

Activation of Yes-Associated Protein Is Indispensable for Transformation of Kidney Fibroblasts into Myofibroblasts during Repeated Administration of Cisplatin


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatment
2.2. Cellular Viability
2.3. Western Blot Analysis
2.4. Immunocytochemistry
2.5. Cell Size
2.6. Senescence-Associated β-Galactosidase (SA-β-gal) ACTIVITY
2.7. Statistical Analysis
3. Results
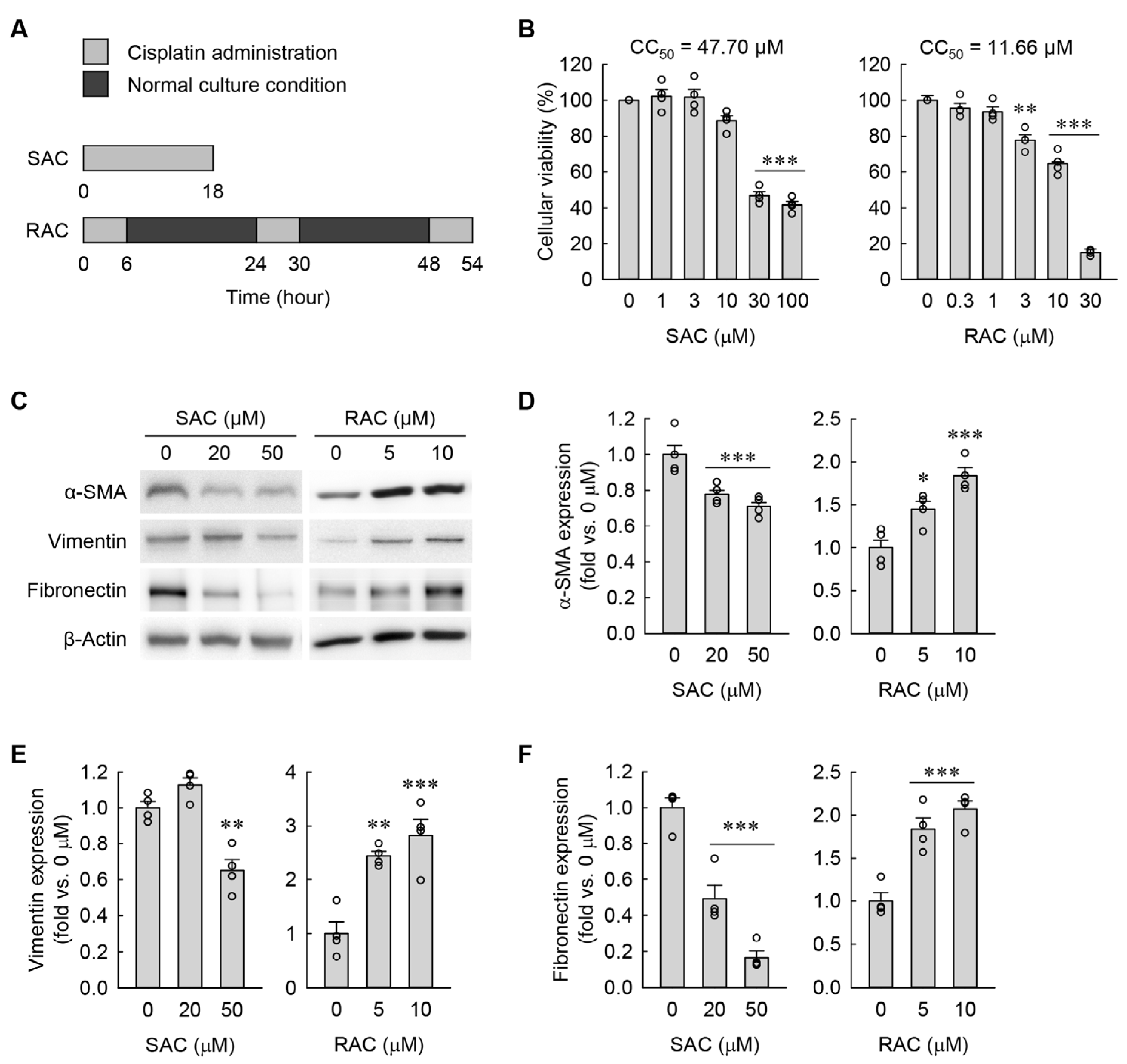
3.1. RAC Transforms Kidney Fibroblasts into Myofibroblasts, but SAC Does Not
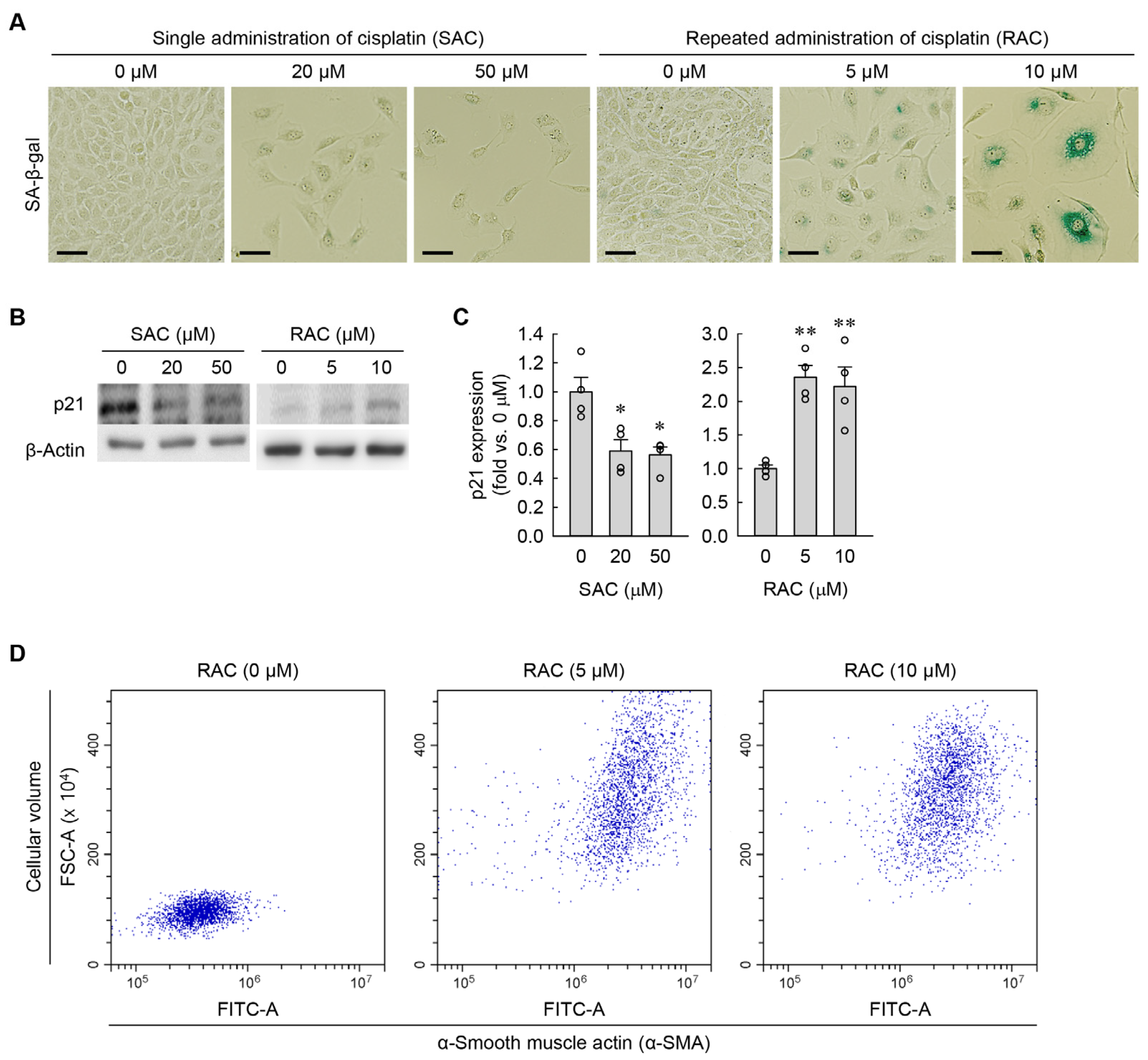
3.2. RAC Enlarges Cell Size and Flattening in Kidney Fibroblasts, but SAC Does Not
3.3. Time-Dependent Change in Cellular Morphology during RAC
3.4. Time-Dependent Changes in Cellular Senescence and Myofibroblast Transformation during RAC
3.5. Cisplatin-Induced JNK Activation Leads to Cellular Hypertrophy but Not to Cellular Senescence and Myofibroblast Formation in Kidney Fibroblasts
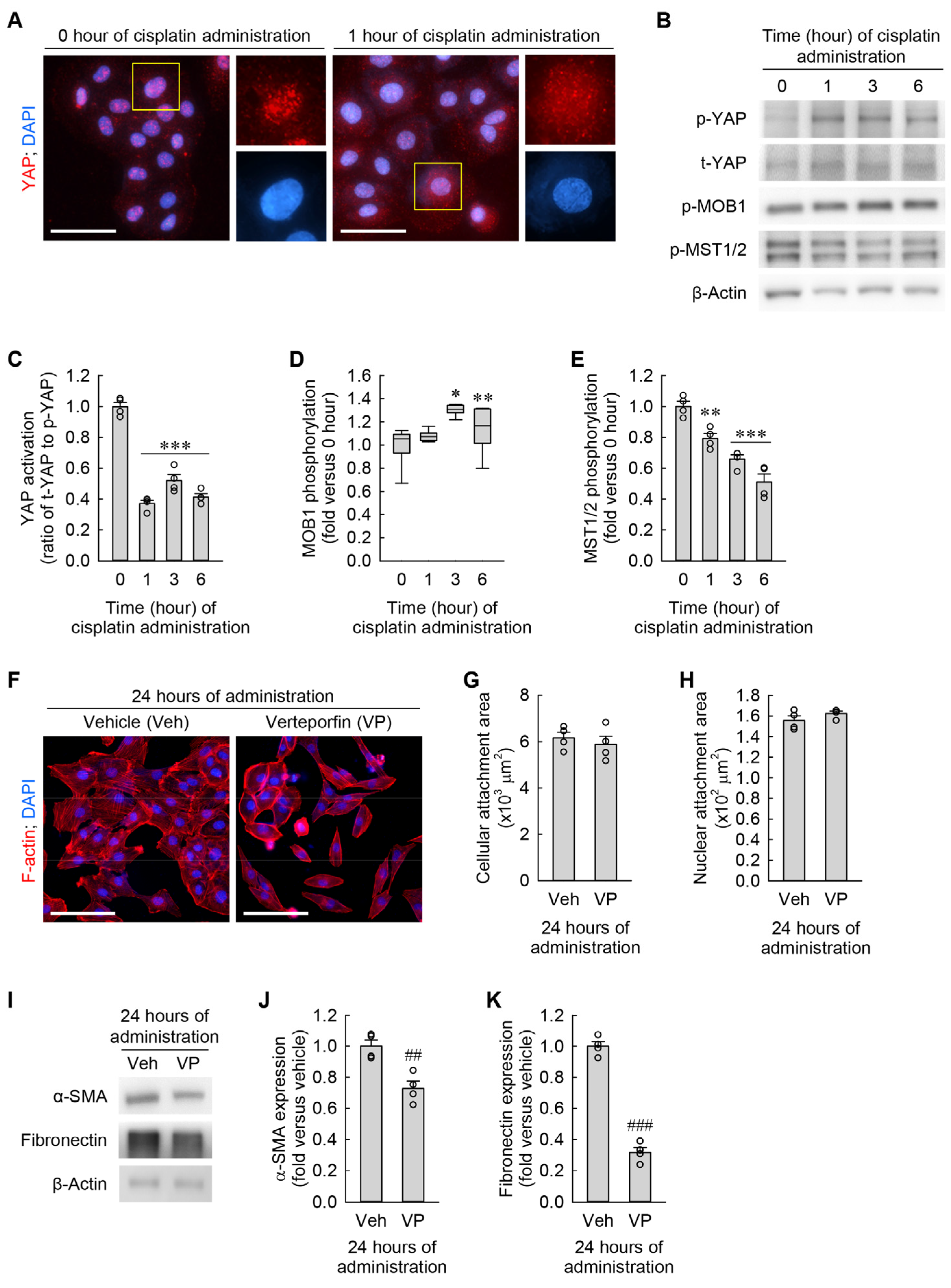
3.6. Cisplatin Attenuates YAP Activation in Kidney Fibroblasts
3.7. YAP Inhibition Prevents Cisplatin-Induced Cellular Hypertrophy, Senescence, and Myofibroblast Transformation in Kidney Fibroblasts
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pabla, N.; Dong, Z. Cisplatin nephrotoxicity: Mechanisms and renoprotective strategies. Kidney Int. 2008, 73, 994–1007. [Google Scholar] [CrossRef]
- Miller, R.P.; Tadagavadi, R.K.; Ramesh, G.; Reeves, W.B. Mechanisms of Cisplatin nephrotoxicity. Toxins 2010, 2, 2490–2518. [Google Scholar] [CrossRef]
- Freyer, D.R.; Brock, P.R.; Chang, K.W.; Dupuis, L.L.; Epelman, S.; Knight, K.; Mills, D.; Phillips, R.; Potter, E.; Risby, D.; et al. Prevention of cisplatin-induced ototoxicity in children and adolescents with cancer: A clinical practice guideline. Lancet Child. Adolesc. Health 2020, 4, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Sears, S.M.; Siskind, L.J. Potential Therapeutic Targets for Cisplatin-Induced Kidney Injury: Lessons from Other Models of AKI and Fibrosis. J. Am. Soc. Nephrol. 2021, 32, 1559–1567. [Google Scholar] [CrossRef] [PubMed]
- Orwick, A.; Sears, S.M.; Sharp, C.N.; Doll, M.A.; Shah, P.P.; Beverly, L.J.; Siskind, L.J. Lung cancer-kidney cross talk induces kidney injury, interstitial fibrosis, and enhances cisplatin-induced nephrotoxicity. Am. J. Physiol. Renal. Physiol. 2023, 324, F287–F300. [Google Scholar] [CrossRef]
- Strutz, F.; Zeisberg, M. Renal fibroblasts and myofibroblasts in chronic kidney disease. J. Am. Soc. Nephrol. 2006, 17, 2992–2998. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Cai, J.; Li, F.; Liu, Z.; Shu, S.; Wang, Y.; Liu, Y.; Tang, C.; Dong, Z. Chronic effects of repeated low-dose cisplatin treatment in mouse kidneys and renal tubular cells. Am. J. Physiol. Renal. Physiol. 2019, 317, F1582–F1592. [Google Scholar] [CrossRef] [PubMed]
- Sharp, C.N.; Doll, M.A.; Dupre, T.V.; Shah, P.P.; Subathra, M.; Siow, D.; Arteel, G.E.; Megyesi, J.; Beverly, L.J.; Siskind, L.J. Repeated administration of low-dose cisplatin in mice induces fibrosis. Am. J. Physiol. Renal. Physiol. 2016, 310, F560–F568. [Google Scholar] [CrossRef]
- Wei, Q.; Dong, G.; Franklin, J.; Dong, Z. The pathological role of Bax in cisplatin nephrotoxicity. Kidney Int. 2007, 72, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.B.; Lee, D.S.; Padanilam, B.J.; Kim, J. Repeated Administration of Cisplatin Transforms Kidney Fibroblasts through G2/M Arrest and Cellular Senescence. Cells 2022, 11, 3472. [Google Scholar] [CrossRef]
- Meran, S.; Steadman, R. Fibroblasts and myofibroblasts in renal fibrosis. Int. J. Exp. Pathol. 2011, 92, 158–167. [Google Scholar] [CrossRef]
- Yu, F.X.; Zhao, B.; Guan, K.L. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 2015, 163, 811–828. [Google Scholar] [CrossRef]
- Gholipour, M.; Tabrizi, A. The role of Hippo signaling pathway in physiological cardiac hypertrophy. Bioimpacts 2020, 10, 251–257. [Google Scholar] [CrossRef]
- Aureille, J.; Buffiere-Ribot, V.; Harvey, B.E.; Boyault, C.; Pernet, L.; Andersen, T.; Bacola, G.; Balland, M.; Fraboulet, S.; Van Landeghem, L.; et al. Nuclear envelope deformation controls cell cycle progression in response to mechanical force. EMBO Rep. 2019, 20, e48084. [Google Scholar] [CrossRef]
- Ramirez, M.T.; Sah, V.P.; Zhao, X.L.; Hunter, J.J.; Chien, K.R.; Brown, J.H. The MEKK-JNK pathway is stimulated by alpha1-adrenergic receptor and ras activation and is associated with in vitro and in vivo cardiac hypertrophy. J. Biol. Chem. 1997, 272, 14057–14061. [Google Scholar] [CrossRef]
- Liang, Q.; Molkentin, J.D. Redefining the roles of p38 and JNK signaling in cardiac hypertrophy: Dichotomy between cultured myocytes and animal models. J. Mol. Cell. Cardiol. 2003, 35, 1385–1394. [Google Scholar] [CrossRef]
- Kim, J.; Padanilam, B.J. Loss of poly(ADP-ribose) polymerase 1 attenuates renal fibrosis and inflammation during unilateral ureteral obstruction. Am. J. Physiol. Renal. Physiol. 2011, 301, F450–F459. [Google Scholar] [CrossRef] [PubMed]
- Moon, D.; Kim, J. Cyclosporin A aggravates hydrogen peroxide-induced cell death in kidney proximal tubule epithelial cells. Anat. Cell Biol. 2019, 52, 312–323. [Google Scholar] [CrossRef] [PubMed]
- Moon, D.; Kim, J.; Yoon, S.P. Yeast extract inhibits the proliferation of renal cell carcinoma cells via regulation of iron metabolism. Mol. Med. Rep. 2019, 20, 3933–3941. [Google Scholar] [CrossRef]
- Moon, D.; Padanilam, B.J.; Jang, H.S.; Kim, J. 2-Mercaptoethanol protects against DNA double-strand breaks after kidney ischemia and reperfusion injury through GPX4 upregulation. Pharmacol. Rep. 2022, 74, 1041–1053. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Moon, D.; Jung, S.; Lee, J.; Kim, J. Cisplatin nephrotoxicity is induced via poly(ADP-ribose) polymerase activation in adult zebrafish and mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2020, 318, R843–R854. [Google Scholar] [CrossRef]
- Kim, J.; Devalaraja-Narashimha, K.; Padanilam, B.J. TIGAR regulates glycolysis in ischemic kidney proximal tubules. Am. J. Physiol. Renal. Physiol. 2015, 308, F298–F308. [Google Scholar] [CrossRef]
- Latham, S.L.; Ehmke, N.; Reinke, P.Y.A.; Taft, M.H.; Eicke, D.; Reindl, T.; Stenzel, W.; Lyons, M.J.; Friez, M.J.; Lee, J.A.; et al. Variants in exons 5 and 6 of ACTB cause syndromic thrombocytopenia. Nat. Commun. 2018, 9, 4250. [Google Scholar] [CrossRef]
- Li, W.L.; Padanilam, B.J.; Kim, J. The Presence of Testis Determines Aristolochic Acid-Induced Nephrotoxicity in Mice. Toxins 2023, 15, 118. [Google Scholar] [CrossRef] [PubMed]
- Blagosklonny, M.V. Cell senescence: Hypertrophic arrest beyond the restriction point. J. Cell. Physiol. 2006, 209, 592–597. [Google Scholar] [CrossRef] [PubMed]
- Feng, M.; Peng, H.; Yao, R.; Zhang, Z.; Mao, G.; Yu, H.; Qiu, Y. Inhibition of cellular communication network factor 1 (CCN1)-driven senescence slows down cartilage inflammaging and osteoarthritis. Bone 2020, 139, 115522. [Google Scholar] [CrossRef]
- Li, Q.; Hagberg, C.E.; Silva Cascales, H.; Lang, S.; Hyvonen, M.T.; Salehzadeh, F.; Chen, P.; Alexandersson, I.; Terezaki, E.; Harms, M.J.; et al. Obesity and hyperinsulinemia drive adipocytes to activate a cell cycle program and senesce. Nat. Med. 2021, 27, 1941–1953. [Google Scholar] [CrossRef] [PubMed]
- Pillai, V.B.; Samant, S.; Hund, S.; Gupta, M.; Gupta, M.P. The nuclear sirtuin SIRT6 protects the heart from developing aging-associated myocyte senescence and cardiac hypertrophy. Aging 2021, 13, 12334–12358. [Google Scholar] [CrossRef]
- Mylonas, K.J.; O’Sullivan, E.D.; Humphries, D.; Baird, D.P.; Docherty, M.H.; Neely, S.A.; Krimpenfort, P.J.; Melk, A.; Schmitt, R.; Ferreira-Gonzalez, S.; et al. Cellular senescence inhibits renal regeneration after injury in mice, with senolytic treatment promoting repair. Sci. Transl. Med. 2021, 13, eabb0203. [Google Scholar] [CrossRef]
- MacKenna, D.A.; Dolfi, F.; Vuori, K.; Ruoslahti, E. Extracellular signal-regulated kinase and c-Jun NH2-terminal kinase activation by mechanical stretch is integrin-dependent and matrix-specific in rat cardiac fibroblasts. J. Clin. Investig. 1998, 101, 301–310. [Google Scholar] [CrossRef]
- Singh, R.; Kaundal, R.K.; Zhao, B.; Bouchareb, R.; Lebeche, D. Resistin induces cardiac fibroblast-myofibroblast differentiation through JAK/STAT3 and JNK/c-Jun signaling. Pharmacol. Res. 2021, 167, 105414. [Google Scholar] [CrossRef]
- Miyamura, N.; Nishina, H. YAP regulates liver size and function. Cell Cycle 2018, 17, 267–268. [Google Scholar] [CrossRef]
- Reginensi, A.; Scott, R.P.; Gregorieff, A.; Bagherie-Lachidan, M.; Chung, C.; Lim, D.S.; Pawson, T.; Wrana, J.; McNeill, H. Yap- and Cdc42-dependent nephrogenesis and morphogenesis during mouse kidney development. PLoS Genet. 2013, 9, e1003380. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Takahashi, M.; Yanagita, M. Pathophysiology of AKI to CKD progression. Semin. Nephrol. 2020, 40, 206–215. [Google Scholar] [CrossRef]
- Sharp, C.N.; Siskind, L.J. Developing better mouse models to study cisplatin-induced kidney injury. Am. J. Physiol. Renal. Physiol. 2017, 313, F835–F841. [Google Scholar] [CrossRef] [PubMed]
- Kim, J. Poly(ADP-ribose) polymerase activation induces high mobility group box 1 release from proximal tubular cells during cisplatin nephrotoxicity. Physiol. Res. 2016, 65, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Long, K.E.; Tang, K.; Padanilam, B.J. Poly(ADP-ribose) polymerase 1 activation is required for cisplatin nephrotoxicity. Kidney Int. 2012, 82, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Wang, Q.; Fan, G.K. The Antiproliferative and Antifibrotic Effects of Cisplatin on Primary Human Vocal Fold Fibroblasts. ORL J. Otorhinolaryngol. Relat. Spec. 2020, 82, 188–200. [Google Scholar] [CrossRef]
- Ramos, C.; Montano, M.; Becerril, C.; Cisneros-Lira, J.; Barrera, L.; Ruiz, V.; Pardo, A.; Selman, M. Acidic fibroblast growth factor decreases alpha-smooth muscle actin expression and induces apoptosis in human normal lung fibroblasts. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 291, L871–L879. [Google Scholar] [CrossRef] [PubMed]
- Nakazono-Kusaba, A.; Takahashi-Yanaga, F.; Morimoto, S.; Furue, M.; Sasaguri, T. Staurosporine-induced cleavage of alpha-smooth muscle actin during myofibroblast apoptosis. J. Investig. Dermatol. 2002, 119, 1008–1013. [Google Scholar] [CrossRef]
- Xie, S.; Swaffer, M.; Skotheim, J.M. Eukaryotic Cell Size Control and Its Relation to Biosynthesis and Senescence. Annu. Rev. Cell Dev. Biol. 2022, 38, 291–319. [Google Scholar] [CrossRef]
- Wilflingseder, J.; Willi, M.; Lee, H.K.; Olauson, H.; Jankowski, J.; Ichimura, T.; Erben, R.; Valerius, M.T.; Hennighausen, L.; Bonventre, J.V. Enhancer and super-enhancer dynamics in repair after ischemic acute kidney injury. Nat. Commun. 2020, 11, 3383. [Google Scholar] [CrossRef] [PubMed]
- Lian, X.J.; Gallouzi, I.E. Oxidative Stress Increases the Number of Stress Granules in Senescent Cells and Triggers a Rapid Decrease in p21waf1/cip1 Translation. J. Biol. Chem. 2009, 284, 8877–8887. [Google Scholar] [CrossRef]
- Nyunoya, T.; Monick, M.M.; Klingelhutz, A.; Yarovinsky, T.O.; Cagley, J.R.; Hunninghake, G.W. Cigarette smoke induces cellular senescence. Am. J. Respir. Cell Mol. Biol. 2006, 35, 681–688. [Google Scholar] [CrossRef]
- Zeng, J.P.; Bi, B.; Chen, L.; Yang, P.; Guo, Y.; Zhou, Y.Q.; Liu, T.Y. Repeated exposure of mouse dermal fibroblasts at a sub-cytotoxic dose of UVB leads to premature senescence: A robust model of cellular photoaging. J. Dermatol. Sci. 2014, 73, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Myrna, K.E.; Pot, S.A.; Murphy, C.J. Meet the corneal myofibroblast: The role of myofibroblast transformation in corneal wound healing and pathology. Vet. Ophthalmol. 2009, 12 (Suppl. S1), 25–27. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Zhou, X.; Jiang, X.; Sun, T. Deletion of ACTA2 in mice promotes angiotensin II induced pathogenesis of thoracic aortic aneurysms and dissections. J. Thorac. Dis. 2018, 10, 4733–4740. [Google Scholar] [CrossRef] [PubMed]
- Bortner, C.D.; Cidlowski, J.A. A necessary role for cell shrinkage in apoptosis. Biochem. Pharmacol. 1998, 56, 1549–1559. [Google Scholar] [CrossRef] [PubMed]
- Benedetti, G.; Fredriksson, L.; Herpers, B.; Meerman, J.; van de Water, B.; de Graauw, M. TNF-alpha-mediated NF-kappaB survival signaling impairment by cisplatin enhances JNK activation allowing synergistic apoptosis of renal proximal tubular cells. Biochem. Pharmacol. 2013, 85, 274–286. [Google Scholar] [CrossRef]
- Sanchez-Perez, I.; Murguia, J.R.; Perona, R. Cisplatin induces a persistent activation of JNK that is related to cell death. Oncogene 1998, 16, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Ohsawa, S.; Igaki, T. Mitochondrial defects trigger proliferation of neighbouring cells via a senescence-associated secretory phenotype in Drosophila. Nat. Commun. 2014, 5, 5264. [Google Scholar] [CrossRef] [PubMed]
- Yosef, R.; Pilpel, N.; Papismadov, N.; Gal, H.; Ovadya, Y.; Vadai, E.; Miller, S.; Porat, Z.; Ben-Dor, S.; Krizhanovsky, V. p21 maintains senescent cell viability under persistent DNA damage response by restraining JNK and caspase signaling. EMBO J. 2017, 36, 2280–2295. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Choo-Wing, R.; Sun, H.; Sureshbabu, A.; Sakurai, R.; Rehan, V.K.; Bhandari, V. A potential role of the JNK pathway in hyperoxia-induced cell death, myofibroblast transdifferentiation and TGF-beta1-mediated injury in the developing murine lung. BMC Cell Biol. 2011, 12, 54. [Google Scholar] [CrossRef]
- Zimmermann, T.; Pommer, M.; Kluge, V.; Chiheb, C.; Muehlich, S.; Bosserhoff, A.K. Detection of Cellular Senescence in Human Primary Melanocytes and Malignant Melanoma Cells In Vitro. Cells 2022, 11, 1489. [Google Scholar] [CrossRef]
- Mugahid, D.; Kalocsay, M.; Liu, X.; Gruver, J.S.; Peshkin, L.; Kirschner, M.W. YAP regulates cell size and growth dynamics via non-cell autonomous mediators. Elife 2020, 9, e53404. [Google Scholar] [CrossRef]
- Vassilev, A.; Kaneko, K.J.; Shu, H.; Zhao, Y.; DePamphilis, M.L. TEAD/TEF transcription factors utilize the activation domain of YAP65, a Src/Yes-associated protein localized in the cytoplasm. Genes Dev. 2001, 15, 1229–1241. [Google Scholar] [CrossRef]
- Xie, Q.; Chen, J.; Feng, H.; Peng, S.; Adams, U.; Bai, Y.; Huang, L.; Li, J.; Huang, J.; Meng, S.; et al. YAP/TEAD-mediated transcription controls cellular senescence. Cancer Res. 2013, 73, 3615–3624. [Google Scholar] [CrossRef]
- Anerillas, C.; Mazan-Mamczarz, K.; Herman, A.B.; Munk, R.; Lam, K.G.; Calvo-Rubio, M.; Garrido, A.; Tsitsipatis, D.; Martindale, J.L.; Altes, G.; et al. The YAP-TEAD complex promotes senescent cell survival by lowering endoplasmic reticulum stress. Nat. Aging 2023, 3, 1237–1250. [Google Scholar] [CrossRef]
- Hoxha, S.; Shepard, A.; Troutman, S.; Diao, H.; Doherty, J.R.; Janiszewska, M.; Witwicki, R.M.; Pipkin, M.E.; Ja, W.W.; Kareta, M.S.; et al. YAP-Mediated Recruitment of YY1 and EZH2 Represses Transcription of Key Cell-Cycle Regulators. Cancer Res. 2020, 80, 2512–2522. [Google Scholar] [CrossRef]
- Zhao, B.; Wei, X.; Li, W.; Udan, R.S.; Yang, Q.; Kim, J.; Xie, J.; Ikenoue, T.; Yu, J.; Li, L.; et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007, 21, 2747–2761. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.; Yu, M.; Xia, R.; Song, K.; Wang, J.; Luo, J.; Chen, G.; Cheng, J. Yap/Taz Deletion in Gli(+) Cell-Derived Myofibroblasts Attenuates Fibrosis. J. Am. Soc. Nephrol. 2017, 28, 3278–3290. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, J.-B.; Padanilam, B.J.; Kim, J. Activation of Yes-Associated Protein Is Indispensable for Transformation of Kidney Fibroblasts into Myofibroblasts during Repeated Administration of Cisplatin. Cells 2024, 13, 1475. https://doi.org/10.3390/cells13171475
Yu J-B, Padanilam BJ, Kim J. Activation of Yes-Associated Protein Is Indispensable for Transformation of Kidney Fibroblasts into Myofibroblasts during Repeated Administration of Cisplatin. Cells. 2024; 13(17):1475. https://doi.org/10.3390/cells13171475
Chicago/Turabian StyleYu, Jia-Bin, Babu J. Padanilam, and Jinu Kim. 2024. "Activation of Yes-Associated Protein Is Indispensable for Transformation of Kidney Fibroblasts into Myofibroblasts during Repeated Administration of Cisplatin" Cells 13, no. 17: 1475. https://doi.org/10.3390/cells13171475