
PDCD10 Is a Key Player in TMZ-Resistance and Tumor Cell Regrowth: Insights into Its Underlying Mechanism in Glioblastoma Cells
 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Generation and Culture of PDCD10 Knockdown GBM Cells
2.2. Generation and Culture of Lentiviral Transduced PDCD10-Overexpressing GBM Cells
2.3. Generation of TMZ-Resistant Variants in ev/shPDCD10-GBM Cells
2.4. Real-Time RT-PCR (RT2-PCR)
2.5. Western Blot
2.6. Evaluation of Cell Viability in TMZ Treatment Phase and in Post-Treatment Phase by MTT Assay
2.7. EdU Proliferation Assay and Cell Cycle Assay by Flow Cytometry
2.8. Colony Formation Assay
2.9. Neurosphere Assay and Characterization of Glioblastoma Stem Cells (GSC)
2.10. Statistics
3. Results
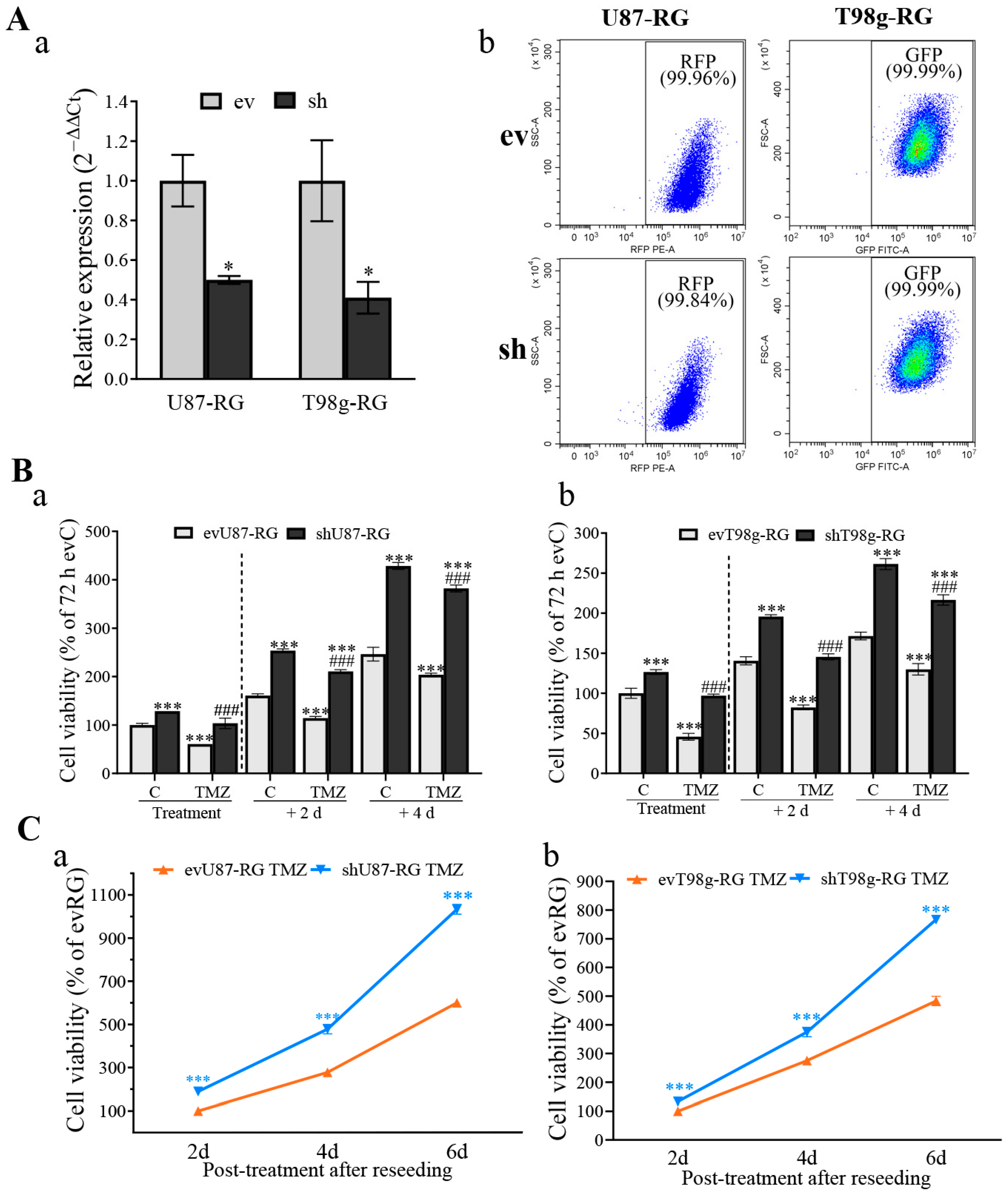
3.1. PDCD10 Knockdown in GBM Cells Leads to TMZ-Resistance and Tumor Cell Regrowth
3.2. PDCD10 Knockdown in RG Cells Increases Cell Viability during TMZ Treatment and Restores Growth Capacity in the Post-Treatment Phase
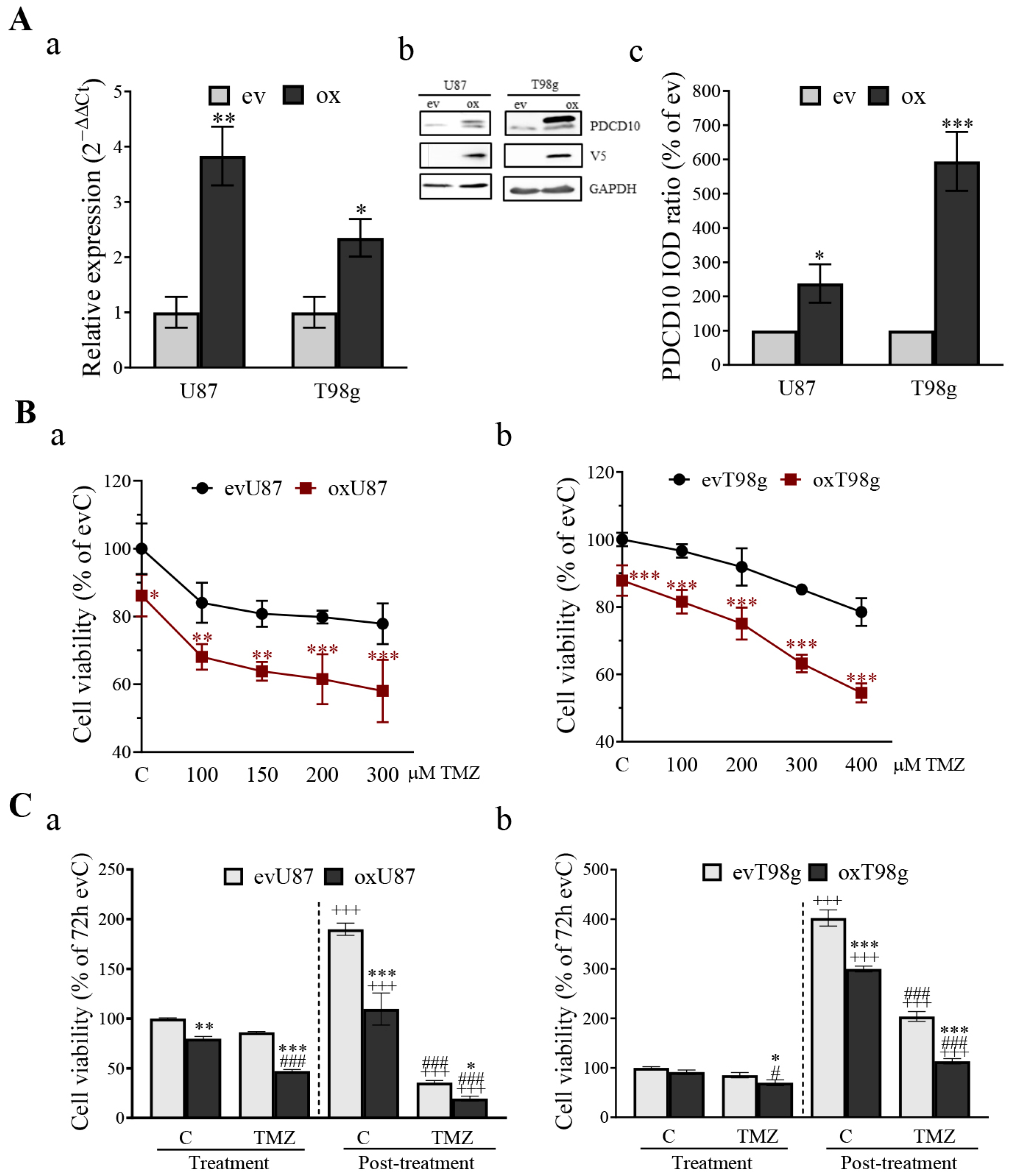
3.3. Overexpression of PDCD10 Sensitizes GBM Cells to TMZ Treatment
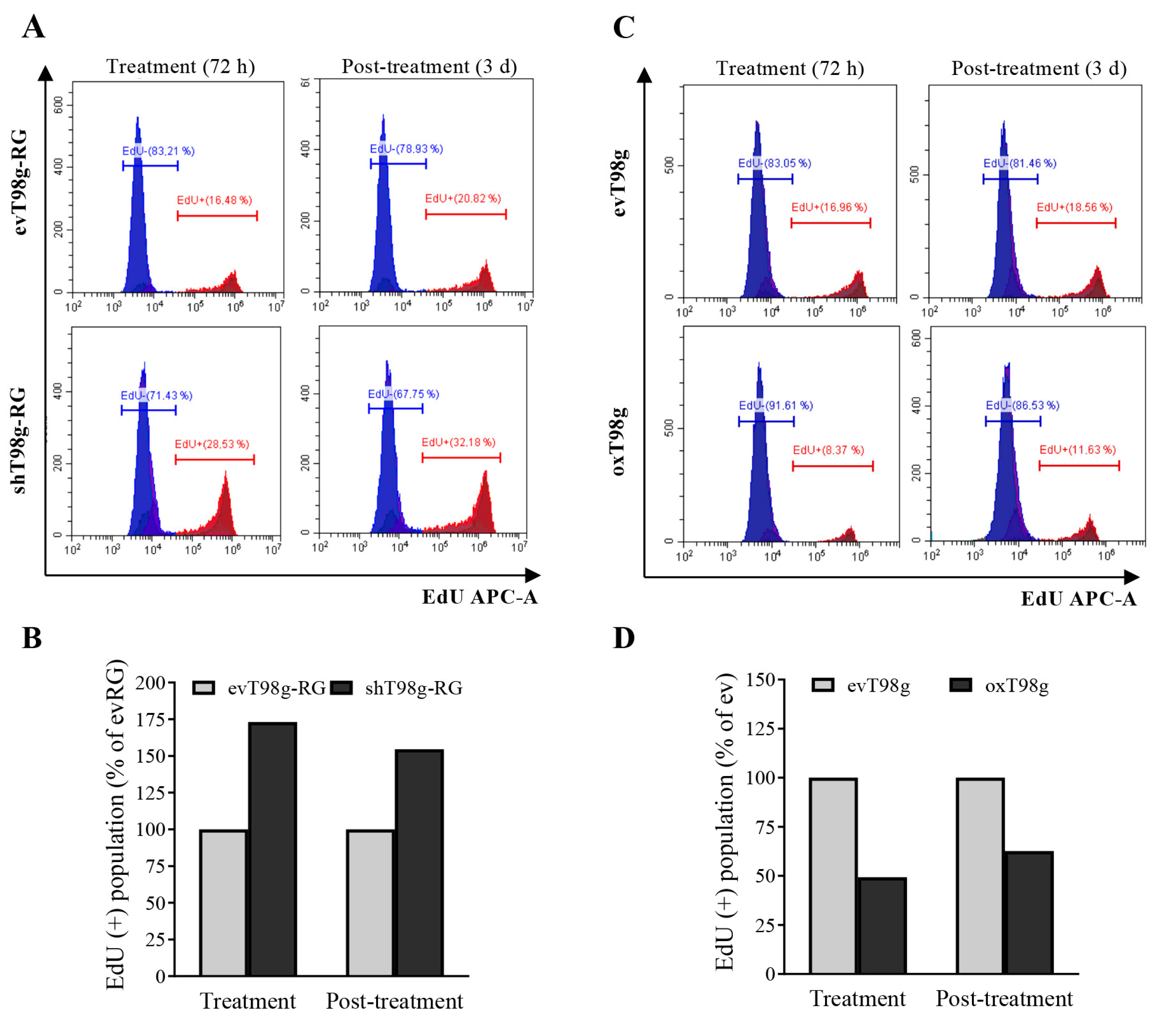
3.4. Alteration of DNA Replication and Cell Cycle in a PDCD10-Dependent Manner in Response to TMZ Treatment
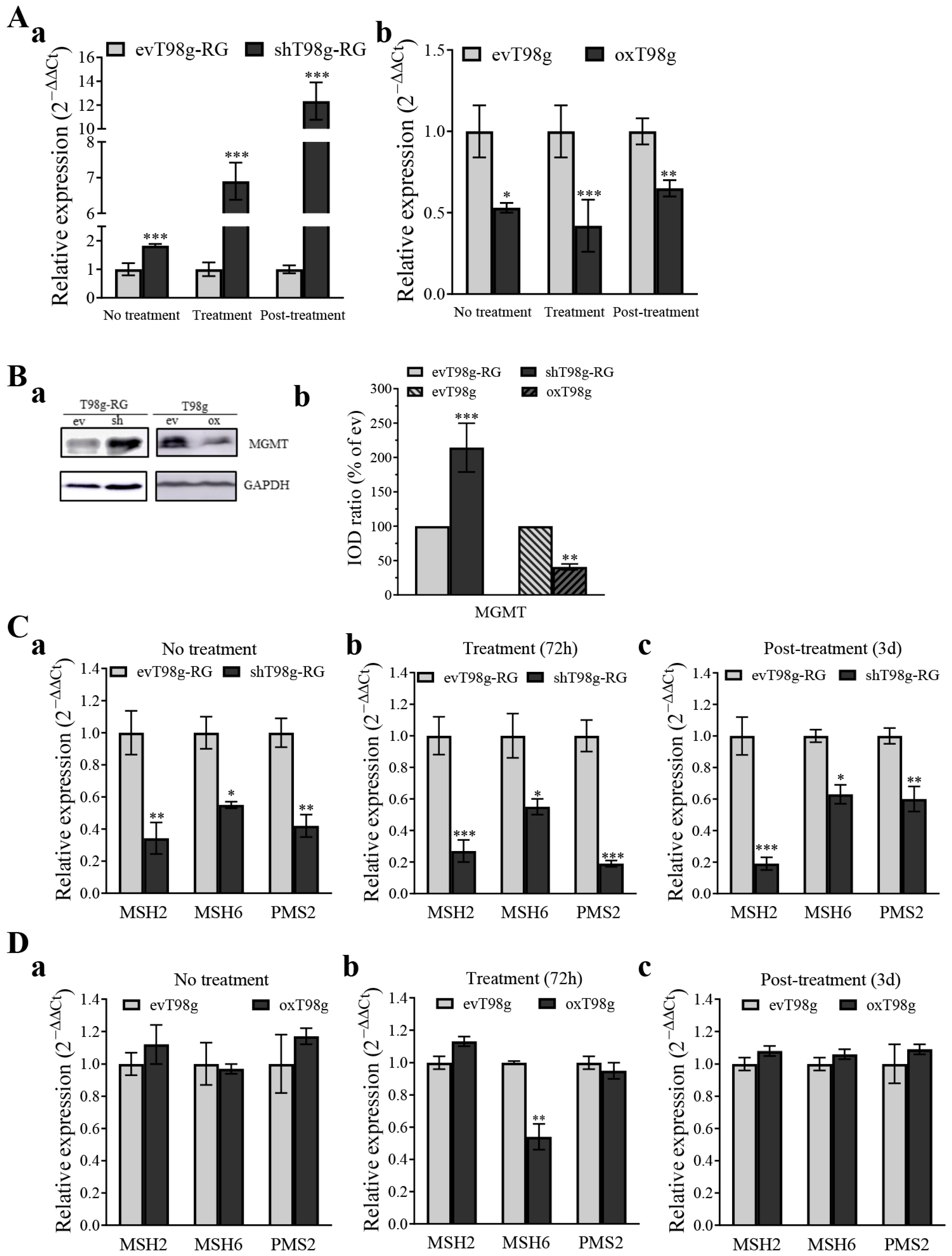
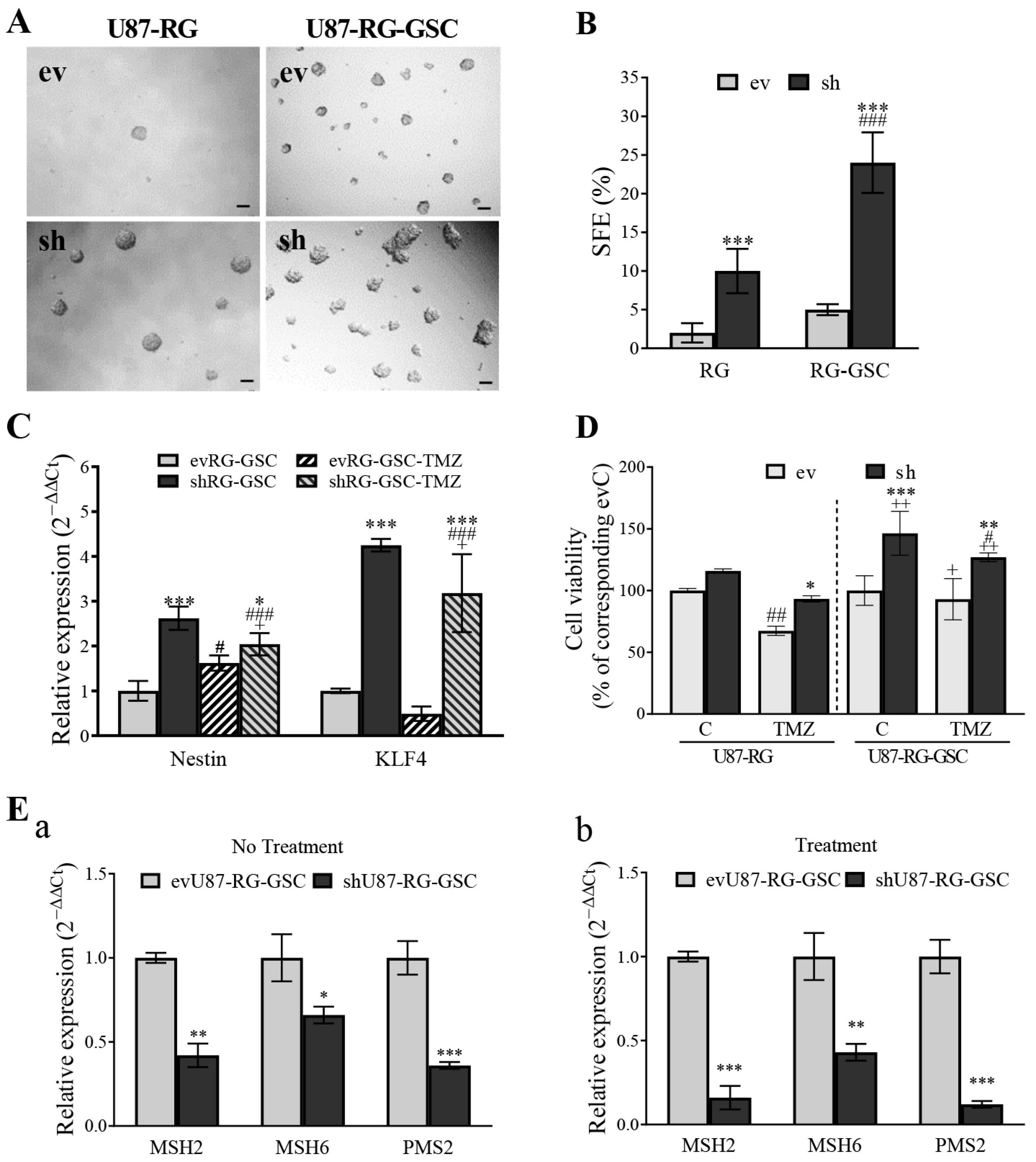
3.5. PDCD10 Knockdown Upregulates MGMT and Downregulates the Expression of DNA Mismatch Repair (MMR) Genes
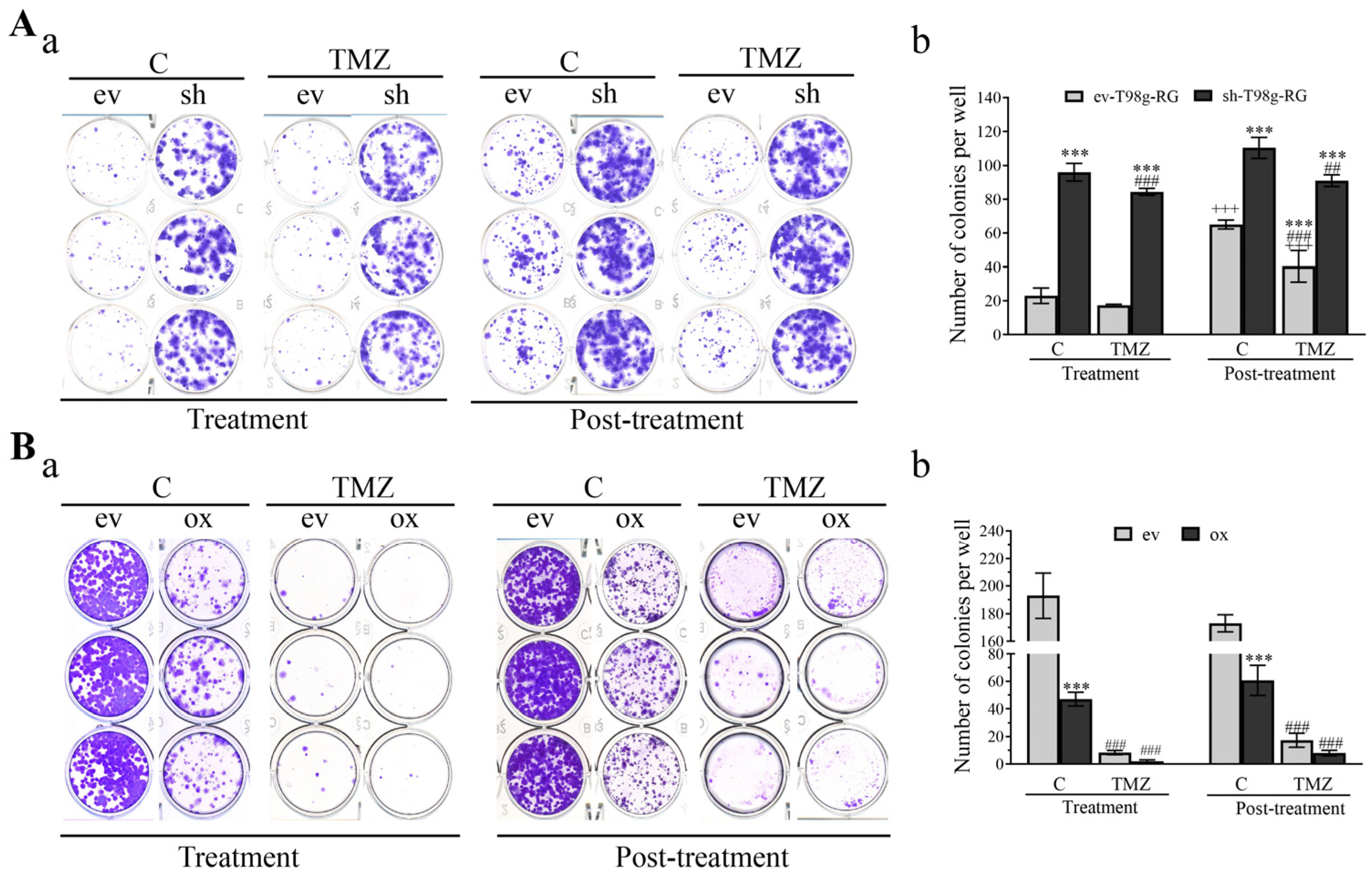
3.6. PDCD10 Knockdown Enhances Colony Formation Capacity of RG Cells
3.7. PDCD10 Knockdown Enhances Stemness Properties of RG Cells in Association with TMZ-Resistance
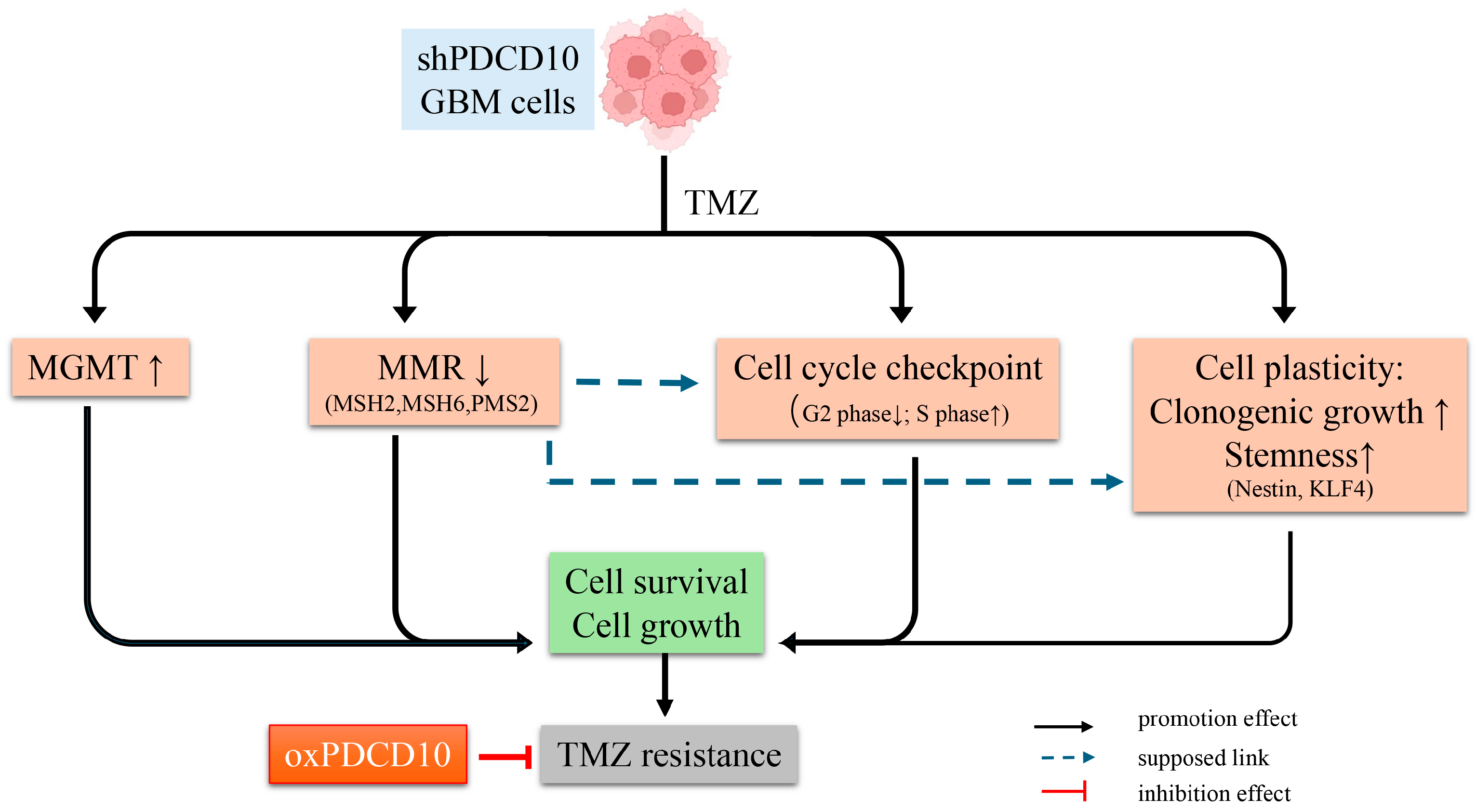
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ostrom, Q.T.; Cioffi, G.; Gittleman, H.; Patil, N.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012–2016. Neuro-Oncology 2019, 21, v1–v100. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Brada, M.; van den Bent, M.J.; Tonn, J.C.; Pentheroudakis, G.; Group, E.G.W. High-grade glioma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2014, 25 (Suppl. S3), iii93–iii101. [Google Scholar] [CrossRef]
- Weller, M.; van den Bent, M.; Preusser, M.; Le Rhun, E.; Tonn, J.C.; Minniti, G.; Bendszus, M.; Balana, C.; Chinot, O.; Dirven, L.; et al. EANO guidelines on the diagnosis and treatment of diffuse gliomas of adulthood. Nat. Rev. Clin. Oncol. 2021, 18, 170–186. [Google Scholar] [CrossRef] [PubMed]
- Rončević, A.; Koruga, N.; Soldo Koruga, A.; Rončević, R.; Rotim, T.; Šimundić, T.; Kretić, D.; Perić, M.; Turk, T.; Štimac, D. Personalized Treatment of Glioblastoma: Current State and Future Perspective. Biomedicines 2023, 11, 1579. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, R.; Perazzoli, G.; Cabeza, L.; Jimenez-Luna, C.; Luque, R.; Prados, J.; Melguizo, C. Temozolomide: An Updated Overview of Resistance Mechanisms, Nanotechnology Advances and Clinical Applications. Curr. Neuropharmacol. 2021, 19, 513–537. [Google Scholar] [CrossRef]
- Singh, N.; Miner, A.; Hennis, L.; Mittal, S. Mechanisms of temozolomide resistance in glioblastoma—A comprehensive review. Cancer Drug Resist. 2021, 4, 17–43. [Google Scholar] [CrossRef]
- Kang, H.; Lee, H.; Kim, D.; Kim, B.; Kang, J.; Kim, H.Y.; Youn, H.; Youn, B. Targeting Glioblastoma Stem Cells to Overcome Chemoresistance: An Overview of Current Therapeutic Strategies. Biomedicines 2022, 10, 1308. [Google Scholar] [CrossRef]
- Merlo, L.M.F.; Pepper, J.W.; Reid, B.J.; Maley, C.C. Cancer as an evolutionary and ecological process. Nat. Rev. Cancer 2006, 6, 924–935. [Google Scholar] [CrossRef]
- Prager, B.C.; Bhargava, S.; Mahadev, V.; Hubert, C.G.; Rich, J.N. Glioblastoma Stem Cells: Driving Resilience through Chaos. Trends Cancer 2020, 6, 223–235. [Google Scholar] [CrossRef]
- Felsberg, J.; Thon, N.; Eigenbrod, S.; Hentschel, B.; Sabel, M.C.; Westphal, M.; Schackert, G.; Kreth, F.W.; Pietsch, T.; Loffler, M.; et al. Promoter methylation and expression of MGMT and the DNA mismatch repair genes MLH1, MSH2, MSH6 and PMS2 in paired primary and recurrent glioblastomas. Int. J. Cancer 2011, 129, 659–670. [Google Scholar] [CrossRef]
- Perazzoli, G.; Prados, J.; Ortiz, R.; Caba, O.; Cabeza, L.; Berdasco, M.; Gonzalez, B.; Melguizo, C. Temozolomide Resistance in Glioblastoma Cell Lines: Implication of MGMT, MMR, P-Glycoprotein and CD133 Expression. PLoS ONE 2015, 10, e0140131. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, Z.; Li, J.; Huang, T.; Wang, Y.; Chang, L.; Zheng, W.; Ma, Y.; Chen, F.; Gong, X.; et al. Genomic analysis of primary and recurrent gliomas reveals clinical outcome related molecular features. Sci. Rep. 2019, 9, 16058. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Chen, Z.; Kim, S.N.; Gan, C.; Ryl, T.; Lesjak, M.S.; Rodemerk, J.; Zhong, R.D.; Wrede, K.; Dammann, P.; et al. Characterization of Temozolomide Resistance Using a Novel Acquired Resistance Model in Glioblastoma Cell Lines. Cancers 2022, 14, 2211. [Google Scholar] [CrossRef] [PubMed]
- Eckerdt, F.; Platanias, L.C. Emerging Role of Glioma Stem Cells in Mechanisms of Therapy Resistance. Cancers 2023, 15, 3458. [Google Scholar] [CrossRef]
- Valentino, M.; Dejana, E.; Malinverno, M. The multifaceted PDCD10/CCM3 gene. Genes. Dis. 2020, 8, 798–813. [Google Scholar] [CrossRef]
- Shi, Z.; Jiao, S.; Zhou, Z. STRIPAK complexes in cell signaling and cancer. Oncogene 2016, 35, 4549–4557. [Google Scholar] [CrossRef]
- Swamy, H.; Glading, A.J. Is Location Everything? Regulation of the Endothelial CCM Signaling Complex. Front. Cardiovasc. Med. 2022, 9, 954780. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, H.; Zhang, Y.; Ma, D. cDNA cloning and expression of an apoptosis-related gene, humanTFAR15 gene. Sci. China C Life Sci. 1999, 42, 323–329. [Google Scholar] [CrossRef]
- Liu, J.; Zhao, K.; Wu, S.; Li, C.; You, C.; Wang, J.; Shu, K.; Lei, T. The Dual Role of PDCD10 in Cancers: A Promising Therapeutic Target. Cancers 2022, 14, 5986. [Google Scholar] [CrossRef]
- Nickel, A.-C.; Wan, X.-Y.; Saban, D.-V.; Weng, Y.-L.; Zhang, S.; Keyvani, K.; Sure, U.; Zhu, Y. Loss of programmed cell death 10 activates tumor cells and leads to temozolomide-resistance in glioblastoma. J. Neurooncol. 2019, 141, 31–41. [Google Scholar] [CrossRef]
- Wan, X.; Saban, D.V.; Kim, S.N.; Weng, Y.; Dammann, P.; Keyvani, K.; Sure, U.; Zhu, Y. PDCD10-Deficiency Promotes Malignant Behaviors and Tumor Growth via Triggering EphB4 Kinase Activity in Glioblastoma. Front. Oncol. 2020, 10, 1377. [Google Scholar] [CrossRef]
- Xu, Y.; Zhu, M. Novel exosomal miR-46146 transfer oxaliplatin chemoresistance in colorectal cancer. Clin. Transl. Oncol. 2020, 22, 1105–1116. [Google Scholar] [CrossRef]
- Fauth, C.; Rostasy, K.; Rath, M.; Gizewski, E.; Lederer, A.G.; Sure, U.; Zschocke, J.; Felbor, U. Highly variable intrafamilial manifestations of a CCM3 mutation ranging from acute childhood cerebral haemorrhage to late-onset meningiomas. Clin. Neurol. Neurosurg. 2015, 128, 41–43. [Google Scholar] [CrossRef]
- Urfali-Mamatoglu, C.; Kazan, H.H.; Gündüz, U. Dual function of programmed cell death 10 (PDCD10) in drug resistance. Biomed. Pharmacother. 2018, 101, 129–136. [Google Scholar] [CrossRef]
- Zhang, Y.; Hu, X.; Miao, X.; Zhu, K.; Cui, S.; Meng, Q.; Sun, J.; Wang, T. MicroRNA-425-5p regulates chemoresistance in colorectal cancer cells via regulation of Programmed Cell Death 10. J. Cell. Mol. Med. 2016, 20, 360–369. [Google Scholar] [CrossRef] [PubMed]
- Lambertz, N.; El Hindy, N.; Kreitschmann-Andermahr, I.; Stein, K.P.; Dammann, P.; Oezkan, N.; Mueller, O.; Sure, U.; Zhu, Y. Downregulation of programmed cell death 10 is associated with tumor cell proliferation, hyperangiogenesis and peritumoral edema in human glioblastoma. BMC Cancer 2015, 15, 759. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Lee, S.Y. Temozolomide resistance in glioblastoma multiforme. Genes Dis. 2016, 3, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Birzu, C.; French, P.; Caccese, M.; Cerretti, G.; Idbaih, A.; Zagonel, V.; Lombardi, G. Recurrent Glioblastoma: From Molecular Landscape to New Treatment Perspectives. Cancers 2021, 13, 47. [Google Scholar] [CrossRef]
- Ali, M.Y.; Oliva, C.R.; Noman, A.S.M.; Allen, B.G.; Goswami, P.C.; Zakharia, Y.; Monga, V.; Spitz, D.R.; Buatti, J.M.; Griguer, C.E. Radioresistance in Glioblastoma and the Development of Radiosensitizers. Cancers 2020, 12, 2511. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Matthews, H.K.; Bertoli, C.; de Bruin, R.A.M. Cell cycle control in cancer. Nat. Rev. Mol. Cell Biol. 2022, 23, 74–88. [Google Scholar] [CrossRef]
- Louvi, A.; Chen, L.; Two, A.M.; Zhang, H.; Min, W.; Günel, M. Loss of cerebral cavernous malformation 3 (Ccm3) in neuroglia leads to CCM and vascular pathology. Proc. Natl. Acad. Sci. USA 2011, 108, 3737–3742. [Google Scholar] [CrossRef] [PubMed]
- You, C.; Erol Sandalcioglu, I.; Dammann, P.; Felbor, U.; Sure, U.; Zhu, Y. Loss of CCM3 impairs DLL4-Notch signalling: Implication in endothelial angiogenesis and in inherited cerebral cavernous malformations. J. Cell. Mol. Med. 2013, 17, 407–418. [Google Scholar] [CrossRef]
- You, C.; Zhao, K.; Dammann, P.; Keyvani, K.; Kreitschmann-Andermahr, I.; Sure, U.; Zhu, Y. EphB4 forward signalling mediates angiogenesis caused by CCM3/PDCD10-ablation. J. Cell. Mol. Med. 2017, 21, 1848–1858. [Google Scholar] [CrossRef] [PubMed]
- Huerta, S.; Harris, D.M.; Jazirehi, A.; Bonavida, B.; Elashoff, D.; Livingston, E.H.; Heber, D. Gene expression profile of metastatic colon cancer cells resistant to cisplatin-induced apoptosis. Int. J. Oncol. 2003, 22, 663–670. [Google Scholar] [CrossRef]
- Yousefi, Y.; Nejati, R.; Eslahi, A.; Alizadeh, F.; Farrokhi, S.; Asoodeh, A.; Mojarrad, M. Enhancing Temozolomide (TMZ) chemosensitivity using CRISPR-dCas9-mediated downregulation of O6-methylguanine DNA methyltransferase (MGMT). J. Neurooncol. 2024, 169, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Rosas-Alonso, R.; Colmenarejo-Fernández, J.; Pernía, O.; Burdiel, M.; Rodríguez-Antolín, C.; Losantos-García, I.; Rubio, T.; Moreno-Velasco, R.; Esteban-Rodríguez, I.; Martínez-Marín, V.; et al. Evaluation of the clinical use of MGMT methylation in extracellular vesicle-based liquid biopsy as a tool for glioblastoma patient management. Sci. Rep. 2024, 14, 11398. [Google Scholar] [CrossRef] [PubMed]
- Cabrini, G.; Fabbri, E.; Lo Nigro, C.; Dechecchi, M.C.; Gambari, R. Regulation of expression of O6-methylguanine-DNA methyltransferase and the treatment of glioblastoma (Review). Int. J. Oncol. 2015, 47, 417–428. [Google Scholar] [CrossRef]
- Touat, M.; Li, Y.Y.; Boynton, A.N.; Spurr, L.F.; Iorgulescu, J.B.; Bohrson, C.L.; Cortes-Ciriano, I.; Birzu, C.; Geduldig, J.E.; Pelton, K.; et al. Mechanisms and therapeutic implications of hypermutation in gliomas. Nature 2020, 580, 517–523. [Google Scholar] [CrossRef]
- Wang, J.; Cazzato, E.; Ladewig, E.; Frattini, V.; Rosenbloom, D.A.-O.; Zairis, S.; Abate, F.; Liu, Z.; Elliott, O.; Shin, Y.J.; et al. Clonal evolution of glioblastoma under therapy. Nat. Genet. 2016, 48, 768–776. [Google Scholar] [CrossRef]
- Auffinger, B.; Tobias, A.L.; Han, Y.; Lee, G.; Guo, D.; Dey, M.; Lesniak, M.S.; Ahmed, A.U. Conversion of differentiated cancer cells into cancer stem-like cells in a glioblastoma model after primary chemotherapy. Cell Death Differ. 2014, 21, 1119–1131. [Google Scholar] [CrossRef]
- Lee, G.; Auffinger, B.; Guo, D.; Hasan, T.; Deheeger, M.; Tobias, A.L.; Kim, J.Y.; Atashi, F.; Zhang, L.; Lesniak, M.S.; et al. Dedifferentiation of Glioma Cells to Glioma Stem-like Cells By Therapeutic Stress-induced HIF Signaling in the Recurrent GBM Model. Mol. Cancer Ther. 2016, 15, 3064–3076. [Google Scholar] [CrossRef] [PubMed]
- Garnier, D.; Meehan, B.; Kislinger, T.; Daniel, P.; Sinha, A.; Abdulkarim, B.; Nakano, I.; Rak, J. Divergent evolution of temozolomide resistance in glioblastoma stem cells is reflected in extracellular vesicles and coupled with radiosensitization. Neuro-Oncology 2018, 20, 236–248. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, H.; Xu, S.; Liu, Z.; Cheng, Q. The adaptive transition of glioblastoma stem cells and its implications on treatments. Signal Transduct. Target. Ther. 2021, 6, 124. [Google Scholar] [CrossRef] [PubMed]
- Bazzoni, R.; Bentivegna, A. Role of Notch Signaling Pathway in Glioblastoma Pathogenesis. Cancers 2019, 11, 292. [Google Scholar] [CrossRef]
- El Hindy, N.; Keyvani, K.; Pagenstecher, A.; Dammann, P.; Sandalcioglu, I.E.; Sure, U.; Zhu, Y. Implications of Dll4-Notch signaling activation in primary glioblastoma multiforme. Neuro-Oncology 2013, 15, 1366–1378. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; He, J.; Xie, K. KLF4 transcription factor in tumorigenesis. Cell Death Discov. 2023, 9, 118. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.K. The Transcription Regulator Kruppel-Like Factor 4 and Its Dual Roles of Oncogene in Glioblastoma and Tumor Suppressor in Neuroblastoma. For. Immunopathol. Dis. Therap 2016, 7, 127–139. [Google Scholar] [CrossRef]
- Riddick, G.; Kotliarova, S.; Rodriguez, V.; Kim, H.S.; Linkous, A.; Storaska, A.J.; Ahn, S.; Walling, J.; Belova, G.; Fine, H.A. A Core Regulatory Circuit in Glioblastoma Stem Cells Links MAPK Activation to a Transcriptional Program of Neural Stem Cell Identity. Sci. Rep. 2017, 7, 43605. [Google Scholar] [CrossRef]
- Malinverno, M.; Maderna, C.; Abu Taha, A.; Corada, M.; Orsenigo, F.; Valentino, M.; Pisati, F.; Fusco, C.; Graziano, P.; Giannotta, M.; et al. Endothelial cell clonal expansion in the development of cerebral cavernous malformations. Nat. Commun. 2019, 10, 2761. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Sequence | Annealing Temperature (°C) |
|---|---|---|
| PDCD10 | 60 | |
| for | TGGCAGCTGATGATGTAGAAG | |
| rev | TCGTGCCTTTTCGTTTAGGT | |
| MGMT | 60 | |
| for | ACCGTTTGCGACTTGGTACTT | |
| rev | GGAGCTTTATTTCGTGCAGACC | |
| MSH2 | 60 | |
| for | TTTACCCGGAGGAGAGACTGC | |
| rev | TGCTCTCCCTTTTTGCCTTTC | |
| MSH6 | 60 | |
| for | AGAGCAATGCAACGTGCAGA | |
| rev | TTTGGCGGCTACTTCGCCTA | |
| PMS2 | 60 | |
| for | ATCGGCGAAGGTTGGAACTC | |
| rev | CGGATGCCTGCTGAAATGAT | |
| Nestin | 60 | |
| for | CTCCAAGAATGGAGGCTGTAGGAA | |
| rev | CCTATGAGATGGAGCAGGCAAGA | |
| KLF4 | 60 | |
| for | GGCTGCGGCAAAACCTACAC | |
| rev | CGGGCGAATTTCCATCCAC | |
| GAPDH | 60 | |
| for | TCACCACCATGGAGAAGGC | |
| rev | GCTAAGCAGTTGGTGGTGCA | |
| RPS13 | 60 | |
| for | CGAAAGCATCTTGAGAGGAACA | |
| rev | TCGAGCCAAACGGTGAATC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, Y.; Kim, S.N.; Chen, Z.-R.; Will, R.; Zhong, R.-D.; Dammann, P.; Sure, U. PDCD10 Is a Key Player in TMZ-Resistance and Tumor Cell Regrowth: Insights into Its Underlying Mechanism in Glioblastoma Cells. Cells 2024, 13, 1442. https://doi.org/10.3390/cells13171442
Zhu Y, Kim SN, Chen Z-R, Will R, Zhong R-D, Dammann P, Sure U. PDCD10 Is a Key Player in TMZ-Resistance and Tumor Cell Regrowth: Insights into Its Underlying Mechanism in Glioblastoma Cells. Cells. 2024; 13(17):1442. https://doi.org/10.3390/cells13171442
Chicago/Turabian StyleZhu, Yuan, Su Na Kim, Zhong-Rong Chen, Rainer Will, Rong-De Zhong, Philipp Dammann, and Ulrich Sure. 2024. "PDCD10 Is a Key Player in TMZ-Resistance and Tumor Cell Regrowth: Insights into Its Underlying Mechanism in Glioblastoma Cells" Cells 13, no. 17: 1442. https://doi.org/10.3390/cells13171442