
A Comparison of Two Versions of the CRISPR-Sirius System for the Live-Cell Visualization of the Borders of Topologically Associating Domains


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plasmid Construction
2.2. Cell Culture and Transient Transfection
2.3. Lentivirus Production and Cell Transduction
2.4. FACS
2.5. Western Blot
2.6. Real-Time PCR
2.7. Live-Cell Microscopy
2.8. Graphical Representation of Hi-C and ChIP-Seq Data and Selection of Appropriate TAD Boundaries for Visualization
2.9. Epigenetic Data
3. Results
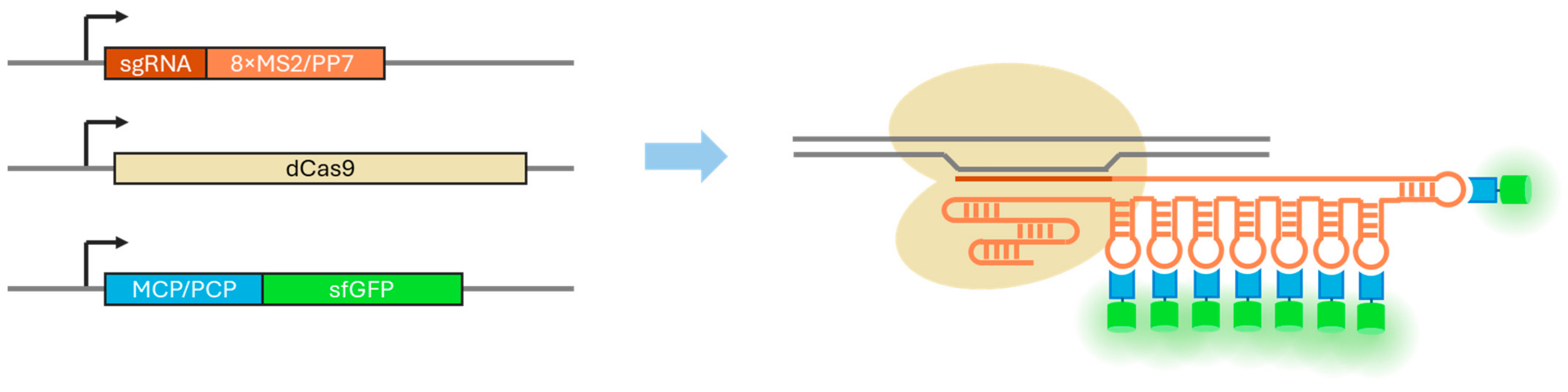
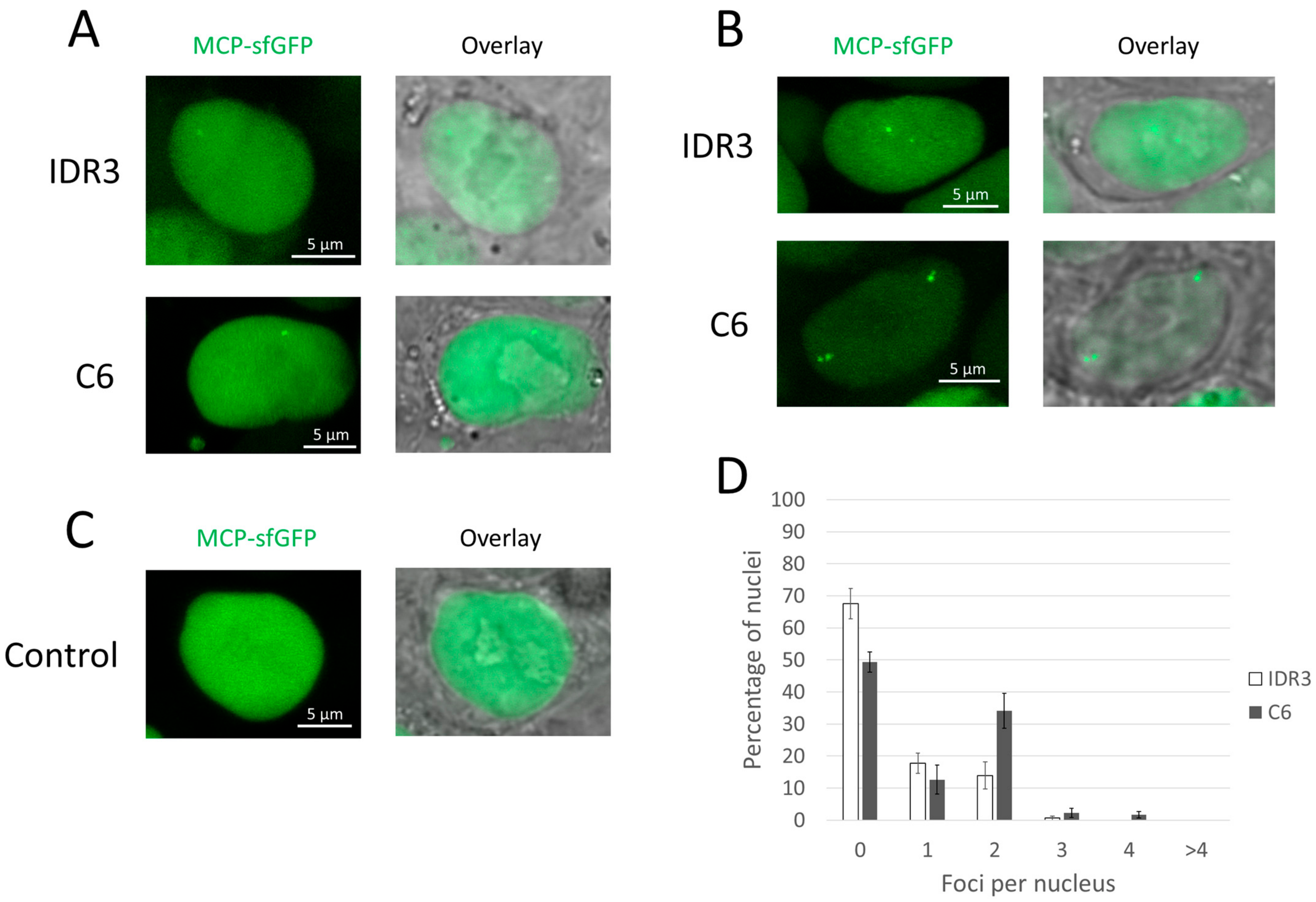
3.1. Evaluation of the PCP Version of the CRISPR-Sirius System
3.2. Evaluation of the MCP Version of the CRISPR-Sirius System
3.3. Evaluation of the Imaging Performance Using a Single Guide RNA per Locus
3.4. Analysis of the Expression of sgRNAs and Stem-Loop-Binding Proteins in Two Versions of the CRISPR-Sirius System
3.5. Expanding the Set of Target Loci—Visualizing the Boundaries of TADs
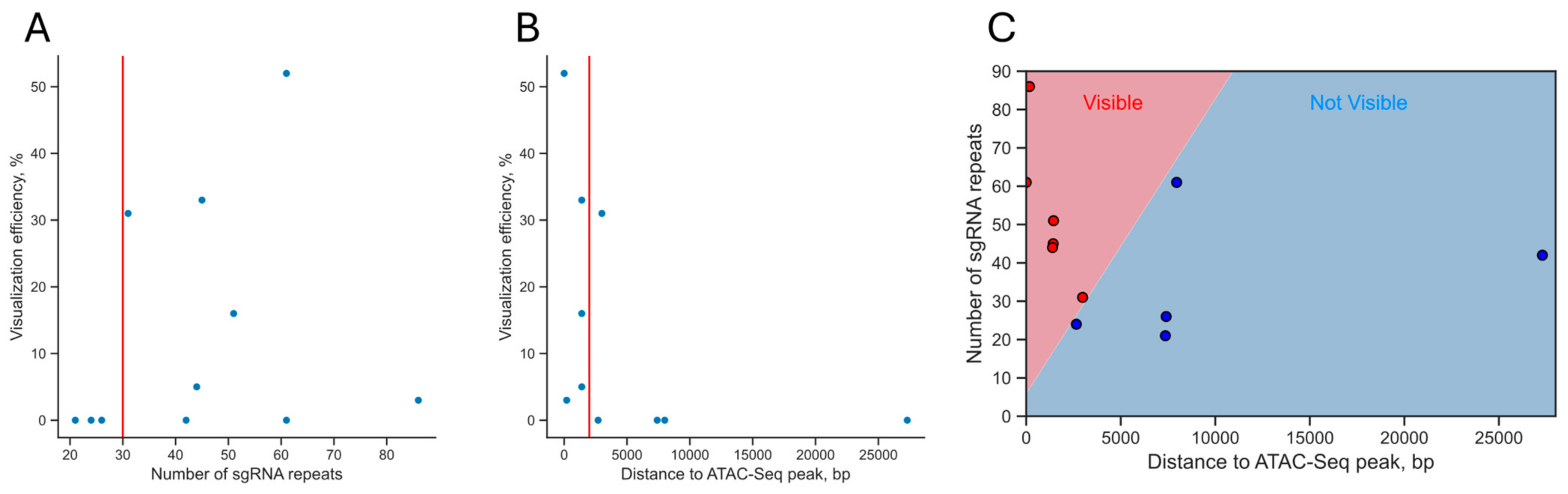
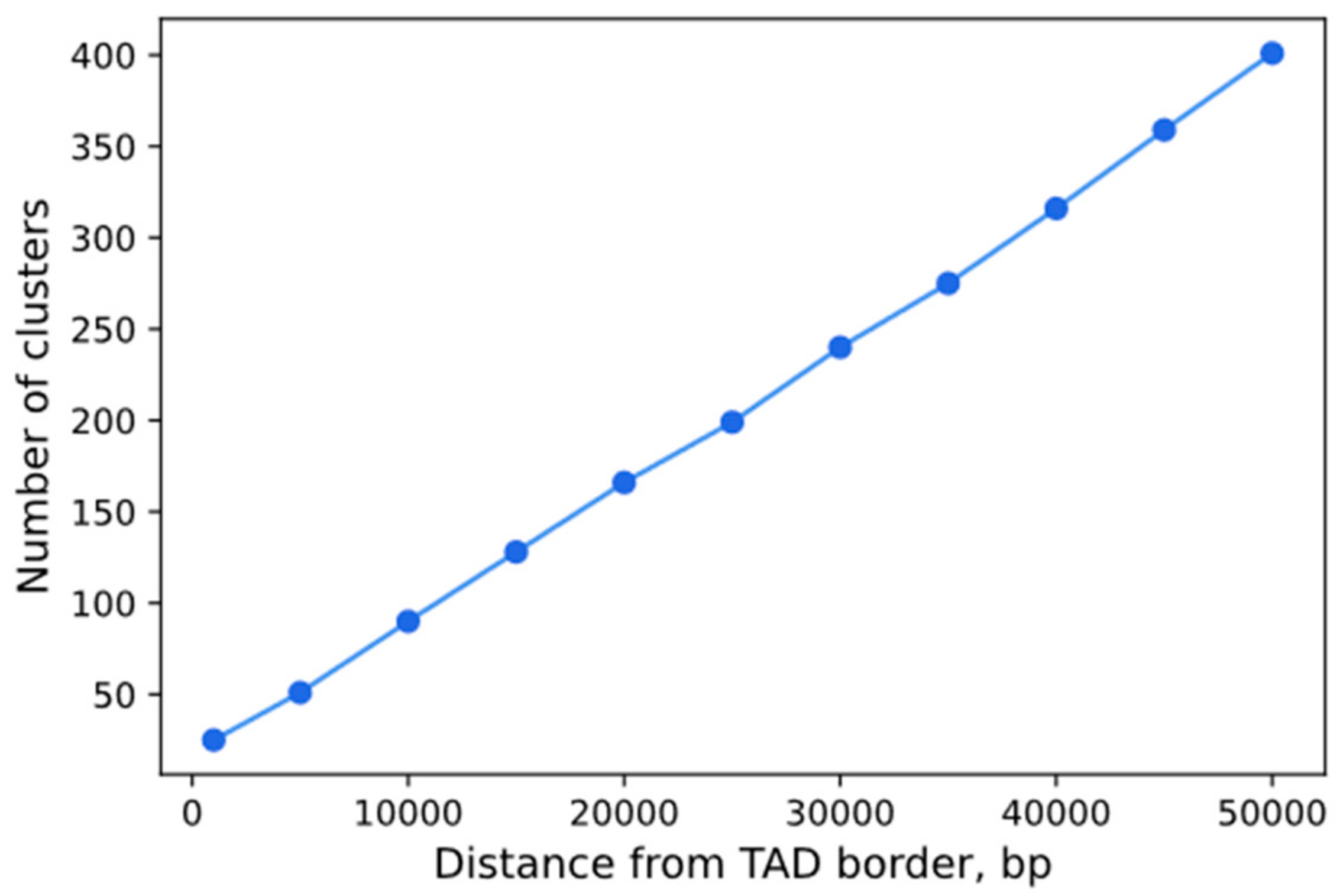
3.6. Analysis of the Dependence of the Visualization Efficiency on the Number of sgRNA Repeats in a Cluster and Epigenetic Factors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huang, S.; Dai, R.; Zhang, Z.; Zhang, H.; Zhang, M.; Li, Z.; Zhao, K.; Xiong, W.; Cheng, S.; Wang, B.; et al. CRISPR/Cas-Based Techniques for Live-Cell Imaging and Bioanalysis. Int. J. Mol. Sci. 2023, 24, 13447. [Google Scholar] [CrossRef] [PubMed]
- Van Staalduinen, J.; van Staveren, T.; Grosveld, F.; Wendt, K.S. Live-cell imaging of chromatin contacts opens a new window into chromatin dynamics. Epigenet. Chromatin 2023, 16, 27. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Hou, Y.; Zhang, X.E.; Gao, Y. Live cell imaging of DNA and RNA with fluorescent signal amplification and background reduction techniques. Front. Cell Dev. Biol. 2023, 11, 1216232. [Google Scholar] [CrossRef] [PubMed]
- Maloshenok, L.G.; Abushinova, G.A.; Ryazanova, A.Y.; Bruskin, S.A.; Zherdeva, V.V. Visualizing the Nucleome Using the CRISPR-Cas9 System: From in vitro to in vivo. Biochemistry 2023, 88, S123–S149. [Google Scholar] [CrossRef]
- Thuma, J.; Chung, Y.C.; Tu, L.C. Advances and challenges in CRISPR-based real-time imaging of dynamic genome organization. Front. Mol. Biosci. 2023, 10, 1173545. [Google Scholar] [CrossRef]
- Viushkov, V.S.; Lomov, N.A.; Rubtsov, M.A.; Vassetzky, Y.S. Visualizing the Genome: Experimental Approaches for Live-Cell Chromatin Imaging. Cells 2022, 11, 4086. [Google Scholar] [CrossRef]
- Sato, H.; Das, S.; Singer, R.H.; Vera, M. Imaging of DNA and RNA in Living Eukaryotic Cells to Reveal Spatiotemporal Dynamics of Gene Expression. Annu. Rev. Biochem. 2020, 89, 159–187. [Google Scholar] [CrossRef]
- Clow, P.A.; Du, M.; Jillette, N.; Taghbalout, A.; Zhu, J.J.; Cheng, A.W. CRISPR-mediated multiplexed live cell imaging of nonrepetitive genomic loci with one guide RNA per locus. Nat. Commun. 2022, 13, 1871. [Google Scholar] [CrossRef]
- Chen, B.; Gilbert, L.A.; Cimini, B.A.; Schnitzbauer, J.; Zhang, W.; Li, G.W.; Park, J.; Blackburn, E.H.; Weissman, J.S.; Qi, L.S.; et al. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell 2013, 155, 1479–1491. [Google Scholar] [CrossRef]
- Tanenbaum, M.E.; Gilbert, L.A.; Qi, L.S.; Weissman, J.S.; Vale, R.D. A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell 2014, 159, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Naseri, A.; Reyes-Gutierrez, P.; Wolfe, S.A.; Zhang, S.; Pederson, T. Multicolor CRISPR labeling of chromosomal loci in human cells. Proc. Natl. Acad. Sci. USA 2015, 112, 3002–3007. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Hu, J.; Almeida, R.; Liu, H.; Balakrishnan, S.; Covill-Cooke, C.; Lim, W.A.; Huang, B. Expanding the CRISPR imaging toolset with Staphylococcus aureus Cas9 for simultaneous imaging of multiple genomic loci. Nucleic Acids Res. 2016, 44, e75. [Google Scholar] [CrossRef]
- Chen, B.; Zou, W.; Xu, H.; Liang, Y.; Huang, B. Efficient labeling and imaging of protein-coding genes in living cells using CRISPR-Tag. Nat. Commun. 2018, 9, 5065. [Google Scholar] [CrossRef]
- Hong, Y.; Lu, G.; Duan, J.; Liu, W.; Zhang, Y. Comparison and optimization of CRISPR/dCas9/gRNA genome-labeling systems for live cell imaging. Genome Biol. 2018, 19, 39. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, N.; Nho, S.H.; Cho, H.; Gantumur, N.; Ra, J.S.; Myung, K.; Kim, H. Background-suppressed live visualization of genomic loci with an improved CRISPR system based on a split fluorophore. Genome Res. 2020, 30, 1306–1316. [Google Scholar] [CrossRef] [PubMed]
- Shao, S.; Zhang, W.; Hu, H.; Xue, B.; Qin, J.; Sun, C.; Sun, Y.; Wei, W.; Sun, Y. Long-term dual-color tracking of genomic loci by modified sgRNAs of the CRISPR/Cas9 system. Nucleic Acids Res. 2016, 44, e86. [Google Scholar] [CrossRef]
- Wang, S.; Su, J.H.; Zhang, F.; Zhuang, X. An RNA-aptamer-based two-color CRISPR labeling system. Sci. Rep. 2016, 6, 26857. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Rocha, P.P.; Luo, V.M.; Raviram, R.; Deng, Y.; Mazzoni, E.O.; Skok, J.A. CRISPR-dCas9 and sgRNA scaffolds enable dual-colour live imaging of satellite sequences and repeat-enriched individual loci. Nat. Commun. 2016, 7, 11707. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Tu, L.C.; Naseri, A.; Huisman, M.; Zhang, S.; Grunwald, D.; Pederson, T. Multiplexed labeling of genomic loci with dCas9 and engineered sgRNAs using CRISPRainbow. Nat. Biotechnol. 2016, 34, 528–530. [Google Scholar] [CrossRef]
- Cheng, A.W.; Jillette, N.; Lee, P.; Plaskon, D.; Fujiwara, Y.; Wang, W.; Taghbalout, A.; Wang, H. Casilio: A versatile CRISPR-Cas9-Pumilio hybrid for gene regulation and genomic labeling. Cell Res. 2016, 26, 254–257. [Google Scholar] [CrossRef]
- Qin, P.; Parlak, M.; Kuscu, C.; Bandaria, J.; Mir, M.; Szlachta, K.; Singh, R.; Darzacq, X.; Yildiz, A.; Adli, M. Live cell imaging of low- and non-repetitive chromosome loci using CRISPR-Cas9. Nat. Commun. 2017, 8, 14725. [Google Scholar] [CrossRef]
- Ma, H.; Tu, L.C.; Naseri, A.; Chung, Y.C.; Grunwald, D.; Zhang, S.; Pederson, T. CRISPR-Sirius: RNA scaffolds for signal amplification in genome imaging. Nat. Methods 2018, 15, 928–931. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Tu, L.C.; Chung, Y.C.; Naseri, A.; Grunwald, D.; Zhang, S.; Pederson, T. Cell cycle-and genomic distance-dependent dynamics of a discrete chromosomal region. J. Cell Biol. 2019, 218, 1467–1477. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.C.; Bisht, M.; Thuma, J.; Tu, L.C. Single-chromosome dynamics reveals locus-dependent dynamics and chromosome territory orientation. J. Cell Sci. 2023, 136, jcs260137. [Google Scholar] [CrossRef] [PubMed]
- Rowley, M.J.; Corces, V.G. Organizational principles of 3D genome architecture. Nat. Rev. Genet. 2018, 19, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Nora, E.P.; Lajoie, B.R.; Schulz, E.G.; Giorgetti, L.; Okamoto, I.; Servant, N.; Piolot, T.; van Berkum, N.L.; Meisig, J.; Sedat, J.; et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 2012, 485, 381–385. [Google Scholar] [CrossRef]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef]
- Rao, S.S.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef]
- Flyamer, I.M.; Gassler, J.; Imakaev, M.; Brandao, H.B.; Ulianov, S.V.; Abdennur, N.; Razin, S.V.; Mirny, L.A.; Tachibana-Konwalski, K. Single-nucleus Hi-C reveals unique chromatin reorganization at oocyte-to-zygote transition. Nature 2017, 544, 110–114. [Google Scholar] [CrossRef]
- Bintu, B.; Mateo, L.J.; Su, J.H.; Sinnott-Armstrong, N.A.; Parker, M.; Kinrot, S.; Yamaya, K.; Boettiger, A.N.; Zhuang, X. Super-resolution chromatin tracing reveals domains and cooperative interactions in single cells. Science 2018, 362, eaau1783. [Google Scholar] [CrossRef]
- Fudenberg, G.; Abdennur, N.; Imakaev, M.; Goloborodko, A.; Mirny, L.A. Emerging Evidence of Chromosome Folding by Loop Extrusion. Cold Spring Harb. Symp. Quant. Biol. 2017, 82, 45–55. [Google Scholar] [CrossRef]
- Fudenberg, G.; Imakaev, M.; Lu, C.; Goloborodko, A.; Abdennur, N.; Mirny, L.A. Formation of Chromosomal Domains by Loop Extrusion. Cell Rep. 2016, 15, 2038–2049. [Google Scholar] [CrossRef] [PubMed]
- Gassler, J.; Brandao, H.B.; Imakaev, M.; Flyamer, I.M.; Ladstatter, S.; Bickmore, W.A.; Peters, J.M.; Mirny, L.A.; Tachibana, K. A mechanism of cohesin-dependent loop extrusion organizes zygotic genome architecture. EMBO J. 2017, 36, 3600–3618. [Google Scholar] [CrossRef]
- Rao, S.S.P.; Huang, S.C.; Glenn St Hilaire, B.; Engreitz, J.M.; Perez, E.M.; Kieffer-Kwon, K.R.; Sanborn, A.L.; Johnstone, S.E.; Bascom, G.D.; Bochkov, I.D.; et al. Cohesin Loss Eliminates All Loop Domains. Cell 2017, 171, 305–320.e324. [Google Scholar] [CrossRef]
- Wutz, G.; Varnai, C.; Nagasaka, K.; Cisneros, D.A.; Stocsits, R.R.; Tang, W.; Schoenfelder, S.; Jessberger, G.; Muhar, M.; Hossain, M.J.; et al. Topologically associating domains and chromatin loops depend on cohesin and are regulated by CTCF, WAPL, and PDS5 proteins. EMBO J. 2017, 36, 3573–3599. [Google Scholar] [CrossRef]
- Nora, E.P.; Goloborodko, A.; Valton, A.L.; Gibcus, J.H.; Uebersohn, A.; Abdennur, N.; Dekker, J.; Mirny, L.A.; Bruneau, B.G. Targeted Degradation of CTCF Decouples Local Insulation of Chromosome Domains from Genomic Compartmentalization. Cell 2017, 169, 930–944.e22. [Google Scholar] [CrossRef]
- Haarhuis, J.H.I.; van der Weide, R.H.; Blomen, V.A.; Yanez-Cuna, J.O.; Amendola, M.; van Ruiten, M.S.; Krijger, P.H.L.; Teunissen, H.; Medema, R.H.; van Steensel, B.; et al. The Cohesin Release Factor WAPL Restricts Chromatin Loop Extension. Cell 2017, 169, 693–707.e14. [Google Scholar] [CrossRef] [PubMed]
- Gabriele, M.; Brandao, H.B.; Grosse-Holz, S.; Jha, A.; Dailey, G.M.; Cattoglio, C.; Hsieh, T.S.; Mirny, L.; Zechner, C.; Hansen, A.S. Dynamics of CTCF- and cohesin-mediated chromatin looping revealed by live-cell imaging. Science 2022, 376, 496–501. [Google Scholar] [CrossRef] [PubMed]
- Mach, P.; Kos, P.I.; Zhan, Y.; Cramard, J.; Gaudin, S.; Tunnermann, J.; Marchi, E.; Eglinger, J.; Zuin, J.; Kryzhanovska, M.; et al. Cohesin and CTCF control the dynamics of chromosome folding. Nat. Genet. 2022, 54, 1907–1918. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Durand, N.C.; Robinson, J.T.; Shamim, M.S.; Machol, I.; Mesirov, J.P.; Lander, E.S.; Aiden, E.L. Juicebox Provides a Visualization System for Hi-C Contact Maps with Unlimited Zoom. Cell Syst. 2016, 3, 99–101. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Sun, Z.; Wang, J.; Huang, H.; Kocher, J.P.; Wang, L. CrossMap: A versatile tool for coordinate conversion between genome assemblies. Bioinformatics 2014, 30, 1006–1007. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdottir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Ernst, J.; Kellis, M. ChromHMM: Automating chromatin-state discovery and characterization. Nat. Methods 2012, 9, 215–216. [Google Scholar] [CrossRef]
- Ernst, J.; Kellis, M. Chromatin-state discovery and genome annotation with ChromHMM. Nat. Protoc. 2017, 12, 2478–2492. [Google Scholar] [CrossRef] [PubMed]
- Buenrostro, J.D.; Giresi, P.G.; Zaba, L.C.; Chang, H.Y.; Greenleaf, W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 2013, 10, 1213–1218. [Google Scholar] [CrossRef]
- Konstantakos, V.; Nentidis, A.; Krithara, A.; Paliouras, G. CRISPR-Cas9 gRNA efficiency prediction: An overview of predictive tools and the role of deep learning. Nucleic Acids Res. 2022, 50, 3616–3637. [Google Scholar] [CrossRef]
- Wang, H.; Nakamura, M.; Abbott, T.R.; Zhao, D.; Luo, K.; Yu, C.; Nguyen, C.M.; Lo, A.; Daley, T.P.; La Russa, M.; et al. CRISPR-mediated live imaging of genome editing and transcription. Science 2019, 365, 1301–1305. [Google Scholar] [CrossRef]
- Motoche-Monar, C.; Ordonez, J.E.; Chang, O.; Gonzales-Zubiate, F.A. gRNA Design: How Its Evolution Impacted on CRISPR/Cas9 Systems Refinement. Biomolecules 2023, 13, 1698. [Google Scholar] [CrossRef]
- Fu, Y.; Sander, J.D.; Reyon, D.; Cascio, V.M.; Joung, J.K. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat. Biotechnol. 2014, 32, 279–284. [Google Scholar] [CrossRef]
- Lin, Y.; Cradick, T.J.; Brown, M.T.; Deshmukh, H.; Ranjan, P.; Sarode, N.; Wile, B.M.; Vertino, P.M.; Stewart, F.J.; Bao, G. CRISPR/Cas9 systems have off-target activity with insertions or deletions between target DNA and guide RNA sequences. Nucleic Acids Res. 2014, 42, 7473–7485. [Google Scholar] [CrossRef] [PubMed]
- Dahlman, J.E.; Abudayyeh, O.O.; Joung, J.; Gootenberg, J.S.; Zhang, F.; Konermann, S. Orthogonal gene knockout and activation with a catalytically active Cas9 nuclease. Nat. Biotechnol. 2015, 33, 1159–1161. [Google Scholar] [CrossRef] [PubMed]
- Kiani, S.; Chavez, A.; Tuttle, M.; Hall, R.N.; Chari, R.; Ter-Ovanesyan, D.; Qian, J.; Pruitt, B.W.; Beal, J.; Vora, S.; et al. Cas9 gRNA engineering for genome editing, activation and repression. Nat. Methods 2015, 12, 1051–1054. [Google Scholar] [CrossRef]
- Zhang, J.P.; Li, X.L.; Neises, A.; Chen, W.; Hu, L.P.; Ji, G.Z.; Yu, J.Y.; Xu, J.; Yuan, W.P.; Cheng, T.; et al. Different Effects of sgRNA Length on CRISPR-mediated Gene Knockout Efficiency. Sci. Rep. 2016, 6, 28566. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.; Wu, S.; Wei, R.; Li, Y.; Jin, J.; Mu, Y.; Zhang, Y.; Kong, Q.; Weng, X.; Liu, Z. The length of guide RNA and target DNA heteroduplex effects on CRISPR/Cas9 mediated genome editing efficiency in porcine cells. J. Vet. Sci. 2019, 20, e23. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Primer Sequence | Task |
|---|---|---|
| T2A_BamH_f | AATAAGGATCCGAGGGCAGAGGAAGTCTTCTAACAT | Replacing the P2A-HSA fragment in the dCas9 vector with the T2A-Puro fragment |
| Puro_Xba_r | AATAATCTAGATAGATCAGGCACCGGGCTT | |
| sfGFP_BamH_f | ATTAAGGATCCATGCGTAAAGGCGAAGAGCT | Replacing the HaloTag with sfGFP in a plasmid with the MCP gene |
| sfGFP_Xho_r | TAATTCTCGAGTTTGTACAGTTCATCCATACCATGCG |
| Target Locus 1 | sgRNA Recognition Sequence |
|---|---|
| IDR3 (chr19: 380,836–382,654) | AGCAGATGTAGG (45) 2 |
| C6 (chr6: 157,310,367–157,314,361) | sg1: GTGAGTGCACAC (22) sg2: TGGGACACTATGATG (39) |
| Target | PCP-sfGFP Version | MCP-sfGFP Version | ||
|---|---|---|---|---|
| Transient Transfection | Stable Transduction | Transient Transfection | Stable Transduction | |
| IDR3 | 6% (n = 130) 1 | 26% (n = 124) | 13% (n = 245) | 33% (n = 138) |
| C6 | 3% (n = 132) | 20% (n = 102) | 9% (n = 127) | 52% (n = 131) |
| Target Locus 1 | sgRNA Recognition Sequence |
|---|---|
| C6 (chr6: 157,310,367–157,314,361) | sg1: GTGAGTGCACAC (22) 2 sg2: TGGGACACTATGATG (39) |
| 6T1_L (chr6: 168,378,356–168,380,872) | sg1: ACTCGGGCTGTG (35) sg2: CTGTGTGGGACT (26) |
| 6T1_R (chr6: 168,849,859–168,850,601) | sg1: GCAGAGGTGGCA (22) sg2: TGTGGGCAGAGG (20) |
| 6T2_L (chr6: 169,781,629–169,782,955 for sg1, and chr6: 169,803,533–169,807,849 for sg2) | sg1: ACCACTCGGAAA (21) sg2: GCTCTGTGTCTG (24) |
| 6T2_R (chr6: 170,500,882–170,504,181 for sg1, and chr6: 170,507,778–170,509,799 for sg2) | sg1: CTGCAGCCATCA (31) sg2: CACTCATTCAGC (26) |
| 4T (chr4: 186,033,845–186,035,464) | sg1: CCCTGAGGGATT (22) sg2: TCTGTACCCTGA (29) |
| 5T (chr5: 1,781,394–1,781,810) | sg1: AGGCTGAGGGTG (21) sg2: AGGGTGAGGCTG (23) |
| 22T (chr22: 47,211,081–47,213,579) | sg1: CATATTTGAGTG (56) sg2: GGACGGTCAGTG (30) |
| Target | PCP-sfGFP Version | MCP-sfGFP Version | Proportion of Cells with Two Signals 2 | Signal-to-Background Ratio (Median) |
|---|---|---|---|---|
| C6 | 20% (n = 102) 1 | 52% (n = 131) | 34% | 2.3 |
| 6T1_L | 0% (n = 118) | 0% (n = 113) | 0% | n.a. |
| 6T1_R | 0% (n = 121) | 0% (n = 166) | 0% | n.a. |
| 6T2_L | 0% (n = 108) | 0% (n = 122) | 0% | n.a. |
| 6T2_R | 0% (n = 134) | 29% (n = 159) | 8% | 1.5 |
| 4T | 0% (n = 127) | 16% (n = 117) | 4% | 1.4 |
| 5T | 0% (n = 123) | 5% (n = 129) | 0% | 1.5 |
| 22T | 0% (n = 158) | 3% (n = 116) | 0% | 1.4 |
| Repeat Cluster | Visualization Efficiency (MCP-sfGFP Version) | Number of sgRNA Repeats | Chromatin Status (ChromHMM18) | Transcription | Hi-C Compartment |
|---|---|---|---|---|---|
| IDR3 | 33% 1 | 45 | Quiescent | No | A |
| C6 | 52% | 61 | Quiescent | Weak | A |
| 6T1_L | 0% | 61 | Quiescent | No | A |
| 6T1_R | 0% | 42 | Quiescent | No | A |
| 6T2_L_sg1 | 0% | 21 | Quiescent | No | A |
| 6T2_L_sg2 | 0% | 24 | Repressed Polycomb/weakly repressed Polycomb | No | A |
| 6T2_R_sg1 | 31% | 31 | Weakly Polycomb repressed | No | A |
| 6T2_R_sg2 | 0% | 26 | Quiescent | No | A |
| 4T | 16% | 51 | Quiescent | No | A |
| 5T | 5% | 44 | Quiescent | No | A |
| 22T | 3% | 86 | Quiescent | No | B |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Viushkov, V.S.; Lomov, N.A.; Rubtsov, M.A. A Comparison of Two Versions of the CRISPR-Sirius System for the Live-Cell Visualization of the Borders of Topologically Associating Domains. Cells 2024, 13, 1440. https://doi.org/10.3390/cells13171440
Viushkov VS, Lomov NA, Rubtsov MA. A Comparison of Two Versions of the CRISPR-Sirius System for the Live-Cell Visualization of the Borders of Topologically Associating Domains. Cells. 2024; 13(17):1440. https://doi.org/10.3390/cells13171440
Chicago/Turabian StyleViushkov, Vladimir S., Nikolai A. Lomov, and Mikhail A. Rubtsov. 2024. "A Comparison of Two Versions of the CRISPR-Sirius System for the Live-Cell Visualization of the Borders of Topologically Associating Domains" Cells 13, no. 17: 1440. https://doi.org/10.3390/cells13171440
APA StyleViushkov, V. S., Lomov, N. A., & Rubtsov, M. A. (2024). A Comparison of Two Versions of the CRISPR-Sirius System for the Live-Cell Visualization of the Borders of Topologically Associating Domains. Cells, 13(17), 1440. https://doi.org/10.3390/cells13171440