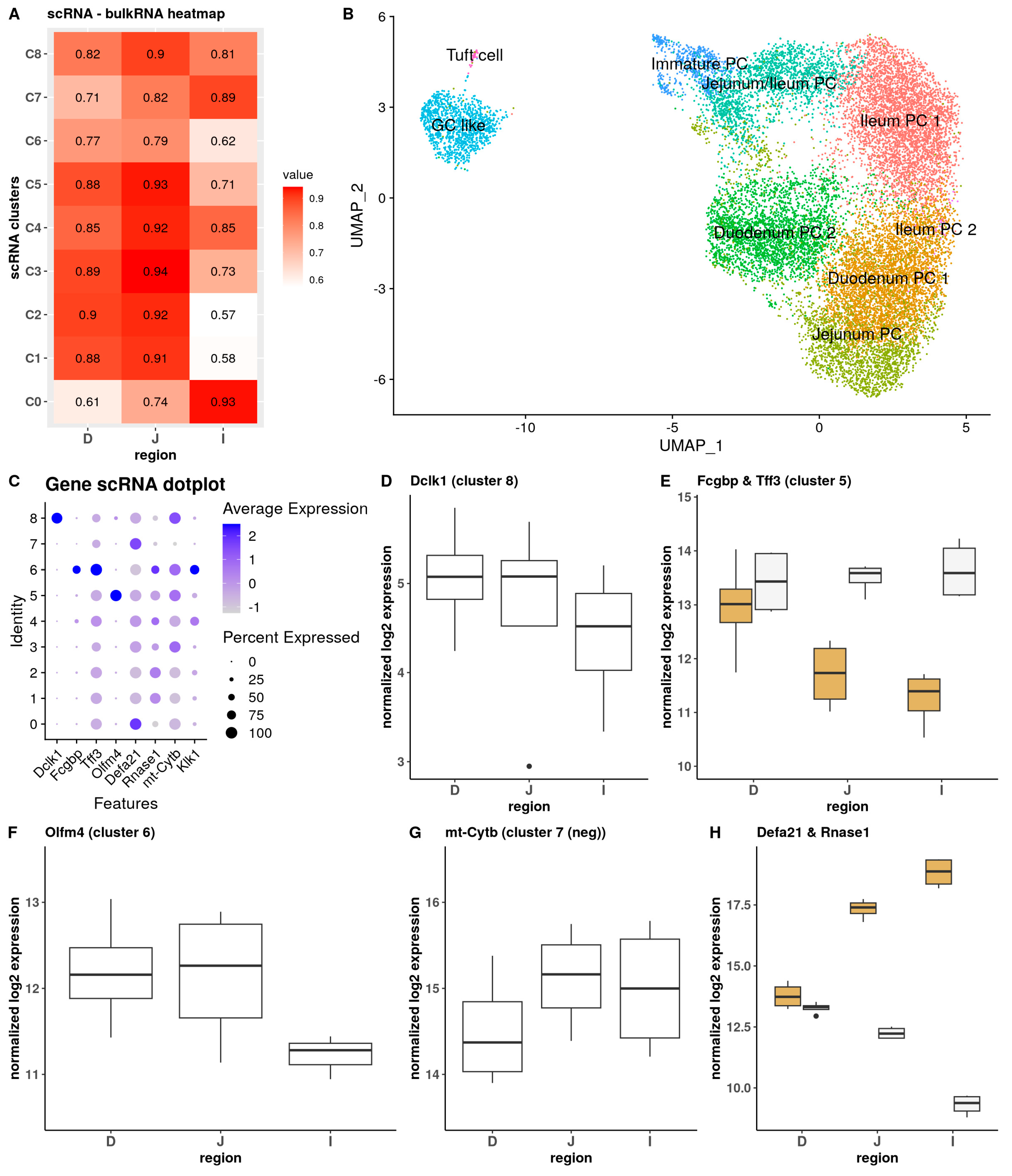
3.3. Integrative Analysis of Bulk and Single-Cell Data Links Specific PC Clusters to Regions in the Small Intestine
3.4. Confirmation of PC Clusters via Spatial Transcriptomics
3.4.1. Spatial Transcriptomic Probe Design to Validate PC Clusters
3.4.2. Simple Co-Location Analysis Confirms the Existence of Tuft Cell-like PCs and GC-like PCs
3.4.3. Cell-Based Analysis Identifies Region-Dependent Paneth Cell Identities along the Small Intestine, but Not within the Crypts Themselves
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Haegebarth, A.; Clevers, H. Wnt signaling, Lgr5, and stem cells in the intestine and skin. Am. J. Pathol. 2009, 174, 715–721. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; van Es, J.H.; Snippert, H.J.; Stange, D.E.; Vries, R.G.; van den Born, M.; Barker, N.; Shroyer, N.F.; van de Wetering, M.; Clevers, H. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature 2011, 469, 415–418. [Google Scholar] [CrossRef]
- Van Der Flier, L.G.; Clevers, H. Stem cells, self-renewal, and differentiation in the intestinal epithelium. Annu. Rev. Physiol. 2009, 71, 241–260. [Google Scholar] [CrossRef] [PubMed]
- Sender, R.; Fuchs, S.; Milo, R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol. 2016, 14, 1–14. [Google Scholar] [CrossRef]
- Yersin, S.; Vonaesch, P. Small intestinal microbiota: From taxonomic composition to metabolism. Trends Microbiol. 2024; Online ahead of print. [Google Scholar] [CrossRef]
- Layer, P.; Go, V.L.W.; DiMagno, E.P. Fate of pancreatic enzymes during small intestinal aboral transit in humans. Am. J. Physiol.—Gastrointest. Liver Physiol. 1986, 251, 475–480. [Google Scholar] [CrossRef]
- Folz, J.; Culver, R.N.; Morales, J.M.; Grembi, J.; Triadafilopoulos, G.; Relman, D.A.; Huang, K.C.; Shalon, D.; Fiehn, O. Human metabolome variation along the upper intestinal tract. Nat. Metab. 2023, 5, 777–788. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Wang, H.; Huang, X.; Kraiczy, J.; Singh, P.N.P.; Ng, C.; Dagdeviren, S.; Houghton, S.; Pellon-Cardenas, O.; Lan, Y.; et al. SATB2 preserves colon stem cell identity and mediates ileum-colon conversion via enhancer remodeling. Cell Stem Cell 2022, 29, 101–115.e10. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Huang, X.; Singh, P.N.P.; Li, S.; Lan, Y.; Deng, M.; Lacko, L.A.; Gomez-Salinero, J.M.; Rafii, S.; Verzi, M.P.; et al. A MTA2-SATB2 chromatin complex restrains colonic plasticity toward small intestine by retaining HNF4A at colonic chromatin. Nat. Commun. 2024, 15, 3595. [Google Scholar] [CrossRef]
- Koziolek, M.; Grimm, M.; Becker, D.; Iordanov, V.; Zou, H.; Shimizu, J.; Wanke, C.; Garbacz, G.; Weitschies, W. Investigation of pH and Temperature Profiles in the GI Tract of Fasted Human Subjects Using the Intellicap® System. J. Pharm. Sci. 2015, 104, 2855–2863. [Google Scholar] [CrossRef]
- Delbaere, K.; Roegiers, I.; Bron, A.; Durif, C.; Van De Wiele, T.; Blanquet-Diot, S.; Marinelli, L. The small intestine: Dining table of host-microbiota meetings. FEMS Microbiol. Rev. 2023, 47, fuad022. [Google Scholar] [CrossRef]
- Kopp, Z.A.; Jain, U.; Van Limbergen, J.; Stadnyk, A.W. Do antimicrobial peptides and complement collaborate in the intestinal mucosa? Front. Immunol. 2015, 6, 17. [Google Scholar] [CrossRef]
- Zhao, L.; Zhang, X.; Zuo, T.; Yu, J. The Composition of Colonic Commensal Bacteria According to Anatomical Localization in Colorectal Cancer. Engineering 2017, 3, 90–97. [Google Scholar] [CrossRef]
- Atuma, C.; Strugala, V.; Allen, A.; Holm, L. The adherent gastrointestinal mucus gel layer: Thickness and physical state in vivo. Am. J. Physiol.—Gastrointest. Liver Physiol. 2001, 280, 922–929. [Google Scholar] [CrossRef]
- Friedman, E.S.; Bittinger, K.; Esipova, T.V.; Hou, L.; Chau, L.; Jiang, J.; Mesaros, C.; Lund, P.J.; Liang, X.; FitzGerald, G.A.; et al. Microbes vs. chemistry in the origin of the anaerobic gut lumen. Proc. Natl. Acad. Sci. USA 2018, 115, 4170–4175. [Google Scholar] [CrossRef] [PubMed]
- Mallory, A.; Kern, F.; Smith, J.; Savage, D. Patterns of bile acids and microflora in the human small intestine: I. Bile acids. Gastroenterology 1973, 64, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Cornes, J.S. Number, size, and distribution of Peyer’s patches in the human small intestine: Part I The development of Peyer’s patches. Gut 1965, 6, 225–229. [Google Scholar] [CrossRef]
- Chikina, A.; Matic Vignjevic, D. At the right time in the right place: How do luminal gradients position the microbiota along the gut? Cells Dev. 2021, 168, 203712. [Google Scholar] [CrossRef]
- Haber, A.L.; Biton, M.; Rogel, N.; Herbst, R.H.; Smillie, C.; Burgin, G.; Delorey, T.M.; Howitt, M.R.; Katz, Y.; Tirosh, I.; et al. A single-cell survey of the small intestinal epithelium Adam. Nature 2018, 551, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Yokoi, Y.; Fukaya, R.; Ohira, S.; Shinozaki, R.; Nishida, T.; Kikuchi, M.; Ayabe, T. Expression and Localization of Paneth Cells and Their α-Defensins in the Small Intestine of Adult Mouse. Front. Immunol. 2020, 11, 570296. [Google Scholar] [CrossRef]
- Mei, X.; Gu, M.; Li, M. Plasticity of Paneth cells and their ability to regulate intestinal stem cells. Stem Cell Res. Ther. 2020, 11, 349. [Google Scholar] [CrossRef]
- Schmitt, M.; Schewe, M.; Sacchetti, A.; Feijtel, D.; van de Geer, W.S.; Teeuwssen, M.; Sleddens, H.F.; Joosten, R.; van Royen, M.E.; van de Werken, H.J.G.; et al. Paneth Cells Respond to Inflammation and Contribute to Tissue Regeneration by Acquiring Stem-like Features through SCF/c-Kit Signaling. Cell Rep. 2018, 24, 2312–2328.e7. [Google Scholar] [CrossRef]
- Wallaeys, C.; Garcia-Gonzalez, N.; Libert, C. Paneth cells as the cornerstones of intestinal and organismal health: A primer. EMBO Mol. Med. 2023, 15, e16427. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Vanlaere, I.; De Rycke, R.; Cauwels, A.; Joosten, L.A.B.; Lubberts, E.; Van Den Berg, W.B.; Libert, C. IL-17 produced by Paneth cells drives TNF-induced shock. J. Exp. Med. 2008, 205, 1755–1761. [Google Scholar] [CrossRef] [PubMed]
- Geiser, J.; Venken, K.J.T.; de Lisle, R.C.; Andrews, G.K. A mouse model of acrodermatitis enteropathica: Loss of intestine zinc transporter ZIP4 (Slc39a4) disrupts the stem cell niche and intestine integrity. PLoS Genet. 2012, 8, e1002766. [Google Scholar] [CrossRef]
- Bel, S.; Pendse, M.; Wang, Y.; Li, Y.; Ruhn, K.A.; Hassell, B.; Leal, T.; Winter, S.E.; Xavier, R.J.; Hooper, L.V. Paneth cells secrete lysozyme via secretory autophagy during bacterial infection of the intestine. Science 2017, 1052, 1047–1052. [Google Scholar] [CrossRef] [PubMed]
- Shankman, L.S.; Fleury, S.T.; Evans, W.B.; Ravichandran, K.S.; Shankman, L.S.; Fleury, S.T.; Evans, W.B.; Penberthy, K.K.; Arandjelovic, S. Efferocytosis by Paneth cells within the intestine. Curr. Biol. 2021, 31, 2469–2476. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 1–21. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Zheng, G.X.Y.; Terry, J.M.; Belgrader, P.; Ryvkin, P.; Bent, Z.W.; Wilson, R.; Ziraldo, S.B.; Wheeler, T.D.; McDermott, G.P.; Zhu, J.; et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017, 8, 14049. [Google Scholar] [CrossRef]
- Nio-Kobayashi, J.; Takahashi-Iwanaga, H.; Iwanaga, T. Immunohistochemical localization of six galectin subtypes in the mouse digestive tract. J. Histochem. Cytochem. 2009, 57, 41–50. [Google Scholar] [CrossRef]
- Kolberg, L.; Raudvere, U.; Kuzmin, I.; Vilo, J.; Peterson, H. gprofiler2—An R package for gene list functional enrichment analysis and namespace conversion toolset g:Profiler. F1000Research 2020, 9, 709. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; Volume 35. [Google Scholar] [CrossRef]
- Metsalu, T.; Vilo, J. ClustVis: A web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. B 2015, 43, W566–W570. [Google Scholar] [CrossRef] [PubMed]
- Schewe, M.; Sacchetti, A.; Schmitt, M.; Fodde, R. The organoid reconstitution assay (ORA) for the functional analysis of intestinal stem and niche cells. J. Vis. Exp. 2017, 2017, 6–9. [Google Scholar] [CrossRef]
- Hao, Y.; Stuart, T.; Kowalski, M.H.; Choudhary, S.; Hoffman, P.; Hartman, A.; Srivastava, A.; Molla, G.; Madad, S.; Fernandez-Granda, C.; et al. Dictionary learning for integrative, multimodal and scalable single-cell analysis. Nat. Biotechnol. 2023, 42, 293–304. [Google Scholar] [CrossRef]
- Deckx, R.J.; Vantrappen, G.R.; Parein, M.M. Localization of lysozyme activity in a Paneth cell granule fraction. BBA—Enzymol. 1967, 139, 204–207. [Google Scholar] [CrossRef]
- Treveil, A.; Sudhakar, P.; Matthews, Z.J.; Wrzesiński, T.; Jones, E.J.; Brooks, J.; Ölbei, M.; Hautefort, I.; Hall, L.J.; Carding, S.R.; et al. Regulatory network analysis of Paneth cell and goblet cell enriched gut organoids using transcriptomics approaches. Mol. Omi. 2020, 16, 39–58. [Google Scholar] [CrossRef]
- Ouellette, A.J.; Satchell, D.P.; Hsieh, M.M.; Hagen, S.J.; Selsted, M.E. Characterization of luminal Paneth cell α-defensins in mouse small intestine: Attenuated antimicrobial activities of peptides with truncated amino termini. J. Biol. Chem. 2000, 275, 33969–33973. [Google Scholar] [CrossRef]
- O’Donnell, A.M.; Nakamura, H.; Puri, P. Tuft Cells: A New Player in Hirschsprung’s Disease. Eur. J. Pediatr. Surg. 2020, 30, 59–63. [Google Scholar] [CrossRef]
- Grunddal, K.V.; Tonack, S.; Egerod, K.L.; Thompson, J.J.; Petersen, N.; Engelstoft, M.S.; Vagne, C.; Keime, C.; Gradwohl, G.; Offermanns, S.; et al. Adhesion receptor ADGRG2/GPR64 is in the GI-tract selectively expressed in mature intestinal tuft cells. Mol. Metab. 2021, 51, 101231. [Google Scholar] [CrossRef]
- Kobayashi, K.; Ogata, H.; Morikawa, M.; Iijima, S.; Harada, N.; Yoshida, T.; Brown, W.R.; Inoue, N.; Hamada, Y.; Ishii, H.; et al. Distribution and partial characterisation of IgG Fc binding protein in various mucin producing cells and body fluids. Gut 2002, 51, 169–177. [Google Scholar] [CrossRef]
- Suemori, S.; Lynch-Devaney, K.; Podolsky, D.K. Identification and characterization of rat intestinal trefoil factor: Tissue- and cell-specific member of the trefoil protein family. Proc. Natl. Acad. Sci. USA 1991, 88, 11017–11021. [Google Scholar] [CrossRef] [PubMed]
- Park, S.W.; Zhen, G.; Verhaeghe, C.; Nakagami, Y.; Nguyenvu, L.T.; Barczak, A.J.; Killeen, N.; Erle, D.J. The protein disulfide isomerase AGR2 is essential for production of intestinal mucus. Proc. Natl. Acad. Sci. USA 2009, 106, 6950–6955. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Edwards, R.; Dizon, D. Disruption of Paneth and goblet cell homeostasis and increased endoplasmic reticulum stress in Agr2−/− mice. Dev. Biol. 2004, 338, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Schuijers, J.; Van Der Flier, L.G.; Van Es, J.; Clevers, H. Robust cre-mediated recombination in small intestinal stem cells utilizing the Olfm4 locus. Stem Cell Rep. 2014, 3, 234–241. [Google Scholar] [CrossRef]
- Barker, N.; Van Es, J.H.; Kuipers, J.; Kujala, P.; Van Den Born, M.; Cozijnsen, M.; Haegebarth, A.; Korving, J.; Begthel, H.; Peters, P.J.; et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 2007, 449, 1003–1007. [Google Scholar] [CrossRef]
- Huttner, K.M.; Selsted, M.E.; Ouellette, A.J. Structure and diversity of the murine cryptdin gene family. Genomics 1993, 19, 448–453. [Google Scholar] [CrossRef]
- Jones, D.E.; Bevins, C.L. Paneth cells of the human small intestine express an antimicrobial peptide gene. J. Biol. Chem. 1992, 267, 23216–23225. [Google Scholar] [CrossRef]
- Amid, C.; Rehaume, L.M.; Brown, K.L.; Gilbert, J.G.R.; Dougan, G.; Hancock, R.E.W.; Harrow, J.L. Manual annotation and analysis of the defensin gene cluster in the C57BL/6J mouse reference genome. BMC Genom. 2009, 10, 606. [Google Scholar] [CrossRef]
- Salzman, N.H. Paneth cell defensins and the regulation of the microbiome. Gut Microbes 2010, 1, 401–406. [Google Scholar] [CrossRef]
- Ebrahim, N.; Shakirova, K.; Dashinimaev, E. PDX1 is the cornerstone of pancreatic β-cell functions and identity. Front. Mol. Biosci. 2022, 9, 1091757. [Google Scholar] [CrossRef]
- Puig, K.L.; Manocha, G.D.; Combs, C.K. Amyloid Precursor Protein Mediated Changes in Intestinal Epithelial Phenotype In Vitro. PLoS ONE 2015, 10, e0119534. [Google Scholar] [CrossRef]
- Im, Y.; Kim, Y. A Comprehensive Overview of RNA Deconvolution Methods and Their Application. Mol. Cells 2023, 46, 99–105. [Google Scholar] [CrossRef]
- van der Flier, L.G.; Haegebarth, A.; Stange, D.E.; van de Wetering, M.; Clevers, H. OLFM4 Is a Robust Marker for Stem Cells in Human Intestine and Marks a Subset of Colorectal Cancer Cells. Gastroenterology 2009, 137, 15–17. [Google Scholar] [CrossRef]
- Shimizu, Y.; Nakamura, K.; Yoshii, A.; Yokoi, Y.; Kikuchi, M.; Shinozaki, R.; Nakamura, S.; Ohira, S.; Sugimoto, R.; Ayabe, T. Paneth cell α-defensin misfolding correlates with dysbiosis and ileitis in Crohn’s disease model mice. Life Sci. Alliance 2020, 3, 1–15. [Google Scholar] [CrossRef]
- Castillo, P.A.; Nonnecke, E.B.; Ossorio, D.T.; Tran, M.T.N.; Goley, S.M.; Lönnerdal, B.; Underwood, M.A.; Bevins, C.L. An Experimental Approach to Rigorously Assess Paneth Cell α-Defensin (Defa) mRNA Expression in C57BL/6 Mice. Sci. Rep. 2019, 9, 13115. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Song, W.; Wang, J.; Wang, T.; Xiong, X.; Qi, Z.; Fu, W.; Yang, X.; Chen, Y.G.; Chen, Y.G. Single-cell transcriptome analysis reveals differential nutrient absorption functions in human intestine. J. Exp. Med. 2020, 217, e20191130. [Google Scholar] [CrossRef]
- Grün, D.; Lyubimova, A.; Kester, L.; Wiebrands, K.; Basak, O.; Sasaki, N.; Clevers, H.; Van Oudenaarden, A. Single-cell messenger RNA sequencing reveals rare intestinal cell types. Nature 2015, 525, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Burclaff, J.; Bliton, R.J.; Breau, K.A.; Ok, M.T.; Gomez-Martinez, I.; Ranek, J.S.; Bhatt, A.P.; Purvis, J.E.; Woosley, J.T.; Magness, S.T. A Proximal-to-Distal Survey of Healthy Adult Human Small Intestine and Colon Epithelium by Single-Cell Transcriptomics. Cell. Mol. Gastroenterol. Hepatol. 2022, 13, 1554–1589. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Bermingham, N.A.; Finegold, M.J.; Zoghbi, H.Y. Requirement of Math1 for secretory cell lineage commitment in the mouse intestine. Science 2001, 294, 2155–2158. [Google Scholar] [CrossRef]
- Kolev, H.M.; Kaestner, K.H. Mammalian Intestinal Development and Differentiation—The State of the Art. Cell. Mol. Gastroenterol. Hepatol. 2023, 16, 809–821. [Google Scholar] [CrossRef]
- Nyström, E.E.L.; Martinez-Abad, B.; Arike, L.; Birchenough, G.M.H.; Nonnecke, E.B.; Castillo, P.A.; Svensson, F.; Bevins, C.L.; Hansson, G.C.; Johansson, M.E.V. An intercrypt subpopulation of goblet cells is essential for colonic mucus barrier function. Science 2021, 372, eabb1590. [Google Scholar] [CrossRef] [PubMed]
- Hickey, J.W.; Becker, W.R.; Nevins, S.A.; Horning, A.; Perez, A.E.; Zhu, C.; Zhu, B.; Wei, B.; Chiu, R.; Chen, D.C.; et al. Organization of the human intestine at single-cell resolution. Nature 2023, 619, 572–584. [Google Scholar] [CrossRef] [PubMed]
- Ramasundara, M.; Leach, S.T.; Lemberg, D.A.; Day, A.S. Defensins and inflammation: The role of defensins in inflammatory bowel disease. J. Gastroenterol. Hepatol. 2009, 24, 202–208. [Google Scholar] [CrossRef]
- Muñoz, M.; Eidenschenk, C.; Ota, N.; Wong, K.; Lohmann, U.; Kühl, A.A.; Wang, X.; Manzanillo, P.; Li, Y.; Rutz, S.; et al. Interleukin-22 Induces Interleukin-18 Expression from Epithelial Cells during Intestinal Infection. Immunity 2015, 42, 321–331. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Timmermans, S.; Wallaeys, C.; Garcia-Gonzalez, N.; Pollaris, L.; Saeys, Y.; Libert, C. Identification and Characterization of Multiple Paneth Cell Types in the Mouse Small Intestine. Cells 2024, 13, 1435. https://doi.org/10.3390/cells13171435
Timmermans S, Wallaeys C, Garcia-Gonzalez N, Pollaris L, Saeys Y, Libert C. Identification and Characterization of Multiple Paneth Cell Types in the Mouse Small Intestine. Cells. 2024; 13(17):1435. https://doi.org/10.3390/cells13171435
Chicago/Turabian StyleTimmermans, Steven, Charlotte Wallaeys, Natalia Garcia-Gonzalez, Lotte Pollaris, Yvan Saeys, and Claude Libert. 2024. "Identification and Characterization of Multiple Paneth Cell Types in the Mouse Small Intestine" Cells 13, no. 17: 1435. https://doi.org/10.3390/cells13171435
APA StyleTimmermans, S., Wallaeys, C., Garcia-Gonzalez, N., Pollaris, L., Saeys, Y., & Libert, C. (2024). Identification and Characterization of Multiple Paneth Cell Types in the Mouse Small Intestine. Cells, 13(17), 1435. https://doi.org/10.3390/cells13171435