Abstract
NAD+-dependent deacetylase sirtuin-1 (Sirt1) belongs to the sirtuins family, known to be longevity regulators, and exerts a key role in the prevention of vascular aging. By aging, the expression levels of Sirt1 decline with a severe impact on vascular function, such as the rise of endothelial dysfunction, which in turn promotes the development of cardiovascular diseases. In this context, the impact of Sirt1 activity in preventing endothelial senescence is particularly important. Given the key role of Sirt1 in counteracting endothelial senescence, great efforts have been made to deepen the knowledge about the intricate cross-talks and interactions of Sirt1 with other molecules, in order to set up possible strategies to boost Sirt1 activity to prevent or treat vascular aging. The aim of this review is to provide a proper background on the regulation and function of Sirt1 in the vascular endothelium and to discuss the recent advances regarding the therapeutic strategies of targeting Sirt1 to counteract vascular aging.
1. Introduction
The term sirtuins stands for Silent Information Regulator-SIRT proteins and includes proteins which are highly conserved class III NAD+-dependent histone deacetylases (HDACs). HDACs deacetylate lysine residues use nicotinamide adenine dinucleotide (NAD+) as a co-enzyme [1]. Traditionally, their deacetylation activity was thought to be directed to histone proteins, although it has been recently demonstrated that sirtuins exert a wide range of enzymatic activity, including deacetylation of non-histone proteins, demalonylation, desuccinylation, demyristoylation, and mono-adenosine diphosphate (ADP)-ribosylation [2]. The mammalian Sirt1 family contains seven enzymes (Sirt1–7) grouped into four classes: class I includes Sirt1–3, class II includes Sirt4, class III includes Sirt5, and class IV includes Sirt6–7 [3]. The intracellular localization of sirtuins is strictly related to their biological function. Indeed, while Sirt1, Sirt6, and Sirt7 are localized in the nucleus, Sirt3, Sirt4, and Sirt5 are located in the mitochondria, and Sirt2 is mainly expressed in the cytoplasm [4]. Due to their deacetylation activity occurring at the post-translational level, sirtuins may modulate a number of cellular pathways, including mitochondrial bioenergetic metabolism, cell cycle progression, homeostasis, DNA repair and antioxidant responses, aging, regulation of transcription, apoptosis, inflammation, and survival [5,6,7]. In this review we focus on the biological functions of Sirt1 and we also provide an insight into its role in preventing endothelial dysfunction, as well as the therapeutic strategies of targeting Sirt1 to counteract vascular aging.
2. Sirt1
Sirt1 was the first sirtuin to be discovered and characterized, and thus has been extensively studied. The human SIRT1 gene is located on the long arm of chromosome 10 in position 21.3 which encodes for a protein composed of 747 amino acids [7]. The structure of Sirt1 is analogous to other mammal sirtuins. The structure of Sirt1 includes a highly conserved NAD+-dependent sirtuins core domain and a nuclear localization signal (KRKKRK) at amino acids 41–46. Moreover, the enzyme also includes a conserved catalytic core of 275 amino acids and an N-terminal nuclear localization signal [8]. The catalytic core is composed of a Rossmann-fold domain coupled with a minor zinc finger domain, in which the zinc ion (Zn2+) coordinates tetrahedrally with the thiol groups belonging to four cysteines. Although the zinc ion does not participate in the catalytic activity, its presence is crucial for Sirt1 activity [1].
The main activity of Sirt1 is the deacetylation of both histones and non-histone proteins. By deacetylating these proteins, the enzyme can influence the DNA-histone interaction, thus controlling gene transcription. Indeed, Sirt1 does not bind DNA directly, but interacts with several factors associated with DNA which facilitate the correct placing of Sirt1, whose activity results in the induction of facultative or constitutive heterochromatin [9]. Sirt1 deacetylates lysines at the N-terminal tails of H3 and H4. Its deacetylation activity is mainly directed to H4K16, and at a lower rate to H3K9, H3K14, H4K8, H4K12. Moreover, it can deacetylate also the linker histone H1 at Lys26 (H1K26) [10]. Among the non-histone proteins, Sirt1 can exert its deacetylation activity also on the well-known tumor suppressor p53. Indeed, the deacetylation of p53 at K382 inhibits its nuclear translocation, thus affecting p53 transcription-dependent and independent apoptosis. Upon deacetylation, p53 shifts onto the outer membrane of mitochondria and triggers the release of the pro-apototic BCL and BAX proteins, which in turn trigger the release of cytochrome c by the mitochondria, thus starting the p53 transcription-independent apoptosis [11].
It has been reported that Sirt1 can also exert its deacetylation activity towards forkhead transcription factor O (FoxO), a key protein for modulating apoptosis, cell differentiation, cell cycle arrest, DNA repair response, and oxidative stress-resistance. The deacetylation of FoxO3 and FoxO4 is vital for aging since it results in a decrease in FOXO-induced apoptosis and in an enhanced FoxO-induced cell cycle arrest [12,13,14]. Another non-histone substrate of Sirt1 activity is peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α), an important controller of transcription factors and also a key regulator of biogenesis of mitochondria [15]. The deacetylation of PGC-1α results in an enhanced biogenesis of mitochondria and is protective against neuronal damage and in ischemic heart disease [16].
Inflammation is an important process involved in body healing, but it also plays a key role in different diseases, including diabetes, cardiovascular disease, cancer, disease of the joints, chronic obstructive pulmonary disease, and allergies [17,18,19,20,21,22,23,24]. The nuclear factor κB (NF-κB) pathway plays a key role in regulating this process [25]. It has been reported that Sirt1 can also deacetylate NF-κB complex which inhibits the NF-κB signaling and increases oxidative metabolism resulting in suppression of inflammation. However, due to an antagonistic crosstalk, NF-κB itself can downregulate Sirt1 activity via miR-34a, IFNγ, and reactive oxygen species (ROS), promoting inflammatory responses as observed in several metabolic- or age-related disorders [26].
Sirt1 intracellular localization may change depending on the age of the cell type; for instance, in young mice, Sirt1 is expressed in the nucleus of cardiomyocytes, whereas in adult mice its expression can be detected both in the nucleus and cytoplasm [27].
Post-translational modifications (PTM) such as phosphorylation, ubiquitination, SUMOylation, and S-nitrosylation, play significant roles in modulating the expression levels, enzymatic activity, and function of Sirt1 [28,29,30]. Sirt1 can undergo phosphorylation in multiple sites at C-terminus and N-terminus triggered by the serine/threonine kinases cyclin-dependent kinase 5 (CDK5), and c-Jun N-terminal kinase (JNK) [31,32,33]. It has been reported that CDK5 can phosphorylate Sirt1 at Ser47 promoting endothelial senescence and thus, inhibition of CDK5 resulted in exerting a protective effect towards the development of cell senescence [34]. The homeodomain-interacting protein kinase 2 (HIPK2), a DNA damage response enzyme, can phosphorylate Sirt1 at Ser682 which suppresses its activity without altering the protein expression level. Moreover, Sirt1 can be phosphorylated also by the Janus kinase 1 (JAK1) at Tyr280 and Tyr301, a PTM which does not modify Sirt1 deacetylase activity but is required for Sirt1 interaction with the transcription factor STAT3 [35,36]. The Sirt1 protein level can be modulated by the activity of the E3 ubiquitin ligase SMURF2. SMURF2 ubiquinates Sirt1, triggering its degradation. Analogously, the E2-conjugating enzyme Ube2v1 triggers Ubc13-mediated Sirt1-ubiquitination and degradation, whereas the ubiquitination-mediated degradation of Sirt1 is prevented when the enzyme is bound to the ring finger protein 219 [37,38,39,40]. It has been reported that Sirt1 can be SUMOylated at Lys734, resulting in an increased catalytic activity and protein stability, whereas mutation of Sirt1 at Lys734 or desumoylation by the nuclear desumoylase Sentrin-specific protease 1 (SENP1) resulted in a reduced deacetylase activity [41]. The deacetylase activity of Sirt1 can be diminished also following S-nitrosylation, at Cys387 and Cys390, resulting in enhanced apoptosis and inflammation by increased acetylation of p53 and p65 [42,43,44]. It has been reported that following stress conditions, Sirt1 is dynamically modified by O-GlcNAcylation at Ser549 in its C-terminus, which boosts its deacetylase activity, protecting cells from stress-induced apoptosis [45]. Among the possible PTMs, the S-glutathionylation occurring at Cys67, Cys268, and Cys623 exerts a negative effect on Sirt1 activity, as well as carbonylation [46,47]. Finally, cystathionine β-synthase and cystathionine ɣ-lyase may be responsible for the indirect S-sulfhydration of 2 CXXC motifs located in the catalytic domain of SIRT1 via hydrogen sulfide generation, which results in an increased protein stability and an enhanced deacetylase activity [48,49].
3. Biosynthesis of NAD+ and Sirt1 Regulation
SIRT1 is known as a stress and energy sensor that can be activated by an increased NAD+/NADH ratio [50]. NAD+ is a vital molecule for a number of cellular reactions and functions such as cellular bioenergetics, metabolism, and survival. Indeed, it is involved in redox reactions, whereas NADH, which is the reduced form of NAD+, works as an electron donor, and participates in a number of reactions as a cosubstrate [51,52]. Since many enzymes utilize NAD+ as substrate, the levels of this molecule are crucial and influence directly and indirectly a large number of metabolic pathways. NAD+ levels influence the activity of the enzymes involved in cell metabolism, DNA repair, regulation of gene expression, mitochondrial activity, redox reactions, inflammation, intracellular molecules trafficking, aging, and apoptosis [53,54]. As a consequence, several pathological conditions are characterized by altered intracellular NAD+ levels, including cardiovascular diseases, obesity, neurodegenerative diseases, cancer, and aging [55,56,57]. In particular, age-related impairments in endothelial cells result in vascular dysfunction which promotes the rise of pathological disorders associated with old age [58].
The biosynthesis of NAD+ can start from amino acid tryptophan (Trp), nicotinic acid (NA), nicotinamide (NAM) and nicotinamide riboside (NR), which involve distinct metabolic pathways to generate the same molecule (Figure 1) [59,60].
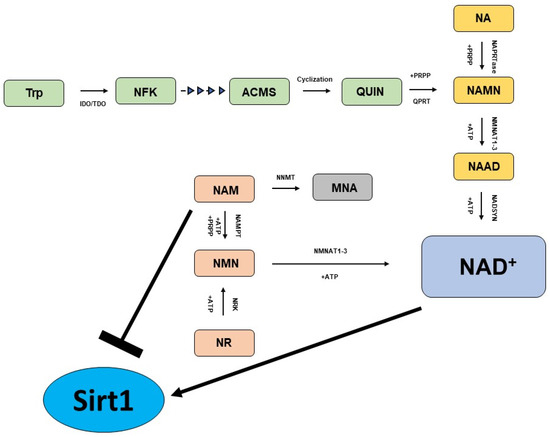
Figure 1.
Key pathways in NAD+ biosynthesis. The Preiss-Handler pathway (yellow); de novo biosynthesis pathway (green); and NAD+ salvage pathway (light brown).
The Preiss–Handler pathway generates NAD+ utilizing NA as a starting molecule. In the Preiss–Handler pathway, the nicotinic acid phosphoribosyltransferase (NAPRTase) transforms NA to nicotinic acid mononucleotide (NAMN), utilizing 5-phosphoribosyl-1-pyrophosphate (PRPP) as a co-substrate and generating pyrophosphate (PPi) as a byproduct. In the next steps, the enzymes nicotinamide mononucleotide adenylyltransferases 1–3 (NMNATs 1–3) catalyze the conversion of NAMN into NA adenine dinucleotide (NAAD), a molecule that is finally converted into NAD+ by the enzyme NAD synthase (NADSYN). NAD+ can also be generated starting from Trp, which is first converted into N-formylkynurenine (NFK) by the enzyme indoleamine 2,3-dioxygenase (IDO) or tryptophan 2,3-dioxygenase (TDO), and upon four subsequent reaction steps it is converted into the unstable molecule α-amino-β-carboxymuconate-ε-semialdehyde (ACMS), that spontaneously undergoes cycling, generating quinolinic acid (QUIN). Subsequently, QUIN is converted into NAMN by the catalytic activity of quinolinate phosphoribosyltransferase (QPRT) using 5-phosphoribosyl-1-pyrophosphate as a cosubstrate. Finally, NAMN can be transformed into NAD+ via the Preiss–Handler pathway. The so-called “NAD+ salvage pathway” is a metabolic route that allows cells to synthesize NAD+ starting from NAM, which is first converted into the intermediate nicotinamide mononucleotide (NMN) by nicotinamide phosphoribosyltransferase (NAMPT), which utilizes ATP and PRPP as a cosubstrate. In the last steps, NMNAT converts NMN to NAD+. The last main metabolic route for the biosynthesis of NAD+ takes place utilizing NR, which undergoes phosphorylation catalyzed by NR kinases (NRK1/NRK2) generating NMN, a molecule that is finally converted to NAD+ by NMNATs in the last step.
It has been reported that specific concentrations of NAM and NAD+ exert a critical impact on cell survival, although excessively elevated concentrations of NAM have been proven to be detrimental [61]. For instance, it has been demonstrated that niacin administration in primary human aortic endothelial cells enhances intracellular NAD+ levels, which in turn activate Sirt1 activity, resulting in improved nitric oxide (NO) bioavailability [62]. There is a strict interplay between NAM and Sirt1 activity. NAM is known to be a potent inhibitor of Sirt1 activity, since it is a product reaction of Sirt1 when the enzyme transfers the acetyl residue from the acetyllysine residue of histones to the ADP-ribose component of NAD+ [63,64]. In this context, the enzyme nicotinamide N-methyltransferase (NNMT) plays a key role. NNMT catalyzes the methylation of NAM, yielding 1-methylnicotinamide (MNA) [65]. By methylating NAM, it reduces the intracellular concentration of NAM, which at high levels would inhibit Sirt1 activity. Indeed, the use of NAM as an inhibitor of both Sirt1 and poly-ADP-ribose polymerases has been proposed for cancer chemoprevention and therapy, especially in those malignancies where NNMT has been reported to be upregulated [66,67,68]. Accordingly, it has been reported that in endothelial cells, the NNMT inhibition resulted in nuclear Sirt1 downregulation and upregulation of the phosphorylated Sirt1 on Ser47, suggesting that the endothelial NNMT/Sirt1 pathway exerts a cytoprotective role protecting endothelial cell viability [69]. In addition, several studies demonstrated a positive effect of both NNMT and MNA, exerting vasoprotective, anti-inflammatory and anti-thrombotic activities [70,71,72]. However, some studies showed that although NAM can act initially as an inhibitor of Sirt1, it might subsequently boost Sirt1 activity due to its conversion to NAD+ through the NAD+ salvage pathway; thus, further studies are required to elucidate these aspects [73,74,75].
4. Endothelial Dysfunction
The endothelium is constituted by a monocellular layer that covers the inner surface of blood vessels. It crucially contributes to maintaining vascular homeostasis through different protective mechanisms, including regulating permeability and vascular tone, and exerts anti-inflammatory, antioxidant, anti-proliferative, and anti-thrombotic functions [76,77,78,79]. Indeed, the endothelium can induce the release of molecules with auto-, para- or endocrine activity, such as NO, prostacyclin, C-type natriuretic peptide (CNP), and endothelium-derived hyperpolarizing factor. By secreting these molecules, the endothelium contributes to inhibiting smooth muscle cell proliferation and migration, platelet adhesion and aggregation, and the fine regulation of biological pathways associated with thrombogenesis [80,81]. Endothelial dysfunction is characterized by a reduced synthesis and/or bioavailability of the vasodilator NO coupled with impairments due to inflammation, senescence, and oxidative stress, thus being a key factor for the development of most cardiovascular diseases (CVDs), including atherosclerosis [82]. Due to aging, several changes occur in the vasculature triggered by systemic endothelial dysfunction and amplified rigidity of large arteries [83,84]. It has been reported that endothelial dysfunction is an initial occurrence of early vascular aging that progresses in aging vessels and that can arise also in the absence of apparent CVDs or established risk factors [85,86]. Thus, the deteriorated endothelial function due to aging is important, not only from a diagnostic and pathophysiological perspective, but is also displays significant therapeutic potential.
5. Sirt1 and Endothelial Aging
Vascular aging involves arteriosclerosis, atherosclerosis, and vascular calcifications and is accelerated by several chronic disorders, and environmental and lifestyle factors [87]. There is established evidence that an early decline in Sirt1 levels is associated with early microvascular dysfunction in adulthood with a consequently increased risk of developing CVD (Figure 2) [88]. A study performed by Guo et al. utilizing endothelial Sirt1-deficient mice demonstrated that downregulation of soluble guanylyl cyclase due to aging and upregulation of cyclooxygenase (COX)-2 in arteries is in part a consequence of the loss of endothelial Sirt1 function. Moreover, it was reported that the overexpression of the enzyme in the endothelium counteracts the impairment of vasodilator responses due to aging and suppresses vasoconstrictor reactions to acetylcholine, enhancing Notch signaling to upregulate soluble guanylyl cyclase-β1 in smooth muscle cells [89].
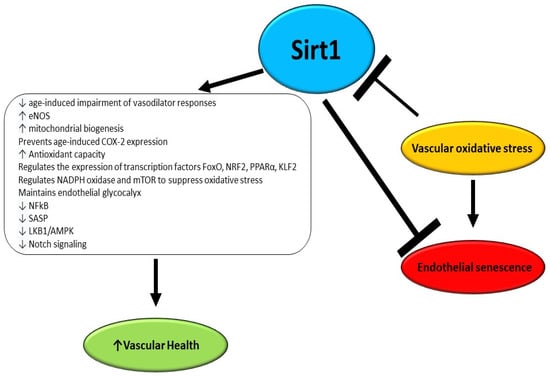
Figure 2.
Effects of Sirt1 as anti-aging and anti-senescence factors in endothelium.
It has been reported that Sirt1 directly impacts the endothelial function of arteries by deacetylating endothelial NO synthase (eNOS), which in turn is activated and preserves vascular homeostasis through NO production. Consistently, the inhibition of Sirt1 in the endothelium of arteries inhibits endothelium-dependent vasodilation and decreases bioavailable NO [90]. Nonetheless, it has not been elucidated yet whether the Sirt1-mediated deacetylation of eNOS has an impact on its phosphorylation status and on the consequent NO release [91]. An elegant study performed by Bai et al. demonstrated that the overexpression of human Sirt1 in the endothelium in eNOS-deficient mice is protective against hypertension and counteracts adverse arterial remodeling occurring in aging vessels [92]. In mice undergoing calorie restriction, an increase was reported in mitochondrial biogenesis, oxygen consumption, and adenosine triphosphate production, and notably a higher expression of Sirt1 was detected. However, in eNOS-knockout mice, calorie restriction-induced expression of Sirt1 was diminished, suggesting a mutual regulatory loop involving the two enzymes in the endothelium [93].
The aging of the vasculature is a progressive event in which an impairment in the relative involvement of NO and other mediators, such as hydrogen peroxide to endothelial-dependent vasodilation and vasoconstriction, occurs. Indeed, while in healthy subjects NO is the main actor for flow-mediated dilation, patients affected by CVDs display a reduced NO-mediated dilation and an augmented hydrogen peroxide contribution [94]. It has been reported that mitochondrial dysfunction occurring in the endothelium results in a weakened/weaker endothelium-dependent vasodilatation as a consequence of diminished NO availability but enhanced generation of hydrogen peroxide [95]. Notably, Sirt1 contributes to modulating mitochondrial biogenesis by enhancing the expression of PGC-1α as well as other genes. Indeed, overexpression of Sirt1 partly reverts the impaired endothelium-dependent vasodilatation in mice characterized by flawed mitochondrial function [96]. During aging, a progressive decline has been observed in Sirt1 expression coupled with an increase of COX-2 which generates vasoconstrictor molecules to boost endothelium-dependent contractions. Interestingly, the upregulation of Sirt1 does not impact eNOS function but it counteracts age-induced COX-2 expression and increases soluble guanylyl cyclase-mediated vasodilatation [89].
ROS play a key role in the onset and progression of several diseases including cardiovascular diseases, diabetes, neurodegenerative diseases, and cancer [97,98,99,100,101,102,103]. In fact, several risk factors for developing a CVD are linked to the rise of oxidative stress, due to an increase in ROS, which promotes vascular aging and endothelial dysfunction and suppresses Sirt1 expression [104,105]. It has been demonstrated that oxidant stimuli including ROS, low-density lipoprotein cholesterol, and high glycaemia can modify the Sirt1 expression level, setting the basis for sustained endothelial dysfunction, since the diminished Sirt1 level is itself a cause of enhanced ROS generation, and contributes to vascular inflammation [31,106,107]. Indeed, Yang et al. recently demonstrated that chronic Sirt1 supplementation ameliorates endothelial function and vascular compliance by boosting eNOS activity and repressing NADPH oxidase (NOX)-related oxidative stress [108]. Consistently, it has been reported that, in animal models, calorie restriction inhibits the decline of Sirt1 levels in arteries and consequent endothelial dysfunction by counteracting oxidative stress [109,110,111,112,113]. Moreover, the strict link between Sirt1 and oxidative stress was confirmed by a study in which the Sirt1 activator SRT1720 was utilized. The activation of Sirt1 was able to revert the endothelial dysfunction by diminishing oxidative stress and inflammation, potentiating also eNOS expression [114,115,116]. Interestingly, Sirt1 activation improves angiogenesis in wounds and facilitates wound healing by promoting angiogenesis, and by protecting vascular endothelial cells from oxidative stress injury [117]. There is strong evidence that Sirt1 regulates glucose and lipid metabolism through its deacetylation activity, and exerts a positive role in ameliorating insulin resistance, which together with obesity is a major cause of endothelial oxidative stress and early vascular aging [118]. Accordingly, it has been reported that the therapeutic modulation of Sirt1 ameliorates obesity- and age-related endothelial dysfunction and protects against high-fat diet-induced metabolic abnormalities [119,120]. Zhou et al. reported that dapagliflozin improves endothelial dysfunction by restoring eNOS activity and NO bioavailability, and decreasing ROS levels via Sirt1 activation in oxidative stress-stimulated endothelial cells [121]. Moreover, it has been reported that Sirt1 can exert a protective effect against oxidative stress by influencing the expression of several nuclear transcription factors such as NF-κB, FoxO, nuclear factor-erythroid-2-related factor 2 (NRF2), peroxisome proliferator-activated receptor alpha (PPAR-α), and Krüppel-like factor 2 (KLF2) [122,123,124,125]. It has been demonstrated that Sirt1 can induce the upregulation of manganese superoxide dismutase thus reducing oxidative stress [126,127]. The ability of Sirt1 to counteract oxidative stress arises also from its capacity of interaction with hydrogen sulfide, due to the inhibition of p66Shc adaptor protein expression levels, through the regulation of NOX, eNOS, and the mechanistic target of rapamycin (mTOR) [126,128,129,130,131]. Finally, Sirt1 contributes to the homeostasis of glycocalyx, which is crucial for flow-induced NO release and inhibits oxidative stress [132,133]. Despite the recent advances achieved in understanding the intricate interactions by which Sirt1 contributes to preventing vascular aging, many mechanisms related to Sirt1 protection against endothelial dysfunction are still far from being fully elucidated, thus requiring deeper insights.
6. Sirt1 and Endothelial Senescence
Endothelial senescence is a pathophysiological process in which the endothelium undergoes structural and functional changes and is crucial for vascular dysfunction, which leads to age-related disease [134,135,136]. Senescent endothelial cells are characterized by changes in cell size and morphology, enhanced lysosomal activity, resistance to apoptosis, decreased NO bioavailability, cell cycle arrest, and reduced proliferation. Moreover, they display augmented senescence-associated-β-galactosidase (SA-β-gal) activity, senescence-associated secretory phenotype (SASP), and increased expression of senescence-associated proteins such as P16 and Sirt1 [134,137]. While aging is characterized by a gradual functional decline, senescence is a cellular response featuring irreversible growth arrest and other phenotypic alterations that include a pro-inflammatory secretome [138,139].
Although endothelial senescence and endothelial dysfunction are mainly initiated by oxidative stress and inflammation, the exact molecular regulatory mechanisms underlying this process remain largely undisclosed [95]. It has been reported that endothelial cell senescence occurs following a number of replication cycles or as a consequence of excessive stress stimulation. Indeed, an accumulation of senescent endothelial cells in arteries during aging has been demonstrated, and in pathological conditions including atherosclerotic plaques and abdominal aortic aneurysm [137,140,141,142,143]. Following senescence, endothelial senescent cells contribute to sustaining a pro-inflammatory and pro-oxidative status and gradually express a SASP that boosts the establishment of a senescent microenvironment that promotes vascular aging [144,145,146,147]. It has been reported that Sirt1 is able to repress the transcriptional activity of NFkB, the main transcription factor for SASP, through the interaction with NFkB and the consequent deacetylation of RelA/p65 at lysine [95,148,149]. Hayakawa et al. reported that Sirt1 can inhibit the expression of SASP factors such as IL-6 and IL-8 through the deacetylation of histones in their promoter regions [150]. Cellular senescence is causally involved in inducing age-related phenotype and thus it should not surprise that removing senescent cells counteracts or delays tissue dysfunction and prolongs lifespan [151]. Endothelial senescence promotes the establishment of endothelial dysfunction, a pro-inflammatory, pro-oxidant and pro-thrombotic condition, and reduces the regeneration capacity of the endothelium, thus playing a crucial role in promoting atherogenesis and age-related vascular disorders [136,152,153,154]. Indeed, human artery endothelial cells that have become senescent arrest the cell cycle preventing further divisions and acquire typical phenotypic traits causing impairment of the angiogenic process, vascular inflammation, and remodeling, and display increased levels of ROS and a decreased NO bioavailability [144,155,156]. In this regard, the accumulation of endothelial senescent cells in the vasculature is a serious clinical problem and several senolytic molecules have been designed in order to mitigate the SASP effects [157,158,159].
It has been reported that Sirt1 displays anti-senescence properties in a wide range of mammalian cells, and that Sirt1 inhibition triggers premature senescence-like replication arrest in human cancer cell lines, characterized by senescence-associated beta-galactosidase activity and increased expression of plasminogen activator inhibitor 1. Moreover, the senescence status was accompanied by an altered activation of some mitogen-activated protein kinase (MAPK) pathways, such as extracellular-regulated protein kinase, c-jun N-terminal kinase and p38 MAPK, in response to epidermal growth factor (EGF) and insulin-like growth factor-I (IGF-I) [160]. Moreover, a study performed by Zheng et al. demonstrated that Sirt1 inhibition by nicotinamide induced a premature senescence of human umbilical vascular endothelial cells (HUVECs), whereas a Sirt1 overexpression triggered by sodium hydrosulfide was able to delay senescence of HUVECs induced by NAM [161]. A recent study performed by Tai et al. showed that the administration of dapagliflozin protects endothelial cells against the rise of senescence by regulating Sirt1 signaling in diabetic mice [162]. In primary porcine aortic endothelial cells (PAECs), which underwent senescence after several passages, an increased phosphorylation of Sirt1 at serine 47 was detected, a post-translational modification that exerts an inhibitory effect on biological functions of Sirt1 in endothelial cells [31]. In line with these findings, the endothelial cells of transgenic mice with endothelial-specific overexpression of Sirt1 were found to be more resistant to paraquat-induced vascular senescence [163]. Senescent PAECs showed enhanced levels of both liver kinase B1 (LKB1), a serine/threonine kinase that also acts as a tumor suppressor, and phosphorylated AMP-activated protein kinase (AMPK), a downstream target of LKB1. This evidence suggests that Sirt1 represses LKB1/AMPK signaling resulting in the promotion of survival and proliferation, and thus inhibiting senescence in endothelial cells [163]. In the nucleus, Sirt1 binds to the DOC domain of HECT and RLD domain containing E3 ubiquitin protein ligase 2 (HERC2) through its amino-terminus, which in turn is responsible for the ubiquitination LKB1 in the nucleus of endothelial cells. Through this mechanism, Sirt1 finely regulates the crosstalk between endothelial and vascular smooth muscle cells counteracting harmful arterial remodeling and maintaining vascular homeostasis [92].
It has been reported that Sirt1 deficiency in endothelial cells promotes the development of fibrosis and induces aberrant secretome, mainly related to activated Notch signaling, which is known to promote senescence, increased permeability and pro-inflammatory responses [89,164,165,166,167]. Indeed, it has been demonstrated that Notch pathway components are upregulated in luminal endothelial cells of atherosclerotic lesions in both mouse and human aortas, as well as in aged but not in young endothelial cells. The upregulation of Notch pathways results in significantly upregulated expression of several molecules implicated in the inflammatory response such as IL-6, IL-8, IL-1α, RANTES and ICAM-1 [168]. In this context, it is noteworthy that Sirt1-deficient endothelial cells are characterized by a marked activation of Notch signaling, proven by the overexpression of delta-like ligand 4 (DLL4), Notch intracellular Domain (NICD) and Notch target genes such as Hey1 and Hes1 [167]. However, although it has been reported that an overexpression of Sirt1 in smooth muscle cells upregulates the Notch signaling to upregulate soluble guanylyl cyclase, which contrasts COX-2, thus preventing vascular aging, it is still unclear whether Sirt1 counteracts the rise of senescence by inhibiting Notch signaling [89].
It is well known that Sirt1 plays a key role in maintaining genome stability which is one of the major mechanisms by which accumulating DNA damage during cell replications leads to accumulation of senescent endothelial cells in arteries [169,170,171]. Indeed, upon DNA damage, the activation of Sirt1 exerts a protective effect against p53-induced senescence or apoptosis by interacting with poly-ADP ribose and forms molecular complexes with key factors such as checkpoint kinase 2 (CHEK2), BRCA1/BRCA2-associated helicase 1, tumor suppressor p53-binding protein 1, Werner helicase, BRG1, and H2AX [172,173,174,175].
Finally, another mechanism by which endothelial cells undergo senescence is the telomere shortening or capping [140,176,177]. It has been reported that Sirt1 is able to interact with telomeric repeat-binding factor 2-interacting protein 1 (TERF2IP), a regulator of both telomere function and NFkB signaling, preventing the nuclear-cytoplasmic shuttle of TERF2IP which results in a suppressed activation of NFkB and an upregulation of COX-2 [31,178]. It has been demonstrated that cyclin-dependent kinase 5 hyperphosphorylation of Sirt1 at the serine 47 residue blocks the antisenescence activity of Sirt1, and plays a pivotal role in the loss-of-Sirt1 function occurring in vascular aging. Indeed, the use of roscovitine, a cyclin-dependent kinase 5 inhibitor, prevents the development of cellular senescence and atherosclerosis in mice [33]. The anti-aging effects of Sirt1 have also been linked to the aging-suppressor protein Klotho. Indeed, it was demonstrated that Klotho deficiency downregulates Sirt1 activity in endothelial cells [179]. Taken together, these pieces of evidence confirm that vascular aging occurs in part also due to endothelial senescence and thus, current efforts are directed to the development of senotherapeutics which can be senolytics, drugs that can remove senescent cells, or senomorphics, which are molecules able to repress their SASP secretome [180,181,182,183,184]. Given the above-mentioned roles of Sirt1, this enzyme is an excellent candidate for preventing cellular senescence, in order to develop Sirt1-based therapies to reverse endothelial cell senescence and vascular aging.
7. Conclusions
Due to its crucial role in vascular aging, Sirt1 represents a promising therapeutic target. Indeed, through its antioxidant and anti-inflammatory effects, and by improving endothelium-dependent vasodilatation, Sirt1 prevents arterial aging. Discovering natural compounds or designing and developing synthetic drugs that could be effective and safe molecules for achieving Sirt1 activation could be an essential strategy for preventing and treating vascular aging. In this context, identifying nutraceutical regimens providing Sirt1-activating agents may also have significant importance for health promotion.
Author Contributions
Conceptualization, R.C. and A.V.; methodology, R.C.; software, S.A., V.P. and A.V.; resources, L.M.; writing—original draft preparation, R.C.; writing—review and editing, R.C., L.M., A.V. and M.E.; visualization, R.C., V.P. and S.A.; supervision, A.V. and M.E. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Finkel, T.; Deng, C.X.; Mostoslavsky, R. Recent progress in the biology and physiology of sirtuins. Nature 2009, 460, 587–591. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.; Sinclair, D.A.; Ellis, J.L.; Steegborn, C. Sirtuin activators and inhibitors: Promises, achievements, and challenges. Pharmacol. Ther. 2018, 188, 140–154. [Google Scholar] [CrossRef]
- Michan, S.; Sinclair, D. Sirtuins in mammals: Insights into their biological function. Biochem. J. 2007, 404, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Houtkooper, R.H.; Pirinen, E.; Auwerx, J. Sirtuins as regulators of metabolism and healthspan. Nat. Rev. Mol. Cell Biol. 2012, 13, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Mei, Z.; Zhang, X.; Yi, J.; Huang, J.; He, J.; Tao, Y. Sirtuins in metabolism, DNA repair and cancer. J. Exp. Clin. Cancer Res. 2016, 35, 182. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.H. Mechanisms and disease implications of sirtuin-mediated autophagic regulation. Exp. Mol. Med. 2019, 51, 1–11. [Google Scholar] [CrossRef]
- Vachharajani, V.T.; Liu, T.; Wang, X.; Hoth, J.J.; Yoza, B.K.; McCall, C.E. Sirtuins Link Inflammation and Metabolism. J. Immunol. Res. 2016, 2016, 8167273. [Google Scholar] [CrossRef]
- Alqarni, M.H.; Foudah, A.I.; Muharram, M.M.; Labrou, N.E. The Pleiotropic Function of Human Sirtuins as Modulators of Metabolic Pathways and Viral Infections. Cells 2021, 10, 460. [Google Scholar] [CrossRef] [PubMed]
- Basova, L.V.; Bortell, N.; Conti, B.; Fox, H.S.; Milner, R.; Marcondes, M.C.G. Age-associated changes in microglia activation and Sirtuin-1- chromatin binding patterns. Aging 2022, 14, 8205–8220. [Google Scholar] [CrossRef] [PubMed]
- Vaquero, A.; Scher, M.; Lee, D.; Erdjument-Bromage, H.; Tempst, P.; Reinberg, D. Human SirT1 interacts with histone H1 and promotes formation of facultative heterochromatin. Mol. Cell 2004, 16, 93–105. [Google Scholar] [CrossRef]
- Yi, J.; Luo, J. SIRT1 and p53, effect on cancer, senescence and beyond. Biochim. Biophys. Acta 2010, 1804, 1684–1689. [Google Scholar] [CrossRef] [PubMed]
- van der Horst, A.; Tertoolen, L.G.; de Vries-Smits, L.M.; Frye, R.A.; Medema, R.H.; Burgering, B.M. FOXO4 is acetylated upon peroxide stress and deacetylated by the longevity protein hSir2(SIRT1). J. Biol. Chem. 2004, 279, 28873–28879. [Google Scholar] [CrossRef] [PubMed]
- Birkenkamp, K.U.; Coffer, P.J. Regulation of cell survival and proliferation by the FOXO (Forkhead box, class O) subfamily of Forkhead transcription factors. Biochem. Soc. Trans. 2003, 31, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Van Der Heide, L.P.; Hoekman, M.F.; Smidt, M.P. The ins and outs of FoxO shuttling: Mechanisms of FoxO translocation and transcriptional regulation. Biochem. J. 2004, 380, 297–309. [Google Scholar] [CrossRef]
- Xia, N.; Strand, S.; Schlufter, F.; Siuda, D.; Reifenberg, G.; Kleinert, H.; Forstermann, U.; Li, H. Role of SIRT1 and FOXO factors in eNOS transcriptional activation by resveratrol. Nitric Oxide 2013, 32, 29–35. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, S.; Li, Y.; Yu, S.; Zhao, Y. SIRT1/PGC-1alpha Signaling Promotes Mitochondrial Functional Recovery and Reduces Apoptosis after Intracerebral Hemorrhage in Rats. Front. Mol. Neurosci. 2017, 10, 443. [Google Scholar]
- Adolph, T.E.; Tilg, H. Western diets and chronic diseases. Nat. Med. 2024, 30, 2133–2147. [Google Scholar] [CrossRef]
- Molinelli, E.; Ceccarelli, G.; Fantone, S.; Di Mercurio, E.; Gambini, D.; Maurizi, A.; Perugini, J.; Tossetta, G.; Brisigotti, V.; De Simoni, E.; et al. Melanoma and subcutaneous adipose tissue: Role of peritumoral adipokines in disease characterization and prognosis. Pigment. Cell Melanoma Res. 2023, 36, 423–430. [Google Scholar] [CrossRef]
- Tossetta, G.; Fantone, S.; Gesuita, R.; Di Renzo, G.C.; Meyyazhagan, A.; Tersigni, C.; Scambia, G.; Di Simone, N.; Marzioni, D. HtrA1 in Gestational Diabetes Mellitus: A Possible Biomarker? Diagnostics 2022, 12, 2705. [Google Scholar] [CrossRef]
- Perugini, J.; Di Mercurio, E.; Tossetta, G.; Severi, I.; Monaco, F.; Reguzzoni, M.; Tomasetti, M.; Dani, C.; Cinti, S.; Giordano, A. Biological Effects of Ciliary Neurotrophic Factor on hMADS Adipocytes. Front. Endocrinol. 2019, 10, 768. [Google Scholar] [CrossRef]
- Altobelli, E.; Latella, G.; Morroni, M.; Licini, C.; Tossetta, G.; Mazzucchelli, R.; Profeta, V.F.; Coletti, G.; Leocata, P.; Castellucci, M.; et al. Low HtrA1 expression in patients with long-standing ulcerative colitis and colorectal cancer. Oncol. Rep. 2017, 38, 418–426. [Google Scholar] [CrossRef]
- Cecati, M.; Sartini, D.; Campagna, R.; Biagini, A.; Ciavattini, A.; Emanuelli, M.; Giannubilo, S.R. Molecular analysis of endometrial inflammation in preterm birth. Cell. Mol. Biol. 2017, 63, 51–57. [Google Scholar] [CrossRef]
- Licini, C.; Tossetta, G.; Avellini, C.; Ciarmela, P.; Lorenzi, T.; Toti, P.; Gesuita, R.; Voltolini, C.; Petraglia, F.; Castellucci, M.; et al. Analysis of cell-cell junctions in human amnion and chorionic plate affected by chorioamnionitis. Histol. Histopathol. 2016, 31, 759–767. [Google Scholar]
- Fantone, S.; Giannubilo, S.R.; Marzioni, D.; Tossetta, G. HTRA family proteins in pregnancy outcome. Tissue Cell 2021, 72, 101549. [Google Scholar] [CrossRef] [PubMed]
- Matsumori, A. Nuclear Factor-kappaB is a Prime Candidate for the Diagnosis and Control of Inflammatory Cardiovascular Disease. Eur. Cardiol. 2023, 18, e40. [Google Scholar] [CrossRef] [PubMed]
- Kauppinen, A.; Suuronen, T.; Ojala, J.; Kaarniranta, K.; Salminen, A. Antagonistic crosstalk between NF-kappaB and SIRT1 in the regulation of inflammation and metabolic disorders. Cell Signal. 2013, 25, 1939–1948. [Google Scholar] [CrossRef]
- Guarente, L. Sirtuins in aging and disease. Cold Spring Harb. Symp. Quant. Biol. 2007, 72, 483–488. [Google Scholar] [CrossRef]
- Revollo, J.R.; Li, X. The ways and means that fine tune Sirt1 activity. Trends Biochem. Sci. 2013, 38, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Flick, F.; Luscher, B. Regulation of sirtuin function by posttranslational modifications. Front. Pharmacol. 2012, 3, 29. [Google Scholar] [CrossRef]
- Porter, K.; Medford, H.M.; McIntosh, C.M.; Marsh, S.A. Cardioprotection requires flipping the ‘posttranslational modification’ switch. Life Sci. 2012, 90, 89–98. [Google Scholar] [CrossRef]
- Bai, B.; Liang, Y.; Xu, C.; Lee, M.Y.; Xu, A.; Wu, D.; Vanhoutte, P.M.; Wang, Y. Cyclin-dependent kinase 5-mediated hyperphosphorylation of sirtuin-1 contributes to the development of endothelial senescence and atherosclerosis. Circulation 2012, 126, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Zschoernig, B.; Mahlknecht, U. Carboxy-terminal phosphorylation of SIRT1 by protein kinase CK2. Biochem. Biophys. Res. Commun. 2009, 381, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Bai, B.; Vanhoutte, P.M.; Wang, Y. Loss-of-SIRT1 function during vascular ageing: Hyperphosphorylation mediated by cyclin-dependent kinase 5. Trends Cardiovasc. Med. 2014, 24, 81–84. [Google Scholar] [CrossRef]
- Wang, S.; Yang, Y.; He, X.; Yang, L.; Wang, J.; Xia, S.; Liu, D.; Liu, S.; Yang, L.; Liu, W.; et al. Cdk5-Mediated Phosphorylation of Sirt1 Contributes to Podocyte Mitochondrial Dysfunction in Diabetic Nephropathy. Antioxid. Redox Signal. 2021, 34, 171–190. [Google Scholar] [CrossRef]
- Conrad, E.; Polonio-Vallon, T.; Meister, M.; Matt, S.; Bitomsky, N.; Herbel, C.; Liebl, M.; Greiner, V.; Kriznik, B.; Schumacher, S.; et al. HIPK2 restricts SIRT1 activity upon severe DNA damage by a phosphorylation-controlled mechanism. Cell Death Differ. 2016, 23, 110–122. [Google Scholar] [CrossRef]
- Wang, W.; Li, F.; Xu, Y.; Wei, J.; Zhang, Y.; Yang, H.; Gao, B.; Yu, G.; Fang, D. JAK1-mediated Sirt1 phosphorylation functions as a negative feedback of the JAK1-STAT3 pathway. J. Biol. Chem. 2018, 293, 11067–11075. [Google Scholar] [CrossRef]
- Yu, L.; Dong, L.; Li, H.; Liu, Z.; Luo, Z.; Duan, G.; Dai, X.; Lin, Z. Ubiquitination-mediated degradation of SIRT1 by SMURF2 suppresses CRC cell proliferation and tumorigenesis. Oncogene 2020, 39, 4450–4464. [Google Scholar] [CrossRef]
- Peng, L.; Yuan, Z.; Li, Y.; Ling, H.; Izumi, V.; Fang, B.; Fukasawa, K.; Koomen, J.; Chen, J.; Seto, E. Ubiquitinated sirtuin 1 (SIRT1) function is modulated during DNA damage-induced cell death and survival. J. Biol. Chem. 2015, 290, 8904–8912. [Google Scholar] [CrossRef] [PubMed]
- Shen, T.; Cai, L.D.; Liu, Y.H.; Li, S.; Gan, W.J.; Li, X.M.; Wang, J.R.; Guo, P.D.; Zhou, Q.; Lu, X.X.; et al. Ube2v1-mediated ubiquitination and degradation of Sirt1 promotes metastasis of colorectal cancer by epigenetically suppressing autophagy. J. Hematol. Oncol. 2018, 11, 95. [Google Scholar] [CrossRef]
- Hwang, J.S.; Kim, E.; Hur, J.; Yoon, T.J.; Seo, H.G. Ring finger protein 219 regulates inflammatory responses by stabilizing sirtuin 1. Br. J. Pharmacol. 2020, 177, 4601–4614. [Google Scholar] [CrossRef]
- Yang, Y.; Fu, W.; Chen, J.; Olashaw, N.; Zhang, X.; Nicosia, S.V.; Bhalla, K.; Bai, W. SIRT1 sumoylation regulates its deacetylase activity and cellular response to genotoxic stress. Nat. Cell Biol. 2007, 9, 1253–1262. [Google Scholar] [CrossRef] [PubMed]
- Kornberg, M.D.; Sen, N.; Hara, M.R.; Juluri, K.R.; Nguyen, J.V.; Snowman, A.M.; Law, L.; Hester, L.D.; Snyder, S.H. GAPDH mediates nitrosylation of nuclear proteins. Nat. Cell Biol. 2010, 12, 1094–1100. [Google Scholar] [CrossRef]
- Shinozaki, S.; Chang, K.; Sakai, M.; Shimizu, N.; Yamada, M.; Tanaka, T.; Nakazawa, H.; Ichinose, F.; Yamada, Y.; Ishigami, A.; et al. Inflammatory stimuli induce inhibitory S-nitrosylation of the deacetylase SIRT1 to increase acetylation and activation of p53 and p65. Sci. Signal. 2014, 7, ra106. [Google Scholar] [CrossRef]
- Kim, Y.M.; Park, E.J.; Kim, H.J.; Chang, K.C. Sirt1 S-nitrosylation induces acetylation of HMGB1 in LPS-activated RAW264.7 cells and endotoxemic mice. Biochem. Biophys. Res. Commun. 2018, 501, 73–79. [Google Scholar] [CrossRef]
- Han, C.; Gu, Y.; Shan, H.; Mi, W.; Sun, J.; Shi, M.; Zhang, X.; Lu, X.; Han, F.; Gong, Q.; et al. O-GlcNAcylation of SIRT1 enhances its deacetylase activity and promotes cytoprotection under stress. Nat. Commun. 2017, 8, 1491. [Google Scholar] [CrossRef] [PubMed]
- Zee, R.S.; Yoo, C.B.; Pimentel, D.R.; Perlman, D.H.; Burgoyne, J.R.; Hou, X.; McComb, M.E.; Costello, C.E.; Cohen, R.A.; Bachschmid, M.M. Redox regulation of sirtuin-1 by S-glutathiolation. Antioxid. Redox Signal. 2010, 13, 1023–1032. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.W.; Yao, H.; Caito, S.; Sundar, I.K.; Rahman, I. Redox regulation of SIRT1 in inflammation and cellular senescence. Free Radic. Biol. Med. 2013, 61, 95–110. [Google Scholar] [CrossRef] [PubMed]
- Du, C.; Lin, X.; Xu, W.; Zheng, F.; Cai, J.; Yang, J.; Cui, Q.; Tang, C.; Cai, J.; Xu, G.; et al. Sulfhydrated Sirtuin-1 Increasing Its Deacetylation Activity Is an Essential Epigenetics Mechanism of Anti-Atherogenesis by Hydrogen Sulfide. Antioxid. Redox Signal. 2019, 30, 184–197. [Google Scholar] [CrossRef]
- Li, J.; Li, M.; Wang, C.; Zhang, S.; Gao, Q.; Wang, L.; Ma, L. NaSH increases SIRT1 activity and autophagy flux through sulfhydration to protect SH-SY5Y cells induced by MPP~. Cell Cycle 2020, 19, 2216–2225. [Google Scholar] [CrossRef]
- Campagna, R.; Vignini, A. NAD(+) Homeostasis and NAD(+)-Consuming Enzymes: Implications for Vascular Health. Antioxidants 2023, 12, 376. [Google Scholar] [CrossRef]
- Rajman, L.; Chwalek, K.; Sinclair, D.A. Therapeutic Potential of NAD-Boosting Molecules: The In Vivo Evidence. Cell Metab. 2018, 27, 529–547. [Google Scholar] [CrossRef]
- Zapata-Perez, R.; Wanders, R.J.A.; van Karnebeek, C.D.M.; Houtkooper, R.H. NAD(+) homeostasis in human health and disease. EMBO Mol. Med. 2021, 13, e13943. [Google Scholar] [CrossRef] [PubMed]
- Bogan, K.L.; Brenner, C. Nicotinic acid, nicotinamide, and nicotinamide riboside: A molecular evaluation of NAD+ precursor vitamins in human nutrition. Annu. Rev. Nutr. 2008, 28, 115–130. [Google Scholar] [CrossRef]
- Chu, X.; Raju, R.P. Regulation of NAD(+) metabolism in aging and disease. Metabolism 2022, 126, 154923. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Lautrup, S.; Hou, Y.; Demarest, T.G.; Croteau, D.L.; Mattson, M.P.; Bohr, V.A. NAD(+) in Aging: Molecular Mechanisms and Translational Implications. Trends Mol. Med. 2017, 23, 899–916. [Google Scholar] [CrossRef] [PubMed]
- Radenkovic, D.; Reason; Verdin, E. Clinical Evidence for Targeting NAD Therapeutically. Pharmaceuticals 2020, 13, 247. [Google Scholar] [CrossRef]
- Zhang, M.; Ying, W. NAD(+) Deficiency Is a Common Central Pathological Factor of a Number of Diseases and Aging: Mechanisms and Therapeutic Implications. Antioxid. Redox Signal. 2019, 30, 890–905. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Bloom, S.I.; Donato, A.J. The role of senescence, telomere dysfunction and shelterin in vascular aging. Microcirculation 2019, 26, e12487. [Google Scholar] [CrossRef]
- Palmer, R.D.; Elnashar, M.M.; Vaccarezza, M. Precursor comparisons for the upregulation of nicotinamide adenine dinucleotide. Novel approaches for better aging. Aging Med. 2021, 4, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Mateuszuk, L.; Campagna, R.; Kutryb-Zajac, B.; Kus, K.; Slominska, E.M.; Smolenski, R.T.; Chlopicki, S. Reversal of endothelial dysfunction by nicotinamide mononucleotide via extracellular conversion to nicotinamide riboside. Biochem. Pharmacol. 2020, 178, 114019. [Google Scholar] [CrossRef]
- Naia, L.; Rosenstock, T.R.; Oliveira, A.M.; Oliveira-Sousa, S.I.; Caldeira, G.L.; Carmo, C.; Laco, M.N.; Hayden, M.R.; Oliveira, C.R.; Rego, A.C. Comparative Mitochondrial-Based Protective Effects of Resveratrol and Nicotinamide in Huntington’s Disease Models. Mol. Neurobiol. 2017, 54, 5385–5399. [Google Scholar] [CrossRef] [PubMed]
- Ganji, S.; Kamanna, S.; Kamanna, V.S.; Kashyap, M.L. Niacin increases human aortic endothelial Sirt1 activity and nitric oxide: Effect on endothelial function and vascular aging. Am. J. Transl. Res. 2023, 15, 6771–6778. [Google Scholar] [PubMed]
- Bitterman, K.J.; Anderson, R.M.; Cohen, H.Y.; Latorre-Esteves, M.; Sinclair, D.A. Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast sir2 and human SIRT1. J. Biol. Chem. 2002, 277, 45099–45107. [Google Scholar] [CrossRef]
- Avalos, J.L.; Bever, K.M.; Wolberger, C. Mechanism of sirtuin inhibition by nicotinamide: Altering the NAD(+) cosubstrate specificity of a Sir2 enzyme. Mol. Cell 2005, 17, 855–868. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, V.; Campagna, R.; Sartini, D.; Emanuelli, M. Nicotinamide N-Methyltransferase as Promising Tool for Management of Gastrointestinal Neoplasms. Biomolecules 2022, 12, 1173. [Google Scholar] [CrossRef]
- Nikas, I.P.; Paschou, S.A.; Ryu, H.S. The Role of Nicotinamide in Cancer Chemoprevention and Therapy. Biomolecules 2020, 10, 477. [Google Scholar] [CrossRef]
- Campagna, R.; Pozzi, V.; Spinelli, G.; Sartini, D.; Milanese, G.; Galosi, A.B.; Emanuelli, M. The Utility of Nicotinamide N-Methyltransferase as a Potential Biomarker to Predict the Oncological Outcomes for Urological Cancers: An Update. Biomolecules 2021, 11, 1214. [Google Scholar] [CrossRef]
- Togni, L.; Mascitti, M.; Sartini, D.; Campagna, R.; Pozzi, V.; Salvolini, E.; Offidani, A.; Santarelli, A.; Emanuelli, M. Nicotinamide N-Methyltransferase in Head and Neck Tumors: A Comprehensive Review. Biomolecules 2021, 11, 1594. [Google Scholar] [CrossRef]
- Campagna, R.; Mateuszuk, L.; Wojnar-Lason, K.; Kaczara, P.; Tworzydlo, A.; Kij, A.; Bujok, R.; Mlynarski, J.; Wang, Y.; Sartini, D.; et al. Nicotinamide N-methyltransferase in endothelium protects against oxidant stress-induced endothelial injury. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 119082. [Google Scholar] [CrossRef]
- Chlopicki, S.; Swies, J.; Mogielnicki, A.; Buczko, W.; Bartus, M.; Lomnicka, M.; Adamus, J.; Gebicki, J. 1-Methylnicotinamide (MNA), a primary metabolite of nicotinamide, exerts anti-thrombotic activity mediated by a cyclooxygenase-2/prostacyclin pathway. Br. J. Pharmacol. 2007, 152, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Bryniarski, K.; Biedron, R.; Jakubowski, A.; Chlopicki, S.; Marcinkiewicz, J. Anti-inflammatory effect of 1-methylnicotinamide in contact hypersensitivity to oxazolone in mice; involvement of prostacyclin. Eur. J. Pharmacol. 2008, 578, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Nejabati, H.R.; Mihanfar, A.; Pezeshkian, M.; Fattahi, A.; Latifi, Z.; Safaie, N.; Valiloo, M.; Jodati, A.R.; Nouri, M. N1-methylnicotinamide (MNAM) as a guardian of cardiovascular system. J. Cell. Physiol. 2018, 233, 6386–6394. [Google Scholar] [CrossRef] [PubMed]
- Maiese, K. New Insights for nicotinamide: Metabolic disease, autophagy, and mTOR. Front. Biosci. (Landmark Ed.) 2020, 25, 1925–1973. [Google Scholar] [CrossRef]
- Maiese, K. Triple play: Promoting neurovascular longevity with nicotinamide, WNT, and erythropoietin in diabetes mellitus. Biomed. Pharmacother. 2008, 62, 218–232. [Google Scholar] [CrossRef]
- Hwang, E.S.; Song, S.B. Nicotinamide is an inhibitor of SIRT1 in vitro, but can be a stimulator in cells. Cell. Mol. Life Sci. 2017, 74, 3347–3362. [Google Scholar] [CrossRef]
- Deanfield, J.E.; Halcox, J.P.; Rabelink, T.J. Endothelial function and dysfunction: Testing and clinical relevance. Circulation 2007, 115, 1285–1295. [Google Scholar] [CrossRef] [PubMed]
- Fantone, S.; Tossetta, G.; Di Simone, N.; Tersigni, C.; Scambia, G.; Marcheggiani, F.; Giannubilo, S.R.; Marzioni, D. CD93 a potential player in cytotrophoblast and endothelial cell migration. Cell Tissue Res. 2022, 387, 123–130. [Google Scholar] [CrossRef]
- Tossetta, G.; Piani, F.; Borghi, C.; Marzioni, D. Role of CD93 in Health and Disease. Cells 2023, 12, 1778. [Google Scholar] [CrossRef]
- Zapotoczny, B.; Braet, F.; Kus, E.; Ginda-Makela, K.; Klejevskaja, B.; Campagna, R.; Chlopicki, S.; Szymonski, M. Actin-spectrin scaffold supports open fenestrae in liver sinusoidal endothelial cells. Traffic 2019, 20, 932–942. [Google Scholar] [CrossRef] [PubMed]
- Forstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837, 837a–837d. [Google Scholar] [CrossRef]
- Calabriso, N.; Massaro, M.; Scoditti, E.; Carluccio, C.; Verri, T.; Carluccio, M.A. Epigenetic Mechanisms in Vascular Inflammation: Modulation of Endothelial Adhesion Molecules and Endothelium-Leukocyte Adhesion. Front. Biosci. (Landmark Ed.) 2023, 28, 194. [Google Scholar] [CrossRef]
- Piani, F.; Tossetta, G.; Cara-Fuentes, G.; Agnoletti, D.; Marzioni, D.; Borghi, C. Diagnostic and Prognostic Role of CD93 in Cardiovascular Disease: A Systematic Review. Biomolecules 2023, 13, 910. [Google Scholar] [CrossRef] [PubMed]
- Regnault, V.; Lacolley, P.; Laurent, S. Arterial Stiffness: From Basic Primers to Integrative Physiology. Annu. Rev. Physiol. 2024, 86, 99–121. [Google Scholar] [CrossRef]
- Cheng, D.C.Y.; Climie, R.E.; Shu, M.; Grieve, S.M.; Kozor, R.; Figtree, G.A. Vascular aging and cardiovascular disease: Pathophysiology and measurement in the coronary arteries. Front. Cardiovasc. Med. 2023, 10, 1206156. [Google Scholar] [CrossRef] [PubMed]
- Seals, D.R.; Jablonski, K.L.; Donato, A.J. Aging and vascular endothelial function in humans. Clin. Sci. 2011, 120, 357–375. [Google Scholar] [CrossRef] [PubMed]
- Widlansky, M.E.; Gokce, N.; Keaney, J.F., Jr.; Vita, J.A. The clinical implications of endothelial dysfunction. J. Am. Coll. Cardiol. 2003, 42, 1149–1160. [Google Scholar] [CrossRef]
- Ungvari, Z.; Tarantini, S.; Sorond, F.; Merkely, B.; Csiszar, A. Mechanisms of Vascular Aging, A Geroscience Perspective: JACC Focus Seminar. J. Am. Coll. Cardiol. 2020, 75, 931–941. [Google Scholar] [CrossRef]
- Rodriguez-Miguelez, P.; Looney, J.; Thomas, J.; Harshfield, G.; Pollock, J.S.; Harris, R.A. Sirt1 during childhood is associated with microvascular function later in life. Am. J. Physiol. Heart Circ. Physiol. 2020, 318, H1371–H1378. [Google Scholar] [CrossRef]
- Guo, Y.; Xu, C.; Man, A.W.C.; Bai, B.; Luo, C.; Huang, Y.; Xu, A.; Vanhoutte, P.M.; Wang, Y. Endothelial SIRT1 prevents age-induced impairment of vasodilator responses by enhancing the expression and activity of soluble guanylyl cyclase in smooth muscle cells. Cardiovasc. Res. 2019, 115, 678–690. [Google Scholar] [CrossRef]
- Mattagajasingh, I.; Kim, C.S.; Naqvi, A.; Yamamori, T.; Hoffman, T.A.; Jung, S.B.; DeRicco, J.; Kasuno, K.; Irani, K. SIRT1 promotes endothelium-dependent vascular relaxation by activating endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA 2007, 104, 14855–14860. [Google Scholar] [CrossRef]
- Habibian, J.; Ferguson, B.S. The Crosstalk between Acetylation and Phosphorylation: Emerging New Roles for HDAC Inhibitors in the Heart. Int. J. Mol. Sci. 2018, 20, 102. [Google Scholar] [CrossRef] [PubMed]
- Bai, B.; Man, A.W.; Yang, K.; Guo, Y.; Xu, C.; Tse, H.F.; Han, W.; Bloksgaard, M.; De Mey, J.G.; Vanhoutte, P.M.; et al. Endothelial SIRT1 prevents adverse arterial remodeling by facilitating HERC2-mediated degradation of acetylated LKB1. Oncotarget 2016, 7, 39065–39081. [Google Scholar] [CrossRef] [PubMed]
- Nisoli, E.; Tonello, C.; Cardile, A.; Cozzi, V.; Bracale, R.; Tedesco, L.; Falcone, S.; Valerio, A.; Cantoni, O.; Clementi, E.; et al. Calorie restriction promotes mitochondrial biogenesis by inducing the expression of eNOS. Science 2005, 310, 314–317. [Google Scholar] [CrossRef]
- Beyer, A.M.; Zinkevich, N.; Miller, B.; Liu, Y.; Wittenburg, A.L.; Mitchell, M.; Galdieri, R.; Sorokin, A.; Gutterman, D.D. Transition in the mechanism of flow-mediated dilation with aging and development of coronary artery disease. Basic Res. Cardiol. 2017, 112, 5. [Google Scholar] [CrossRef]
- Donato, A.J.; Morgan, R.G.; Walker, A.E.; Lesniewski, L.A. Cellular and molecular biology of aging endothelial cells. J. Mol. Cell. Cardiol. 2015, 89, 122–135. [Google Scholar] [CrossRef]
- Nagar, H.; Jung, S.B.; Ryu, M.J.; Choi, S.J.; Piao, S.; Song, H.J.; Kang, S.K.; Shin, N.; Kim, D.W.; Jin, S.A.; et al. CR6-Interacting Factor 1 Deficiency Impairs Vascular Function by Inhibiting the Sirt1-Endothelial Nitric Oxide Synthase Pathway. Antioxid. Redox Signal. 2017, 27, 234–249. [Google Scholar] [CrossRef] [PubMed]
- Moris, D.; Spartalis, M.; Spartalis, E.; Karachaliou, G.S.; Karaolanis, G.I.; Tsourouflis, G.; Tsilimigras, D.I.; Tzatzaki, E.; Theocharis, S. The role of reactive oxygen species in the pathophysiology of cardiovascular diseases and the clinical significance of myocardial redox. Ann. Transl. Med. 2017, 5, 326. [Google Scholar] [CrossRef]
- Fantone, S.; Piani, F.; Olivieri, F.; Rippo, M.R.; Sirico, A.; Di Simone, N.; Marzioni, D.; Tossetta, G. Role of SLC7A11/xCT in Ovarian Cancer. Int. J. Mol. Sci. 2024, 25, 587. [Google Scholar] [CrossRef]
- Szczesny-Malysiak, E.; Stojak, M.; Campagna, R.; Grosicki, M.; Jamrozik, M.; Kaczara, P.; Chlopicki, S. Bardoxolone Methyl Displays Detrimental Effects on Endothelial Bioenergetics, Suppresses Endothelial ET-1 Release, and Increases Endothelial Permeability in Human Microvascular Endothelium. Oxid. Med. Cell. Longev. 2020, 2020, 4678252. [Google Scholar] [CrossRef]
- Yan, R.; Zhang, X.; Xu, W.; Li, J.; Sun, Y.; Cui, S.; Xu, R.; Li, W.; Jiao, L.; Wang, T. ROS-Induced Endothelial Dysfunction in the Pathogenesis of Atherosclerosis. Aging Dis. 2024, 16, 1–19. [Google Scholar]
- Tossetta, G.; Fantone, S.; Marzioni, D.; Mazzucchelli, R. Cellular Modulators of the NRF2/KEAP1 Signaling Pathway in Prostate Cancer. Front. Biosci. (Landmark Ed.) 2023, 28, 143. [Google Scholar] [CrossRef] [PubMed]
- Tossetta, G.; Fantone, S.; Goteri, G.; Giannubilo, S.R.; Ciavattini, A.; Marzioni, D. The Role of NQO1 in Ovarian Cancer. Int. J. Mol. Sci. 2023, 24, 7839. [Google Scholar] [CrossRef] [PubMed]
- Bacchetti, T.; Campagna, R.; Sartini, D.; Cecati, M.; Morresi, C.; Bellachioma, L.; Martinelli, E.; Rocchetti, G.; Lucini, L.; Ferretti, G.; et al. C. spinosa L. subsp. rupestris Phytochemical Profile and Effect on Oxidative Stress in Normal and Cancer Cells. Molecules 2022, 27, 6488. [Google Scholar] [CrossRef] [PubMed]
- Sazdova, I.; Hadzi-Petrushev, N.; Keremidarska-Markova, M.; Stojchevski, R.; Sopi, R.; Shileiko, S.; Mitrokhin, V.; Gagov, H.; Avtanski, D.; Lubomirov, L.T.; et al. SIRT-associated attenuation of cellular senescence in vascular wall. Mech. Ageing Dev. 2024, 220, 111943. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.T.; Lin, L.Y.; Chen, C.; Chen, J.W. CCL4 contributes to aging related angiogenic insufficiency through activating oxidative stress and endothelial inflammation. Angiogenesis 2024, 27, 475–499. [Google Scholar] [CrossRef]
- Leal, D.P.; Goncalinho, G.H.F.; Tavoni, T.M.; Kuwabara, K.L.; Paccanaro, A.P.; Freitas, F.R.; Strunz, C.M.C.; Cesar, L.A.M.; Maranhao, R.C.; Mansur, A.P. The Interplay of Sirtuin-1, LDL-Cholesterol, and HDL Function: A Randomized Controlled Trial Comparing the Effects of Energy Restriction and Atorvastatin on Women with Premature Coronary Artery Disease. Antioxidants 2022, 11, 2363. [Google Scholar] [CrossRef]
- Mortuza, R.; Chen, S.; Feng, B.; Sen, S.; Chakrabarti, S. High glucose induced alteration of SIRTs in endothelial cells causes rapid aging in a p300 and FOXO regulated pathway. PLoS ONE 2013, 8, e54514. [Google Scholar] [CrossRef]
- Yang, K.; Velagapudi, S.; Akhmedov, A.; Kraler, S.; Lapikova-Bryhinska, T.; Schmiady, M.O.; Wu, X.; Geng, L.; Camici, G.G.; Xu, A.; et al. Chronic SIRT1 supplementation in diabetic mice improves endothelial function by suppressing oxidative stress. Cardiovasc. Res. 2023, 119, 2190–2201. [Google Scholar] [CrossRef]
- Rogina, B.; Tissenbaum, H.A. SIRT1, resveratrol and aging. Front. Genet. 2024, 15, 1393181. [Google Scholar] [CrossRef]
- Rippe, C.; Lesniewski, L.; Connell, M.; LaRocca, T.; Donato, A.; Seals, D. Short-term calorie restriction reverses vascular endothelial dysfunction in old mice by increasing nitric oxide and reducing oxidative stress. Aging Cell 2010, 9, 304–312. [Google Scholar] [CrossRef]
- Donato, A.J.; Walker, A.E.; Magerko, K.A.; Bramwell, R.C.; Black, A.D.; Henson, G.D.; Lawson, B.R.; Lesniewski, L.A.; Seals, D.R. Life-long caloric restriction reduces oxidative stress and preserves nitric oxide bioavailability and function in arteries of old mice. Aging Cell 2013, 12, 772–783. [Google Scholar] [CrossRef]
- Hajializadeh, Z.; Khaksari, M. Cardioprotective effects of calorie restriction against inflammation and apoptosis in ovariectomized obese rats: Role of classical estrogen receptors and SIRT1. Obes. Res. Clin. Pract. 2023, 17, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Perry, C.A.; Gadde, K.M. The Role of Calorie Restriction in the Prevention of Cardiovascular Disease. Curr. Atheroscler. Rep. 2022, 24, 235–242. [Google Scholar] [CrossRef]
- Gano, L.B.; Donato, A.J.; Pasha, H.M.; Hearon, C.M., Jr.; Sindler, A.L.; Seals, D.R. The SIRT1 activator SRT1720 reverses vascular endothelial dysfunction, excessive superoxide production, and inflammation with aging in mice. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H1754–H1763. [Google Scholar] [CrossRef] [PubMed]
- Li, R.L.; Lu, Z.Y.; Huang, J.J.; Qi, J.; Hu, A.; Su, Z.X.; Zhang, L.; Li, Y.; Shi, Y.Q.; Hao, C.N.; et al. SRT1720, a SIRT1 specific activator, protected H2O2-induced senescent endothelium. Am. J. Transl. Res. 2016, 8, 2876–2888. [Google Scholar]
- Dong, C.; Li, Z.; Wang, X.; Zou, D.; Duan, H.; Zhao, C.; Zhou, Q.; Shi, W. SRT1720 attenuates UVA-induced corneal endothelial damage via inhibition of oxidative stress and cellular apoptosis. Exp. Eye Res. 2023, 231, 109464. [Google Scholar] [CrossRef]
- Li, X.; Wu, G.; Han, F.; Wang, K.; Bai, X.; Jia, Y.; Li, Z.; Cai, W.; Zhang, W.; Su, L.; et al. SIRT1 activation promotes angiogenesis in diabetic wounds by protecting endothelial cells against oxidative stress. Arch. Biochem. Biophys. 2019, 661, 117–124. [Google Scholar] [CrossRef]
- Cao, Y.; Jiang, X.; Ma, H.; Wang, Y.; Xue, P.; Liu, Y. SIRT1 and insulin resistance. J. Diabetes Complicat. 2016, 30, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Mengozzi, A.; Costantino, S.; Paneni, F.; Duranti, E.; Nannipieri, M.; Mancini, R.; Lai, M.; La Rocca, V.; Puxeddu, I.; Antonioli, L.; et al. Targeting SIRT1 Rescues Age- and Obesity-Induced Microvascular Dysfunction in Ex Vivo Human Vessels. Circ. Res. 2022, 131, 476–491. [Google Scholar] [CrossRef]
- Canto, C.; Houtkooper, R.H.; Pirinen, E.; Youn, D.Y.; Oosterveer, M.H.; Cen, Y.; Fernandez-Marcos, P.J.; Yamamoto, H.; Andreux, P.A.; Cettour-Rose, P.; et al. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012, 15, 838–847. [Google Scholar] [CrossRef]
- Zhou, Y.; Tai, S.; Zhang, N.; Fu, L.; Wang, Y. Dapagliflozin prevents oxidative stress-induced endothelial dysfunction via sirtuin 1 activation. Biomed. Pharmacother. 2023, 165, 115213. [Google Scholar] [CrossRef] [PubMed]
- Ungvari, Z.; Tarantini, S.; Nyul-Toth, A.; Kiss, T.; Yabluchanskiy, A.; Csipo, T.; Balasubramanian, P.; Lipecz, A.; Benyo, Z.; Csiszar, A. Nrf2 dysfunction and impaired cellular resilience to oxidative stressors in the aged vasculature: From increased cellular senescence to the pathogenesis of age-related vascular diseases. Geroscience 2019, 41, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Yan, Z.Q.; Zhao, D.; Chen, S.G.; Gao, L.Z.; Zhang, P.; Shen, B.R.; Han, H.C.; Qi, Y.X.; Jiang, Z.L. SIRT1 and FOXO Mediate Contractile Differentiation of Vascular Smooth Muscle Cells under Cyclic Stretch. Cell. Physiol. Biochem. 2015, 37, 1817–1829. [Google Scholar] [CrossRef]
- Brunet, A.; Sweeney, L.B.; Sturgill, J.F.; Chua, K.F.; Greer, P.L.; Lin, Y.; Tran, H.; Ross, S.E.; Mostoslavsky, R.; Cohen, H.Y.; et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 2004, 303, 2011–2015. [Google Scholar] [CrossRef]
- Gracia-Sancho, J.; Villarreal, G., Jr.; Zhang, Y.; Garcia-Cardena, G. Activation of SIRT1 by resveratrol induces KLF2 expression conferring an endothelial vasoprotective phenotype. Cardiovasc. Res. 2010, 85, 514–519. [Google Scholar] [CrossRef]
- Tanno, M.; Kuno, A.; Yano, T.; Miura, T.; Hisahara, S.; Ishikawa, S.; Shimamoto, K.; Horio, Y. Induction of manganese superoxide dismutase by nuclear translocation and activation of SIRT1 promotes cell survival in chronic heart failure. J. Biol. Chem. 2010, 285, 8375–8382. [Google Scholar] [CrossRef] [PubMed]
- Meng, T.; Qin, W.; Liu, B. SIRT1 Antagonizes Oxidative Stress in Diabetic Vascular Complication. Front. Endocrinol. 2020, 11, 568861. [Google Scholar] [CrossRef]
- Zhu, L.; Duan, W.; Wu, G.; Zhang, D.; Wang, L.; Chen, D.; Chen, Z.; Yang, B. Protective effect of hydrogen sulfide on endothelial cells through Sirt1-FoxO1-mediated autophagy. Ann. Transl. Med. 2020, 8, 1586. [Google Scholar] [CrossRef]
- Zhou, S.; Chen, H.Z.; Wan, Y.Z.; Zhang, Q.J.; Wei, Y.S.; Huang, S.; Liu, J.J.; Lu, Y.B.; Zhang, Z.Q.; Yang, R.F.; et al. Repression of P66Shc expression by SIRT1 contributes to the prevention of hyperglycemia-induced endothelial dysfunction. Circ. Res. 2011, 109, 639–648. [Google Scholar] [CrossRef]
- Zarzuelo, M.J.; Lopez-Sepulveda, R.; Sanchez, M.; Romero, M.; Gomez-Guzman, M.; Ungvary, Z.; Perez-Vizcaino, F.; Jimenez, R.; Duarte, J. SIRT1 inhibits NADPH oxidase activation and protects endothelial function in the rat aorta: Implications for vascular aging. Biochem. Pharmacol. 2013, 85, 1288–1296. [Google Scholar] [CrossRef]
- Kanagy, N.L.; Szabo, C.; Papapetropoulos, A. Vascular biology of hydrogen sulfide. Am. J. Physiol. Cell Physiol. 2017, 312, C537–C549. [Google Scholar] [CrossRef]
- Lipphardt, M.; Song, J.W.; Goligorsky, M.S. Sirtuin 1 and endothelial glycocalyx. Pflug. Arch. 2020, 472, 991–1002. [Google Scholar] [CrossRef]
- Zhang, X.; Sun, D.; Song, J.W.; Zullo, J.; Lipphardt, M.; Coneh-Gould, L.; Goligorsky, M.S. Endothelial cell dysfunction and glycocalyx—A vicious circle. Matrix Biol. 2018, 71–72, 421–431. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Aroor, A.R.; Jia, C.; Sowers, J.R. Endothelial cell senescence in aging-related vascular dysfunction. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1802–1809. [Google Scholar] [CrossRef]
- Bloom, S.I.; Islam, M.T.; Lesniewski, L.A.; Donato, A.J. Mechanisms and consequences of endothelial cell senescence. Nat. Rev. Cardiol. 2023, 20, 38–51. [Google Scholar] [CrossRef]
- Hwang, H.J.; Kim, N.; Herman, A.B.; Gorospe, M.; Lee, J.S. Factors and Pathways Modulating Endothelial Cell Senescence in Vascular Aging. Int. J. Mol. Sci. 2022, 23, 10135. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Konja, D.; Singh, S.; Zhang, B.; Wang, Y. Endothelial Senescence: From Macro- to Micro-Vasculature and Its Implications on Cardiovascular Health. Int. J. Mol. Sci. 2024, 25, 1978. [Google Scholar] [CrossRef]
- Mylonas, A.; O’Loghlen, A. Cellular Senescence and Ageing: Mechanisms and Interventions. Front. Aging 2022, 3, 866718. [Google Scholar] [CrossRef] [PubMed]
- Schmeer, C.; Kretz, A.; Wengerodt, D.; Stojiljkovic, M.; Witte, O.W. Dissecting Aging and Senescence-Current Concepts and Open Lessons. Cells 2019, 8, 1446. [Google Scholar] [CrossRef]
- Gao, Z.; Santos, R.B.; Rupert, J.; Van Drunen, R.; Yu, Y.; Eckel-Mahan, K.; Kolonin, M.G. Endothelial-specific telomerase inactivation causes telomere-independent cell senescence and multi-organ dysfunction characteristic of aging. Aging Cell 2024, 23, e14138. [Google Scholar] [CrossRef]
- Childs, B.G.; Durik, M.; Baker, D.J.; van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, I.; Minamino, T. Cellular Senescence in Arterial Diseases. J. Lipid Atheroscler. 2020, 9, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Cafueri, G.; Parodi, F.; Pistorio, A.; Bertolotto, M.; Ventura, F.; Gambini, C.; Bianco, P.; Dallegri, F.; Pistoia, V.; Pezzolo, A.; et al. Endothelial and smooth muscle cells from abdominal aortic aneurysm have increased oxidative stress and telomere attrition. PLoS ONE 2012, 7, e35312. [Google Scholar] [CrossRef]
- Clayton, Z.S.; Rossman, M.J.; Mahoney, S.A.; Venkatasubramanian, R.; Maurer, G.S.; Hutton, D.A.; VanDongen, N.S.; Greenberg, N.T.; Longtine, A.G.; Ludwig, K.R.; et al. Cellular Senescence Contributes to Large Elastic Artery Stiffening and Endothelial Dysfunction With Aging: Amelioration With Senolytic Treatment. Hypertension 2023, 80, 2072–2087. [Google Scholar] [CrossRef] [PubMed]
- Wada, Y.; Umeno, R.; Nagasu, H.; Kondo, M.; Tokuyama, A.; Kadoya, H.; Kidokoro, K.; Taniguchi, S.; Takahashi, M.; Sasaki, T.; et al. Endothelial Dysfunction Accelerates Impairment of Mitochondrial Function in Ageing Kidneys via Inflammasome Activation. Int. J. Mol. Sci. 2021, 22, 9269. [Google Scholar] [CrossRef] [PubMed]
- Venturini, W.; Olate-Briones, A.; Valenzuela, C.; Mendez, D.; Fuentes, E.; Cayo, A.; Mancilla, D.; Segovia, R.; Brown, N.E.; Moore-Carrasco, R. Platelet Activation Is Triggered by Factors Secreted by Senescent Endothelial HMEC-1 Cells In Vitro. Int. J. Mol. Sci. 2020, 21, 3287. [Google Scholar] [CrossRef]
- Rossman, M.J.; Kaplon, R.E.; Hill, S.D.; McNamara, M.N.; Santos-Parker, J.R.; Pierce, G.L.; Seals, D.R.; Donato, A.J. Endothelial cell senescence with aging in healthy humans: Prevention by habitual exercise and relation to vascular endothelial function. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H890–H895. [Google Scholar] [CrossRef]
- Osorio, F.G.; Lopez-Otin, C.; Freije, J.M. NF-kB in premature aging. Aging 2012, 4, 726–727. [Google Scholar] [CrossRef]
- Yang, X.D.; Tajkhorshid, E.; Chen, L.F. Functional interplay between acetylation and methylation of the RelA subunit of NF-kappaB. Mol. Cell. Biol. 2010, 30, 2170–2180. [Google Scholar] [CrossRef]
- Hayakawa, T.; Iwai, M.; Aoki, S.; Takimoto, K.; Maruyama, M.; Maruyama, W.; Motoyama, N. SIRT1 suppresses the senescence-associated secretory phenotype through epigenetic gene regulation. PLoS ONE 2015, 10, e0116480. [Google Scholar] [CrossRef]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; van de Sluis, B.; Kirkland, J.L.; van Deursen, J.M. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Vaitkevicius, P.V.; Fleg, J.L.; Engel, J.H.; O’Connor, F.C.; Wright, J.G.; Lakatta, L.E.; Yin, F.C.; Lakatta, E.G. Effects of age and aerobic capacity on arterial stiffness in healthy adults. Circulation 1993, 88, 1456–1462. [Google Scholar] [CrossRef] [PubMed]
- Bu, L.L.; Yuan, H.H.; Xie, L.L.; Guo, M.H.; Liao, D.F.; Zheng, X.L. New Dawn for Atherosclerosis: Vascular Endothelial Cell Senescence and Death. Int. J. Mol. Sci. 2023, 24, 15160. [Google Scholar] [CrossRef]
- Shang, D.; Liu, H.; Tu, Z. Pro-inflammatory cytokines mediating senescence of vascular endothelial cells in atherosclerosis. Fundam. Clin. Pharmacol. 2023, 37, 928–936. [Google Scholar] [CrossRef]
- Incalza, M.A.; D’Oria, R.; Natalicchio, A.; Perrini, S.; Laviola, L.; Giorgino, F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vascul. Pharmacol. 2018, 100, 1–19. [Google Scholar] [CrossRef]
- Hayashi, T.; Yano, K.; Matsui-Hirai, H.; Yokoo, H.; Hattori, Y.; Iguchi, A. Nitric oxide and endothelial cellular senescence. Pharmacol. Ther. 2008, 120, 333–339. [Google Scholar] [CrossRef]
- Childs, B.G.; Gluscevic, M.; Baker, D.J.; Laberge, R.M.; Marquess, D.; Dananberg, J.; van Deursen, J.M. Senescent cells: An emerging target for diseases of ageing. Nat. Rev. Drug Discov. 2017, 16, 718–735. [Google Scholar] [CrossRef] [PubMed]
- Ge, M.; Hu, L.; Ao, H.; Zi, M.; Kong, Q.; He, Y. Senolytic targets and new strategies for clearing senescent cells. Mech. Ageing Dev. 2021, 195, 111468. [Google Scholar] [CrossRef]
- Birch, J.; Gil, J. Senescence and the SASP: Many therapeutic avenues. Genes Dev. 2020, 34, 1565–1576. [Google Scholar] [CrossRef]
- Ota, H.; Tokunaga, E.; Chang, K.; Hikasa, M.; Iijima, K.; Eto, M.; Kozaki, K.; Akishita, M.; Ouchi, Y.; Kaneki, M. Sirt1 inhibitor, Sirtinol, induces senescence-like growth arrest with attenuated Ras-MAPK signaling in human cancer cells. Oncogene 2006, 25, 176–185. [Google Scholar] [CrossRef]
- Zheng, M.; Qiao, W.; Cui, J.; Liu, L.; Liu, H.; Wang, Z.; Yan, C. Hydrogen sulfide delays nicotinamide-induced premature senescence via upregulation of SIRT1 in human umbilical vein endothelial cells. Mol. Cell. Biochem. 2014, 393, 59–67. [Google Scholar] [CrossRef]
- Tai, S.; Zhou, Y.; Fu, L.; Ding, H.; Zhou, Y.; Yin, Z.; Yang, R.; Liu, Z.; Zhou, S. Dapagliflozin impedes endothelial cell senescence by activating the SIRT1 signaling pathway in type 2 diabetes. Heliyon 2023, 9, e19152. [Google Scholar] [CrossRef] [PubMed]
- Zu, Y.; Liu, L.; Lee, M.Y.; Xu, C.; Liang, Y.; Man, R.Y.; Vanhoutte, P.M.; Wang, Y. SIRT1 promotes proliferation and prevents senescence through targeting LKB1 in primary porcine aortic endothelial cells. Circ. Res. 2010, 106, 1384–1393. [Google Scholar] [CrossRef] [PubMed]
- Lipphardt, M.; Dihazi, H.; Muller, G.A.; Goligorsky, M.S. Fibrogenic Secretome of Sirtuin 1-Deficient Endothelial Cells: Wnt, Notch and Glycocalyx Rheostat. Front. Physiol. 2018, 9, 1325. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, D.; Fredette, N.; Rostama, B.; Tang, Y.; Vary, C.P.; Liaw, L.; Urs, S. RhoA-mediated signaling in Notch-induced senescence-like growth arrest and endothelial barrier dysfunction. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 876–882. [Google Scholar] [CrossRef]
- Rodriguez-Vita, J.; Fischer, A. Notch signaling facilitates crossing of endothelial barriers by tumor cells. Mol. Cell. Oncol. 2017, 4, e1311828. [Google Scholar] [CrossRef]
- Kida, Y.; Zullo, J.A.; Goligorsky, M.S. Endothelial sirtuin 1 inactivation enhances capillary rarefaction and fibrosis following kidney injury through Notch activation. Biochem. Biophys. Res. Commun. 2016, 478, 1074–1079. [Google Scholar] [CrossRef]
- Liu, Z.J.; Tan, Y.; Beecham, G.W.; Seo, D.M.; Tian, R.; Li, Y.; Vazquez-Padron, R.I.; Pericak-Vance, M.; Vance, J.M.; Goldschmidt-Clermont, P.J.; et al. Notch activation induces endothelial cell senescence and pro-inflammatory response: Implication of Notch signaling in atherosclerosis. Atherosclerosis 2012, 225, 296–303. [Google Scholar] [CrossRef]
- Gonfloni, S.; Iannizzotto, V.; Maiani, E.; Bellusci, G.; Ciccone, S.; Diederich, M. P53 and Sirt1: Routes of metabolism and genome stability. Biochem. Pharmacol. 2014, 92, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Utani, K.; Aladjem, M.I. Extra View: Sirt1 Acts As A Gatekeeper Of Replication Initiation To Preserve Genomic Stability. Nucleus 2018, 9, 261–267. [Google Scholar] [CrossRef]
- Wu, H.; Roks, A.J. Genomic instability and vascular aging: A focus on nucleotide excision repair. Trends Cardiovasc. Med. 2014, 24, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Luo, K.; Liu, T.; Lou, Z. Regulation of SIRT1 activity by genotoxic stress. Genes Dev. 2012, 26, 791–796. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Casta, A.; Wang, R.; Lozada, E.; Fan, W.; Kane, S.; Ge, Q.; Gu, W.; Orren, D.; Luo, J. Regulation of WRN protein cellular localization and enzymatic activities by SIRT1-mediated deacetylation. J. Biol. Chem. 2008, 283, 7590–7598. [Google Scholar] [CrossRef] [PubMed]
- Kuno, A.; Hosoda, R.; Tsukamoto, M.; Sato, T.; Sakuragi, H.; Ajima, N.; Saga, Y.; Tada, K.; Taniguchi, Y.; Iwahara, N.; et al. SIRT1 in the cardiomyocyte counteracts doxorubicin-induced cardiotoxicity via regulating histone H2AX. Cardiovasc. Res. 2023, 118, 3360–3373. [Google Scholar] [CrossRef]
- Wang, R.H.; Zheng, Y.; Kim, H.S.; Xu, X.; Cao, L.; Luhasen, T.; Lee, M.H.; Xiao, C.; Vassilopoulos, A.; Chen, W.; et al. Interplay among BRCA1, SIRT1, and Survivin during BRCA1-associated tumorigenesis. Mol. Cell 2008, 32, 11–20. [Google Scholar] [CrossRef]
- Minamino, T.; Miyauchi, H.; Yoshida, T.; Ishida, Y.; Yoshida, H.; Komuro, I. Endothelial cell senescence in human atherosclerosis: Role of telomere in endothelial dysfunction. Circulation 2002, 105, 1541–1544. [Google Scholar] [CrossRef]
- Morgan, R.G.; Ives, S.J.; Lesniewski, L.A.; Cawthon, R.M.; Andtbacka, R.H.; Noyes, R.D.; Richardson, R.S.; Donato, A.J. Age-related telomere uncapping is associated with cellular senescence and inflammation independent of telomere shortening in human arteries. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H251–H258. [Google Scholar] [CrossRef]
- Kotla, S.; Vu, H.T.; Ko, K.A.; Wang, Y.; Imanishi, M.; Heo, K.S.; Fujii, Y.; Thomas, T.N.; Gi, Y.J.; Mazhar, H.; et al. Endothelial senescence is induced by phosphorylation and nuclear export of telomeric repeat binding factor 2-interacting protein. JCI Insight 2019, 4, e124867. [Google Scholar] [CrossRef]
- Gao, D.; Zuo, Z.; Tian, J.; Ali, Q.; Lin, Y.; Lei, H.; Sun, Z. Activation of SIRT1 Attenuates Klotho Deficiency-Induced Arterial Stiffness and Hypertension by Enhancing AMP-Activated Protein Kinase Activity. Hypertension 2016, 68, 1191–1199. [Google Scholar] [CrossRef]
- von Kobbe, C. Targeting senescent cells: Approaches, opportunities, challenges. Aging 2019, 11, 12844–12861. [Google Scholar] [CrossRef]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The Achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef]
- Suda, M.; Paul, K.H.; Minamino, T.; Miller, J.D.; Lerman, A.; Ellison-Hughes, G.M.; Tchkonia, T.; Kirkland, J.L. Senescent Cells: A Therapeutic Target in Cardiovascular Diseases. Cells 2023, 12, 1296. [Google Scholar] [CrossRef]
- Fan, T.; Du, Y.; Zhang, M.; Zhu, A.R.; Zhang, J. Senolytics Cocktail Dasatinib and Quercetin Alleviate Human Umbilical Vein Endothelial Cell Senescence via the TRAF6-MAPK-NF-kappaB Axis in a YTHDF2-Dependent Manner. Gerontology 2022, 68, 920–934. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Kim, S.Y. Endothelial senescence in vascular diseases: Current understanding and future opportunities in senotherapeutics. Exp. Mol. Med. 2023, 55, 1–12. [Google Scholar] [CrossRef]
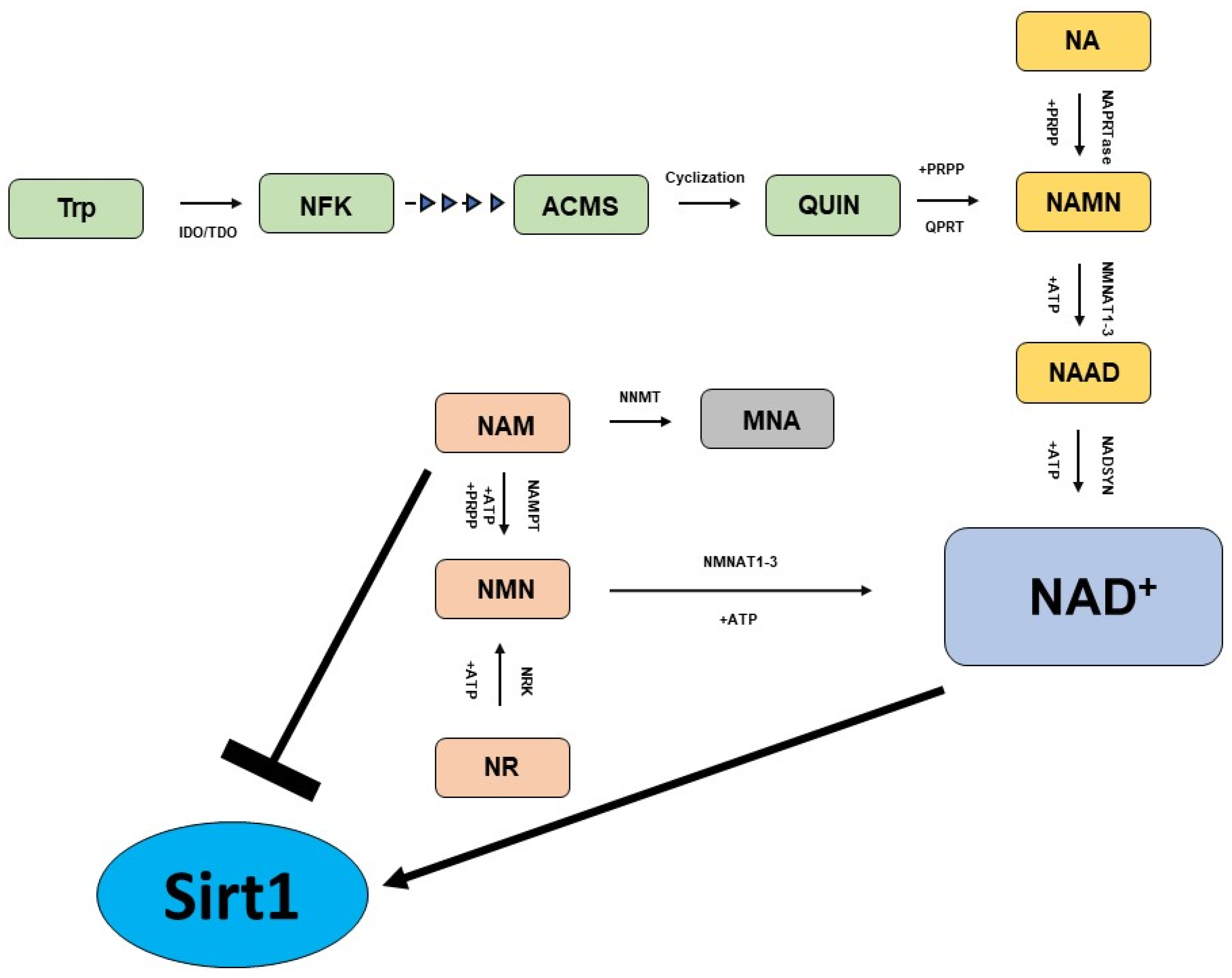
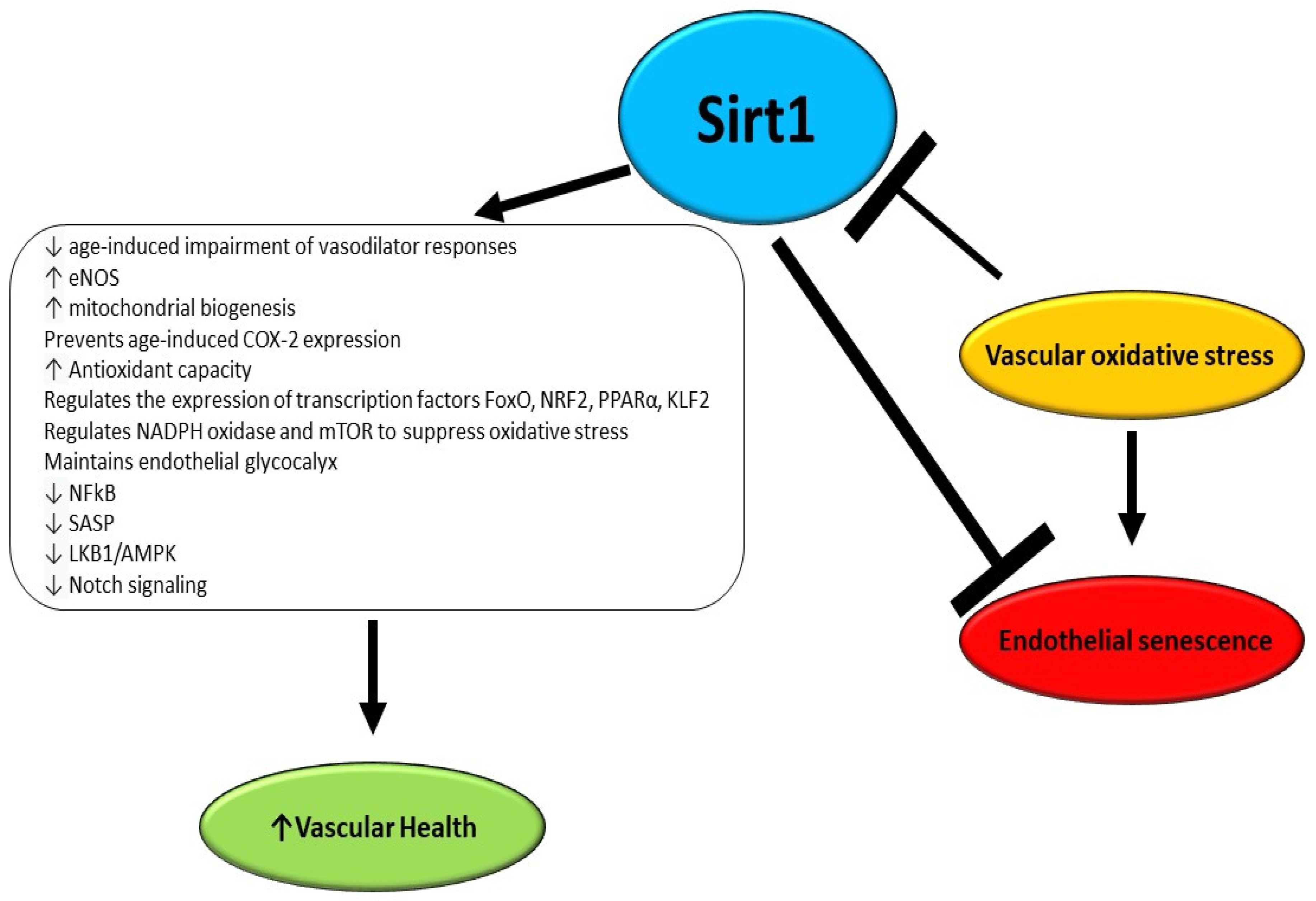
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).