
A Comparison of Two Versions of the CRISPR-Sirius System for the Live-Cell Visualization of the Borders of Topologically Associating Domains


Abstract
1. Introduction
2. Materials and Methods
2.1. Plasmid Construction
2.2. Cell Culture and Transient Transfection
2.3. Lentivirus Production and Cell Transduction
2.4. FACS
2.5. Western Blot
2.6. Real-Time PCR
2.7. Live-Cell Microscopy
2.8. Graphical Representation of Hi-C and ChIP-Seq Data and Selection of Appropriate TAD Boundaries for Visualization
2.9. Epigenetic Data
3. Results
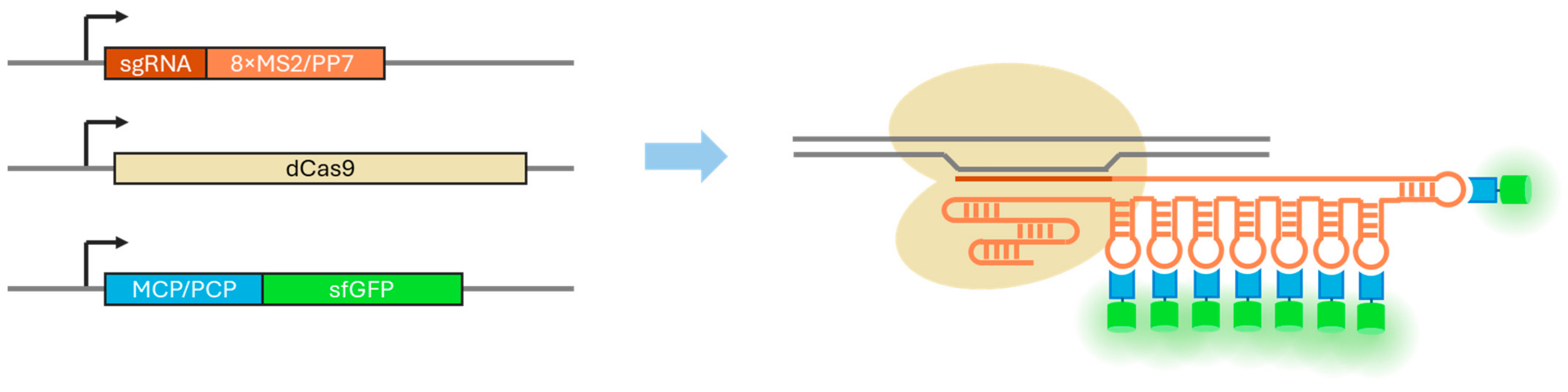
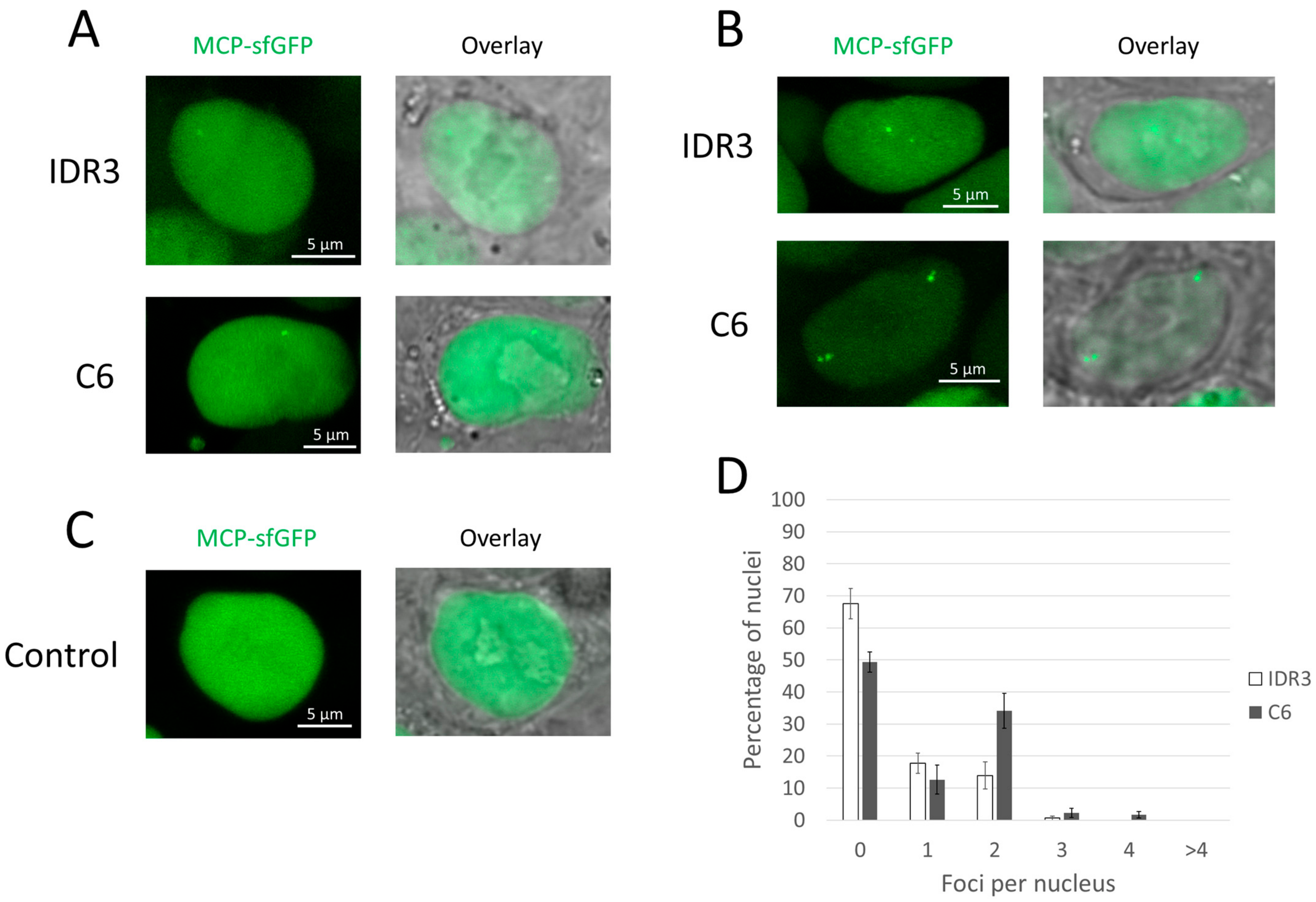
3.1. Evaluation of the PCP Version of the CRISPR-Sirius System
3.2. Evaluation of the MCP Version of the CRISPR-Sirius System
3.3. Evaluation of the Imaging Performance Using a Single Guide RNA per Locus
3.4. Analysis of the Expression of sgRNAs and Stem-Loop-Binding Proteins in Two Versions of the CRISPR-Sirius System
3.5. Expanding the Set of Target Loci—Visualizing the Boundaries of TADs
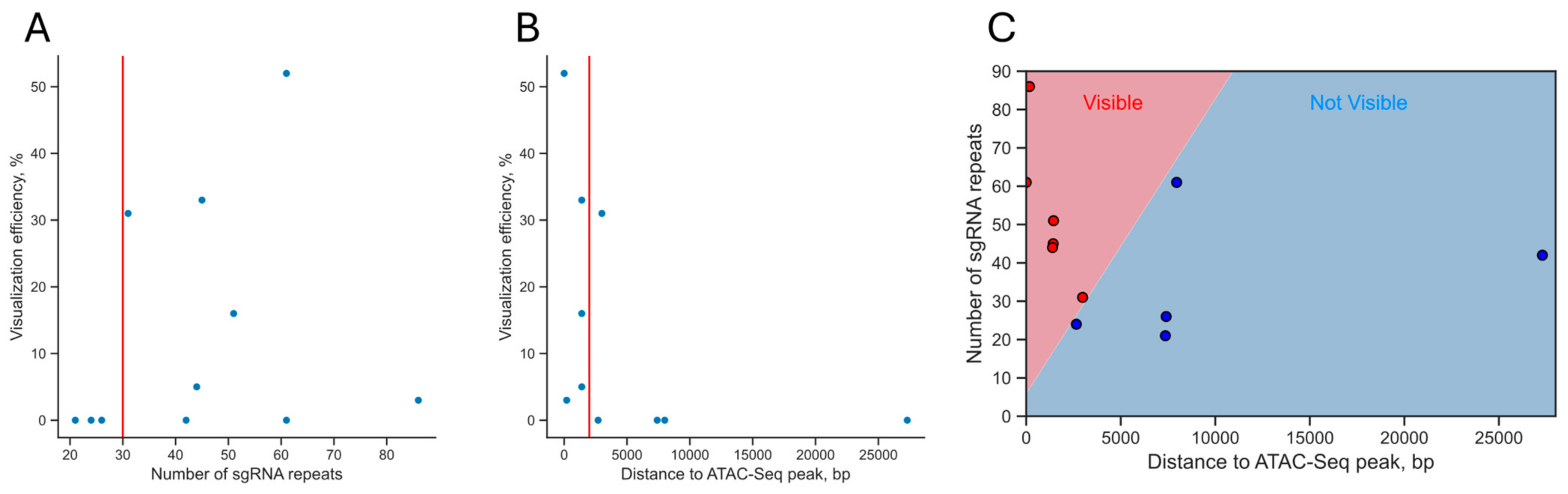
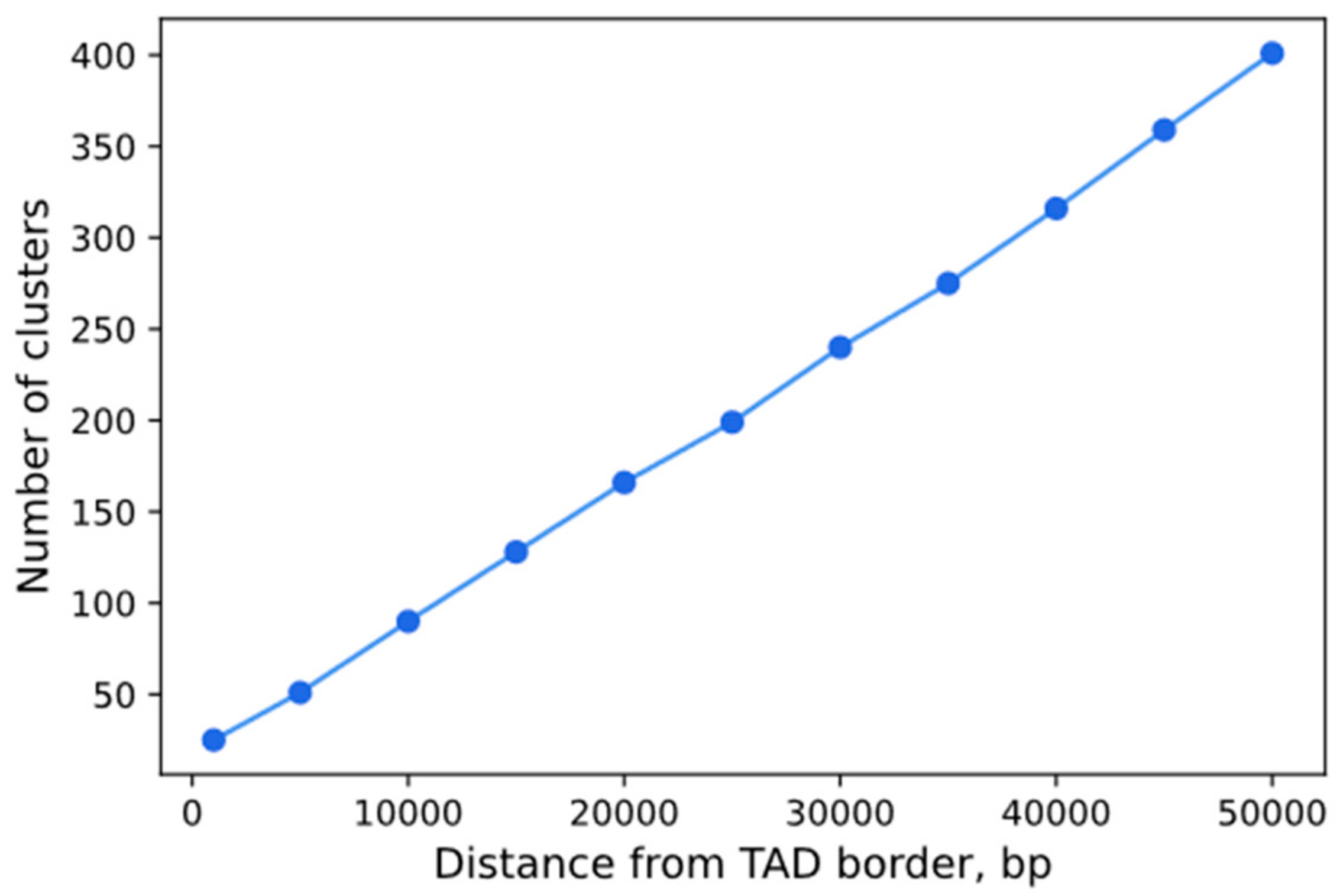
3.6. Analysis of the Dependence of the Visualization Efficiency on the Number of sgRNA Repeats in a Cluster and Epigenetic Factors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huang, S.; Dai, R.; Zhang, Z.; Zhang, H.; Zhang, M.; Li, Z.; Zhao, K.; Xiong, W.; Cheng, S.; Wang, B.; et al. CRISPR/Cas-Based Techniques for Live-Cell Imaging and Bioanalysis. Int. J. Mol. Sci. 2023, 24, 13447. [Google Scholar] [CrossRef] [PubMed]
- Van Staalduinen, J.; van Staveren, T.; Grosveld, F.; Wendt, K.S. Live-cell imaging of chromatin contacts opens a new window into chromatin dynamics. Epigenet. Chromatin 2023, 16, 27. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Hou, Y.; Zhang, X.E.; Gao, Y. Live cell imaging of DNA and RNA with fluorescent signal amplification and background reduction techniques. Front. Cell Dev. Biol. 2023, 11, 1216232. [Google Scholar] [CrossRef] [PubMed]
- Maloshenok, L.G.; Abushinova, G.A.; Ryazanova, A.Y.; Bruskin, S.A.; Zherdeva, V.V. Visualizing the Nucleome Using the CRISPR-Cas9 System: From in vitro to in vivo. Biochemistry 2023, 88, S123–S149. [Google Scholar] [CrossRef]
- Thuma, J.; Chung, Y.C.; Tu, L.C. Advances and challenges in CRISPR-based real-time imaging of dynamic genome organization. Front. Mol. Biosci. 2023, 10, 1173545. [Google Scholar] [CrossRef]
- Viushkov, V.S.; Lomov, N.A.; Rubtsov, M.A.; Vassetzky, Y.S. Visualizing the Genome: Experimental Approaches for Live-Cell Chromatin Imaging. Cells 2022, 11, 4086. [Google Scholar] [CrossRef]
- Sato, H.; Das, S.; Singer, R.H.; Vera, M. Imaging of DNA and RNA in Living Eukaryotic Cells to Reveal Spatiotemporal Dynamics of Gene Expression. Annu. Rev. Biochem. 2020, 89, 159–187. [Google Scholar] [CrossRef]
- Clow, P.A.; Du, M.; Jillette, N.; Taghbalout, A.; Zhu, J.J.; Cheng, A.W. CRISPR-mediated multiplexed live cell imaging of nonrepetitive genomic loci with one guide RNA per locus. Nat. Commun. 2022, 13, 1871. [Google Scholar] [CrossRef]
- Chen, B.; Gilbert, L.A.; Cimini, B.A.; Schnitzbauer, J.; Zhang, W.; Li, G.W.; Park, J.; Blackburn, E.H.; Weissman, J.S.; Qi, L.S.; et al. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell 2013, 155, 1479–1491. [Google Scholar] [CrossRef]
- Tanenbaum, M.E.; Gilbert, L.A.; Qi, L.S.; Weissman, J.S.; Vale, R.D. A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell 2014, 159, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Naseri, A.; Reyes-Gutierrez, P.; Wolfe, S.A.; Zhang, S.; Pederson, T. Multicolor CRISPR labeling of chromosomal loci in human cells. Proc. Natl. Acad. Sci. USA 2015, 112, 3002–3007. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Hu, J.; Almeida, R.; Liu, H.; Balakrishnan, S.; Covill-Cooke, C.; Lim, W.A.; Huang, B. Expanding the CRISPR imaging toolset with Staphylococcus aureus Cas9 for simultaneous imaging of multiple genomic loci. Nucleic Acids Res. 2016, 44, e75. [Google Scholar] [CrossRef]
- Chen, B.; Zou, W.; Xu, H.; Liang, Y.; Huang, B. Efficient labeling and imaging of protein-coding genes in living cells using CRISPR-Tag. Nat. Commun. 2018, 9, 5065. [Google Scholar] [CrossRef]
- Hong, Y.; Lu, G.; Duan, J.; Liu, W.; Zhang, Y. Comparison and optimization of CRISPR/dCas9/gRNA genome-labeling systems for live cell imaging. Genome Biol. 2018, 19, 39. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, N.; Nho, S.H.; Cho, H.; Gantumur, N.; Ra, J.S.; Myung, K.; Kim, H. Background-suppressed live visualization of genomic loci with an improved CRISPR system based on a split fluorophore. Genome Res. 2020, 30, 1306–1316. [Google Scholar] [CrossRef] [PubMed]
- Shao, S.; Zhang, W.; Hu, H.; Xue, B.; Qin, J.; Sun, C.; Sun, Y.; Wei, W.; Sun, Y. Long-term dual-color tracking of genomic loci by modified sgRNAs of the CRISPR/Cas9 system. Nucleic Acids Res. 2016, 44, e86. [Google Scholar] [CrossRef]
- Wang, S.; Su, J.H.; Zhang, F.; Zhuang, X. An RNA-aptamer-based two-color CRISPR labeling system. Sci. Rep. 2016, 6, 26857. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Rocha, P.P.; Luo, V.M.; Raviram, R.; Deng, Y.; Mazzoni, E.O.; Skok, J.A. CRISPR-dCas9 and sgRNA scaffolds enable dual-colour live imaging of satellite sequences and repeat-enriched individual loci. Nat. Commun. 2016, 7, 11707. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Tu, L.C.; Naseri, A.; Huisman, M.; Zhang, S.; Grunwald, D.; Pederson, T. Multiplexed labeling of genomic loci with dCas9 and engineered sgRNAs using CRISPRainbow. Nat. Biotechnol. 2016, 34, 528–530. [Google Scholar] [CrossRef]
- Cheng, A.W.; Jillette, N.; Lee, P.; Plaskon, D.; Fujiwara, Y.; Wang, W.; Taghbalout, A.; Wang, H. Casilio: A versatile CRISPR-Cas9-Pumilio hybrid for gene regulation and genomic labeling. Cell Res. 2016, 26, 254–257. [Google Scholar] [CrossRef]
- Qin, P.; Parlak, M.; Kuscu, C.; Bandaria, J.; Mir, M.; Szlachta, K.; Singh, R.; Darzacq, X.; Yildiz, A.; Adli, M. Live cell imaging of low- and non-repetitive chromosome loci using CRISPR-Cas9. Nat. Commun. 2017, 8, 14725. [Google Scholar] [CrossRef]
- Ma, H.; Tu, L.C.; Naseri, A.; Chung, Y.C.; Grunwald, D.; Zhang, S.; Pederson, T. CRISPR-Sirius: RNA scaffolds for signal amplification in genome imaging. Nat. Methods 2018, 15, 928–931. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Tu, L.C.; Chung, Y.C.; Naseri, A.; Grunwald, D.; Zhang, S.; Pederson, T. Cell cycle-and genomic distance-dependent dynamics of a discrete chromosomal region. J. Cell Biol. 2019, 218, 1467–1477. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.C.; Bisht, M.; Thuma, J.; Tu, L.C. Single-chromosome dynamics reveals locus-dependent dynamics and chromosome territory orientation. J. Cell Sci. 2023, 136, jcs260137. [Google Scholar] [CrossRef] [PubMed]
- Rowley, M.J.; Corces, V.G. Organizational principles of 3D genome architecture. Nat. Rev. Genet. 2018, 19, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Nora, E.P.; Lajoie, B.R.; Schulz, E.G.; Giorgetti, L.; Okamoto, I.; Servant, N.; Piolot, T.; van Berkum, N.L.; Meisig, J.; Sedat, J.; et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 2012, 485, 381–385. [Google Scholar] [CrossRef]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef]
- Rao, S.S.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef]
- Flyamer, I.M.; Gassler, J.; Imakaev, M.; Brandao, H.B.; Ulianov, S.V.; Abdennur, N.; Razin, S.V.; Mirny, L.A.; Tachibana-Konwalski, K. Single-nucleus Hi-C reveals unique chromatin reorganization at oocyte-to-zygote transition. Nature 2017, 544, 110–114. [Google Scholar] [CrossRef]
- Bintu, B.; Mateo, L.J.; Su, J.H.; Sinnott-Armstrong, N.A.; Parker, M.; Kinrot, S.; Yamaya, K.; Boettiger, A.N.; Zhuang, X. Super-resolution chromatin tracing reveals domains and cooperative interactions in single cells. Science 2018, 362, eaau1783. [Google Scholar] [CrossRef]
- Fudenberg, G.; Abdennur, N.; Imakaev, M.; Goloborodko, A.; Mirny, L.A. Emerging Evidence of Chromosome Folding by Loop Extrusion. Cold Spring Harb. Symp. Quant. Biol. 2017, 82, 45–55. [Google Scholar] [CrossRef]
- Fudenberg, G.; Imakaev, M.; Lu, C.; Goloborodko, A.; Abdennur, N.; Mirny, L.A. Formation of Chromosomal Domains by Loop Extrusion. Cell Rep. 2016, 15, 2038–2049. [Google Scholar] [CrossRef] [PubMed]
- Gassler, J.; Brandao, H.B.; Imakaev, M.; Flyamer, I.M.; Ladstatter, S.; Bickmore, W.A.; Peters, J.M.; Mirny, L.A.; Tachibana, K. A mechanism of cohesin-dependent loop extrusion organizes zygotic genome architecture. EMBO J. 2017, 36, 3600–3618. [Google Scholar] [CrossRef]
- Rao, S.S.P.; Huang, S.C.; Glenn St Hilaire, B.; Engreitz, J.M.; Perez, E.M.; Kieffer-Kwon, K.R.; Sanborn, A.L.; Johnstone, S.E.; Bascom, G.D.; Bochkov, I.D.; et al. Cohesin Loss Eliminates All Loop Domains. Cell 2017, 171, 305–320.e324. [Google Scholar] [CrossRef]
- Wutz, G.; Varnai, C.; Nagasaka, K.; Cisneros, D.A.; Stocsits, R.R.; Tang, W.; Schoenfelder, S.; Jessberger, G.; Muhar, M.; Hossain, M.J.; et al. Topologically associating domains and chromatin loops depend on cohesin and are regulated by CTCF, WAPL, and PDS5 proteins. EMBO J. 2017, 36, 3573–3599. [Google Scholar] [CrossRef]
- Nora, E.P.; Goloborodko, A.; Valton, A.L.; Gibcus, J.H.; Uebersohn, A.; Abdennur, N.; Dekker, J.; Mirny, L.A.; Bruneau, B.G. Targeted Degradation of CTCF Decouples Local Insulation of Chromosome Domains from Genomic Compartmentalization. Cell 2017, 169, 930–944.e22. [Google Scholar] [CrossRef]
- Haarhuis, J.H.I.; van der Weide, R.H.; Blomen, V.A.; Yanez-Cuna, J.O.; Amendola, M.; van Ruiten, M.S.; Krijger, P.H.L.; Teunissen, H.; Medema, R.H.; van Steensel, B.; et al. The Cohesin Release Factor WAPL Restricts Chromatin Loop Extension. Cell 2017, 169, 693–707.e14. [Google Scholar] [CrossRef] [PubMed]
- Gabriele, M.; Brandao, H.B.; Grosse-Holz, S.; Jha, A.; Dailey, G.M.; Cattoglio, C.; Hsieh, T.S.; Mirny, L.; Zechner, C.; Hansen, A.S. Dynamics of CTCF- and cohesin-mediated chromatin looping revealed by live-cell imaging. Science 2022, 376, 496–501. [Google Scholar] [CrossRef] [PubMed]
- Mach, P.; Kos, P.I.; Zhan, Y.; Cramard, J.; Gaudin, S.; Tunnermann, J.; Marchi, E.; Eglinger, J.; Zuin, J.; Kryzhanovska, M.; et al. Cohesin and CTCF control the dynamics of chromosome folding. Nat. Genet. 2022, 54, 1907–1918. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Durand, N.C.; Robinson, J.T.; Shamim, M.S.; Machol, I.; Mesirov, J.P.; Lander, E.S.; Aiden, E.L. Juicebox Provides a Visualization System for Hi-C Contact Maps with Unlimited Zoom. Cell Syst. 2016, 3, 99–101. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Sun, Z.; Wang, J.; Huang, H.; Kocher, J.P.; Wang, L. CrossMap: A versatile tool for coordinate conversion between genome assemblies. Bioinformatics 2014, 30, 1006–1007. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdottir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Ernst, J.; Kellis, M. ChromHMM: Automating chromatin-state discovery and characterization. Nat. Methods 2012, 9, 215–216. [Google Scholar] [CrossRef]
- Ernst, J.; Kellis, M. Chromatin-state discovery and genome annotation with ChromHMM. Nat. Protoc. 2017, 12, 2478–2492. [Google Scholar] [CrossRef] [PubMed]
- Buenrostro, J.D.; Giresi, P.G.; Zaba, L.C.; Chang, H.Y.; Greenleaf, W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 2013, 10, 1213–1218. [Google Scholar] [CrossRef]
- Konstantakos, V.; Nentidis, A.; Krithara, A.; Paliouras, G. CRISPR-Cas9 gRNA efficiency prediction: An overview of predictive tools and the role of deep learning. Nucleic Acids Res. 2022, 50, 3616–3637. [Google Scholar] [CrossRef]
- Wang, H.; Nakamura, M.; Abbott, T.R.; Zhao, D.; Luo, K.; Yu, C.; Nguyen, C.M.; Lo, A.; Daley, T.P.; La Russa, M.; et al. CRISPR-mediated live imaging of genome editing and transcription. Science 2019, 365, 1301–1305. [Google Scholar] [CrossRef]
- Motoche-Monar, C.; Ordonez, J.E.; Chang, O.; Gonzales-Zubiate, F.A. gRNA Design: How Its Evolution Impacted on CRISPR/Cas9 Systems Refinement. Biomolecules 2023, 13, 1698. [Google Scholar] [CrossRef]
- Fu, Y.; Sander, J.D.; Reyon, D.; Cascio, V.M.; Joung, J.K. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat. Biotechnol. 2014, 32, 279–284. [Google Scholar] [CrossRef]
- Lin, Y.; Cradick, T.J.; Brown, M.T.; Deshmukh, H.; Ranjan, P.; Sarode, N.; Wile, B.M.; Vertino, P.M.; Stewart, F.J.; Bao, G. CRISPR/Cas9 systems have off-target activity with insertions or deletions between target DNA and guide RNA sequences. Nucleic Acids Res. 2014, 42, 7473–7485. [Google Scholar] [CrossRef] [PubMed]
- Dahlman, J.E.; Abudayyeh, O.O.; Joung, J.; Gootenberg, J.S.; Zhang, F.; Konermann, S. Orthogonal gene knockout and activation with a catalytically active Cas9 nuclease. Nat. Biotechnol. 2015, 33, 1159–1161. [Google Scholar] [CrossRef] [PubMed]
- Kiani, S.; Chavez, A.; Tuttle, M.; Hall, R.N.; Chari, R.; Ter-Ovanesyan, D.; Qian, J.; Pruitt, B.W.; Beal, J.; Vora, S.; et al. Cas9 gRNA engineering for genome editing, activation and repression. Nat. Methods 2015, 12, 1051–1054. [Google Scholar] [CrossRef]
- Zhang, J.P.; Li, X.L.; Neises, A.; Chen, W.; Hu, L.P.; Ji, G.Z.; Yu, J.Y.; Xu, J.; Yuan, W.P.; Cheng, T.; et al. Different Effects of sgRNA Length on CRISPR-mediated Gene Knockout Efficiency. Sci. Rep. 2016, 6, 28566. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.; Wu, S.; Wei, R.; Li, Y.; Jin, J.; Mu, Y.; Zhang, Y.; Kong, Q.; Weng, X.; Liu, Z. The length of guide RNA and target DNA heteroduplex effects on CRISPR/Cas9 mediated genome editing efficiency in porcine cells. J. Vet. Sci. 2019, 20, e23. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Primer Sequence | Task |
|---|---|---|
| T2A_BamH_f | AATAAGGATCCGAGGGCAGAGGAAGTCTTCTAACAT | Replacing the P2A-HSA fragment in the dCas9 vector with the T2A-Puro fragment |
| Puro_Xba_r | AATAATCTAGATAGATCAGGCACCGGGCTT | |
| sfGFP_BamH_f | ATTAAGGATCCATGCGTAAAGGCGAAGAGCT | Replacing the HaloTag with sfGFP in a plasmid with the MCP gene |
| sfGFP_Xho_r | TAATTCTCGAGTTTGTACAGTTCATCCATACCATGCG |
| Target Locus 1 | sgRNA Recognition Sequence |
|---|---|
| IDR3 (chr19: 380,836–382,654) | AGCAGATGTAGG (45) 2 |
| C6 (chr6: 157,310,367–157,314,361) | sg1: GTGAGTGCACAC (22) sg2: TGGGACACTATGATG (39) |
| Target | PCP-sfGFP Version | MCP-sfGFP Version | ||
|---|---|---|---|---|
| Transient Transfection | Stable Transduction | Transient Transfection | Stable Transduction | |
| IDR3 | 6% (n = 130) 1 | 26% (n = 124) | 13% (n = 245) | 33% (n = 138) |
| C6 | 3% (n = 132) | 20% (n = 102) | 9% (n = 127) | 52% (n = 131) |
| Target Locus 1 | sgRNA Recognition Sequence |
|---|---|
| C6 (chr6: 157,310,367–157,314,361) | sg1: GTGAGTGCACAC (22) 2 sg2: TGGGACACTATGATG (39) |
| 6T1_L (chr6: 168,378,356–168,380,872) | sg1: ACTCGGGCTGTG (35) sg2: CTGTGTGGGACT (26) |
| 6T1_R (chr6: 168,849,859–168,850,601) | sg1: GCAGAGGTGGCA (22) sg2: TGTGGGCAGAGG (20) |
| 6T2_L (chr6: 169,781,629–169,782,955 for sg1, and chr6: 169,803,533–169,807,849 for sg2) | sg1: ACCACTCGGAAA (21) sg2: GCTCTGTGTCTG (24) |
| 6T2_R (chr6: 170,500,882–170,504,181 for sg1, and chr6: 170,507,778–170,509,799 for sg2) | sg1: CTGCAGCCATCA (31) sg2: CACTCATTCAGC (26) |
| 4T (chr4: 186,033,845–186,035,464) | sg1: CCCTGAGGGATT (22) sg2: TCTGTACCCTGA (29) |
| 5T (chr5: 1,781,394–1,781,810) | sg1: AGGCTGAGGGTG (21) sg2: AGGGTGAGGCTG (23) |
| 22T (chr22: 47,211,081–47,213,579) | sg1: CATATTTGAGTG (56) sg2: GGACGGTCAGTG (30) |
| Target | PCP-sfGFP Version | MCP-sfGFP Version | Proportion of Cells with Two Signals 2 | Signal-to-Background Ratio (Median) |
|---|---|---|---|---|
| C6 | 20% (n = 102) 1 | 52% (n = 131) | 34% | 2.3 |
| 6T1_L | 0% (n = 118) | 0% (n = 113) | 0% | n.a. |
| 6T1_R | 0% (n = 121) | 0% (n = 166) | 0% | n.a. |
| 6T2_L | 0% (n = 108) | 0% (n = 122) | 0% | n.a. |
| 6T2_R | 0% (n = 134) | 29% (n = 159) | 8% | 1.5 |
| 4T | 0% (n = 127) | 16% (n = 117) | 4% | 1.4 |
| 5T | 0% (n = 123) | 5% (n = 129) | 0% | 1.5 |
| 22T | 0% (n = 158) | 3% (n = 116) | 0% | 1.4 |
| Repeat Cluster | Visualization Efficiency (MCP-sfGFP Version) | Number of sgRNA Repeats | Chromatin Status (ChromHMM18) | Transcription | Hi-C Compartment |
|---|---|---|---|---|---|
| IDR3 | 33% 1 | 45 | Quiescent | No | A |
| C6 | 52% | 61 | Quiescent | Weak | A |
| 6T1_L | 0% | 61 | Quiescent | No | A |
| 6T1_R | 0% | 42 | Quiescent | No | A |
| 6T2_L_sg1 | 0% | 21 | Quiescent | No | A |
| 6T2_L_sg2 | 0% | 24 | Repressed Polycomb/weakly repressed Polycomb | No | A |
| 6T2_R_sg1 | 31% | 31 | Weakly Polycomb repressed | No | A |
| 6T2_R_sg2 | 0% | 26 | Quiescent | No | A |
| 4T | 16% | 51 | Quiescent | No | A |
| 5T | 5% | 44 | Quiescent | No | A |
| 22T | 3% | 86 | Quiescent | No | B |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Viushkov, V.S.; Lomov, N.A.; Rubtsov, M.A. A Comparison of Two Versions of the CRISPR-Sirius System for the Live-Cell Visualization of the Borders of Topologically Associating Domains. Cells 2024, 13, 1440. https://doi.org/10.3390/cells13171440
Viushkov VS, Lomov NA, Rubtsov MA. A Comparison of Two Versions of the CRISPR-Sirius System for the Live-Cell Visualization of the Borders of Topologically Associating Domains. Cells. 2024; 13(17):1440. https://doi.org/10.3390/cells13171440
Chicago/Turabian StyleViushkov, Vladimir S., Nikolai A. Lomov, and Mikhail A. Rubtsov. 2024. "A Comparison of Two Versions of the CRISPR-Sirius System for the Live-Cell Visualization of the Borders of Topologically Associating Domains" Cells 13, no. 17: 1440. https://doi.org/10.3390/cells13171440
APA StyleViushkov, V. S., Lomov, N. A., & Rubtsov, M. A. (2024). A Comparison of Two Versions of the CRISPR-Sirius System for the Live-Cell Visualization of the Borders of Topologically Associating Domains. Cells, 13(17), 1440. https://doi.org/10.3390/cells13171440