Abstract
Immunotherapy represents a transformative shift in cancer treatment. Among myriad immune-based approaches, chimeric antigen receptor (CAR) T-cell therapy has shown promising results in treating hematological malignancies. Despite aggressive treatment options, the prognosis for patients with malignant brain tumors remains poor. Research leveraging CAR T-cell therapy for brain tumors has surged in recent years. Pre-clinical models are crucial in evaluating the safety and efficacy of these therapies before they advance to clinical trials. However, current models recapitulate the human tumor environment to varying degrees. Novel in vitro and in vivo techniques offer the opportunity to validate CAR T-cell therapies but also have limitations. By evaluating the strengths and weaknesses of various pre-clinical glioma models, this review aims to provide a roadmap for the development and pre-clinical testing of CAR T-cell therapies for brain tumors.
1. Introduction
1.1. Background
The development of chimeric antigen receptor (CAR) T-cell therapy for cancer has progressed dramatically since its first generation in 1993 [1,2,3,4]. CAR T-cell therapy is based upon the expression of CARs on T cells, which are engineered to target specific target antigens and trigger T-cell activation pathways upon binding. By directly recognizing target antigens, CAR T cells do not require traditional mechanisms of antigen presentation; therefore, they act in an HLA-independent manner. This recognition is mediated by the extracellular antigen-binding domain of the CAR, which activates the intracellular signaling domains in the T cell. CAR T-cell therapy has been successful in treating hematological cancers but has had limited efficacy against solid tumors [5]. Indeed, all currently FDA-approved CAR T-cell therapies are treatments for forms of leukemia, lymphoma and multiple myeloma [6]. Solid tumors are difficult malignancies to treat due to tumor heterogeneity, an unfavorable tumor microenvironment (TME) and concomitant toxicities [7,8,9]. Recently, the first immune cell therapy for solid tumors was approved by the FDA. Amtagvi (lifileucel), developed by Iovance Biotherapeutics, is a tumor-infiltrating lymphocyte (TIL) cancer therapy that has shown success in treating melanoma [10].
Malignant brain tumors encompass a wide range of subtypes and classifications [11]. For most of these, available treatments are non-curative and have many unwanted side-effects. CAR T-cell therapies have recently shown promise in treating brain tumors [12,13]. Despite this, they face significant barriers to further success—such as tumor heterogeneity, a hostile TME and impaired delivery across the blood–brain barrier (BBB)—that may be addressed in the pre-clinical research stage. Despite these challenges, research in this field has made several advances in recent years, including combination therapies, armored CAR T cells and multi-targeting constructs. These platforms require robust pre-clinical models to test efficacy and recapitulate the most salient aspects of their target malignancies (Figure 1).
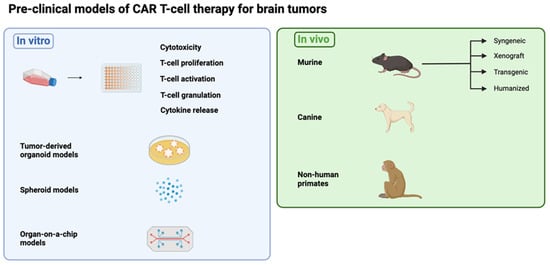
Figure 1.
Overview of the pre-clinical models discussed in this review. (Figure created with BioRender.com (accessed on 3 April 2024)).
Pre-clinical models allow for the screening and evaluation of candidate therapies, particularly for toxicity and efficacy prior to clinical translation. The bulk of pre-clinical work is typically conducted in murine models, which have several strengths and limitations depending on the specific approach. Common models, such as genetically engineered murine models (GEMMs), transgenic mice, and canine and primate models, offer new perspectives. Careful selection and combination of both in vitro and in vivo testing must be considered. This should be based on factors such as the recapitulation of the human TME and the ability to optimally account for associated toxicities in humans. This review describes the different types of currently available pre-clinical models for gliomas, including their qualities and limitations, that can be leveraged to develop new CAR T-cell therapies for tumors in the brain. Similar experimental considerations could be applied to models for TIL therapy.
1.2. CAR T-Cell Therapy for Glioma
Recent advances have shown great promise for CAR T-cell therapy for solid brain tumors [14]. CAR T cells have been shown to cross the BBB and, in some instances, to do so more effectively than other therapeutic strategies, such as monoclonal antibodies or vaccines [15,16]. Another benefit of CAR T-cell therapy is the ability to directly kill tumor cells without the need for an endogenous immune response. Currently, many CAR T cells are in pre-clinical development for solid brain tumors, aiming to successfully translate into clinical trials [17,18].
The most prevalent and lethal type of malignant brain tumor is glioblastoma (GBM). It is classified as a grade 4 tumor according to the World Health Organization (WHO) grading system. The median survival for patients diagnosed with glioblastoma is close to 15 months, highlighting the urgency of developing novel therapies [19]. GBM is the most aggressive type of glioma, which is a broader category of brain tumors. Gliomas are thought to arise from glial cells, which are supportive cells that surround and provide critical support for neurons in the brain, spine or related progenitor cells. Not all gliomas are considered malignant, as some grow slowly and are not considered to be cancerous. Anaplastic gliomas, diffuse intrinsic pontine gliomas (DIPGs) and glioblastomas are some examples of malignant gliomas.
There are several limitations in the treatment of glioma with CAR T cells. First, the identification of adequate tumor-associated antigens (TAAs) has been difficult, as GBMs are heterogeneous and promote antigen escape. Moreover, such targets are often not exclusively found in tumor tissue. This carries the theoretical risk of on-target/off-tumor toxicity, which can be life-threatening [20,21]. Secondly, the hostile TME for CAR T cells represents a significant barrier. A number of tumor-associated changes, including hypoxia, immune infiltration, the Warburg effect and hostile metabolism, combine to promote tumor survival while also inhibiting the efficacy of T cell-based therapies [17].
Prior pre-clinical and clinical studies of CAR T cells that target solid brain tumors have demonstrated the potential for therapeutic benefit. Promising results have been achieved in treating diffuse intrinsic pontine glioma with CAR T cells targeting GD2 [22]. Another target, B7-H3, was shown to have encouraging outcomes in a clinical trial [23]. Another study leveraging CAR T-cell therapy for patients with high-grade glioma targeted EGFRvIII engineered to secrete T cell-engaging antibodies that bind to wild-type EGFR [24,25,26,27]. Tumors regressed in all three patients treated with this therapy. Previously, this unique approach had been tested using several different xenograft models and pre-clinical assays to assess safety, efficacy and toxicity [28].
While CAR T-cell therapy can be highly effective, it can result in life-threatening toxicities. This is of great importance, as pre-clinical models often do not effectively predict these. Toxicities can present with many symptoms. Cytokine Release Syndrome (CRS) and Immune effector Cell-Associated Neurotoxicity Syndrome (ICANS) are most commonly found in patients [29]. Syngeneic murine models have not accurately recapitulated the toxicities observed in patients treated with CAR T-cell therapy, which is reported in 37–93% of patients across various studies [30]. Among pre-clinical publications of CAR T-cell therapy for glioma, very few report on toxicity, despite its observation in the clinical setting [31].
2. Overview of CAR T-Cell Generation
The correct generation of CAR T cells is crucial to effectively implement these pre-clinical models for glioma. Here, we discuss the choice of good candidates as TAAs for CAR T-cell therapy, as well as how researchers can create or obtain their own CAR T-cell therapies.
2.1. Choosing CAR T Cells
CAR T cells should be engineered to target antigens that are overexpressed on glioma cells specifically. Common targets include EGFRvIII, IL13Rα2, HER2 and GD2. As previously discussed, it is challenging to find adequate glioma TAAs for CAR T-cell therapy. In glioma, TAAs are often not tumor-specific but can also be found in small amounts in healthy tissue. Consequently, CAR T-cell therapies targeting these antigens carry the theoretical risk of harm to non-tumor tissues. Researchers are developing new methods to address these challenges and effectively eliminate glioma cells, such as CAR T cells secreting BiTEs or dual TAA targeting [28,32].
2.2. Generating CAR T Cells
Detailed protocols are available in the literature that comprehensively describe various processes of generating customized CAR T cells [33,34]. This can be a rigorous process involving multiple precise steps, and often, collaboration with experts in cell therapy and gene engineering is necessary in order to successfully accomplish these aims. First, an antigen recognition domain should be designed that is specific to the chosen antigen and translated in tandem with a spacer/linker, as well as transmembrane and signaling domains [35]. Several iterations of CARs have been designed, and new generations are actively being engineered [36]. The CAR gene must be subsequently incorporated into a gene delivery platform. The typical delivery strategy is viral transduction due to its high transduction efficiency. Lentiviral transduction is most commonly used for CAR T cells [37].
However, non-viral alternatives are actively being explored to mitigate the higher immunogenicity and manufacturing cost associated with viral vectors [38]. These include the Sleeping Beauty (SB) transposon system, designer nucleases, CRISPR/Cas9 systems, electroporation and nanoparticles [39]. The development and delivery of CAR T-cell therapies could become more accessible and efficient through the use of these methods [40]. Using CRISPR/Cas9, researchers demonstrated high levels of safety and efficiency for anti-CD19 PD1 CAR T cells in pre-clinical models and a phase I clinical trial [41]. SB-engineered CAR T cells were also successfully employed in a phase I/II clinical trial without severe toxicities [42].
Depending on the selected model, T cells should also be derived from the appropriate origin. For example, when leveraging a syngeneic mouse model, where both the immune system and target tumor cells are murine, CAR T cells should be made with murine T cells. However, when using a xenograft mouse model with human target tumor cells, human CAR T cells should be used.
Another possible source of CAR T cells for researchers is transgenic mouse lines developed by the Jackson Laboratory (JAX). These mice are genetically modified so that T cells express specific human or murine proteins, facilitating pre-clinical models. However, as CAR T-cell targets for glioma are delicate and new strategies are continuously being explored, researchers generally generate their own CAR T cells [43]. Researchers have developed unique mouse strains, for example, those from which all T cells continuously express EGFRvIII and Her2 CAR [44,45]. Making murine CAR T cells can be a laborious process and may lead to batches of differing quality. This strategy could serve as a useful source of reliable murine CAR T cells that can be used in syngeneic mouse models.
3. Overview of Glioma Tumor Models for CAR T-Cell Therapy
In this section, we discuss several glioma models that have been used for the testing of CAR T-cell therapy.
3.1. Cell Lines
3.1.1. Murine Glioma Models
Murine cell lines for malignant glioma are used in syngeneic mouse models. Because these models have an intact immune system, they require cells to be injected into mice of an analogous genetic background. This offers a significant advantage in addressing questions to which the endogenous immune environment may be relevant. Different strategies exist to generate glioma cell lines, including chemical induction, viral induction, the use of Sleeping Beauty transposons and the isolation of spontaneously arising tumors.
Chemically induced glioma tumor models include GL261 and CT-2A. These tumor lines were created using carcinogenic chemical compounds. The first brain tumor was induced in a mouse via intracranial delivery of 20-methylcholanthrene in the C3H mouse strain in 1939 [46]. These types of models are especially vulnerable to genetic drift and high mutational burdens [47]. Chemically induced models also frequently lack mismatch repair (MMR) genes, which make cells more robust against alkylating agents such as temozolomide. Virally induced tumor cell lines are used less frequently and maintain high immunogenicity similar to chemically induced models [48]. These are most commonly engineered by injecting lentiviral retroviral vectors into mice that express oncogenes [49].
In comparison, spontaneous tumor models are relatively rare and can be challenging to cultivate [50]. The VM mouse strain is unique, as its inbred properties result in a relatively high incidence (1.5%) of spontaneous brain neoplasms. As these tumors develop more naturally, they mimic the invasive growth characteristics of human GBM. VM-M3 and SMA-560 are two examples of glioma cell lines that were derived from this mouse strain [51,52].
As previously mentioned, transposons may be leveraged to genetically engineer various cell types. The SB transposon system is part of the Tc1/mariner class and can recognize specific DNA sequences known as inverted repeat/direct repeat (IR/DR) sites. After recognition, they are able to ‘cut and paste’ to integrate DNA transposons into the host genome at these sites. This method has been employed to generate novel glioma models in mice by inserting genetic lesions into the genome of stem cells along the lateral ventricle of neonatal mice [53,54,55,56]. By altering the murine genome, the etiology and histopathology of human GBM are closely recapitulated [57]. These tumor cell lines can be engineered to express tumor-specific antigens such as EGFRvIII, IL13Rα2 and GD2 to create CAR T-cell glioma models. Below, we discuss the most relevant murine glioma cell lines to be used for CAR T-cell therapy research.
GL261
The GL261 cell line originated in 1970 through chemical induction, when methylcholanthrene pellets were surgically implanted into the brains of C57BL/6 mice [58]. Despite its widespread use, a disadvantage of GL261 is its robust immunogenicity compared to human GBM [59,60]. GL261 cells have elevated MHC I expression and tumor mutational burden (TMB). The TMB from GL261 is almost 5000 mutations per Mb, while human GBM has an average of 2.7 mutations per Mb [61]. High TMB has been shown to correlate with better responses in models of immune checkpoint blockade, which may, in part, be responsible for the disparity between pre-clinical promise and success in clinical trials for these agents [62,63]. Furthermore, GL261 cells contain mutations of p53 and K-Ras, leading to elevated levels of c-Myc, which is not typically observed in human GBM [59].
CT-2A
CT-2A was created in 1992 by chemical induction using 20-methylcholanthrene [64]. It is believed to be more aggressive than other established glioma cell lines [65]. Similar to GL261, CT-2A is used extensively in GBM research, given its ability to mimic intra-tumoral heterogeneity, as well as the radio- and chemoresistance that is associated with GBM [66].
SMA-560
SMA-560 was not generated with chemical induction but was, instead, derived from spontaneous tumors in 1980 [52,67]. SMA-560 is moderately immunogenic due to modest expression of MHC I [68]. The murine background of this cell line is VM/Dk. SMA-560 is moderately used—but far less frequently than GL261 and CT2A—and is, therefore, also, perhaps, less well characterized [69].
SB28
SB28 was generated in 2014 using sleeping beauty (SB) transposon constructs that induced de novo gliomas in neonatal C57BL/6 mice [54,70]. SB28 has been shown to be low in immunogenicity and is negative for MHC I [71]. Furthermore, compared to all other known murine GBM cell lines, SB28 displays one of the most accurate TMEs relative to human GBM. SB28 is almost fully resistant to treatment with immune checkpoint blockade and has a lower mutational load [62].
005
The 005 murine GBM cell line was derived from glioma stem cells after lentiviral transduction with H-Ras and AKT in Trp53+/− C57BL/6 mice in 2009 [72]. Relative advantages of this model include its reduced immunogenicity and histological similarities to human GBM with regard to heterogeneity and invasiveness. Recently, 005 tumor models, when compared to GL261, CT-2A and Mut-3, were found to most closely resemble the immune-phenotypic environment of human GBM specimens [73].
4C8
In 1999, another new murine glioma cell line was developed utilizing glioma cells from a spontaneous glioma grown in a transgenic B6D2F1 mouse [74]. 4C8 has proven to be a highly cellular tumor with aggressive invasion into cerebral ventricles and meninges (Table 1).

Table 1.
Syngeneic glioma cell lines.
3.1.2. Human Cell Lines for GBM
Human cell lines for GBM can be used both in vitro and in vivo in xenograft models. Xenograft models of GBM involve the engraftment of human GBM cells into immunodeficient mice. The following three types of immunodeficient are primarily used: (1) nude mice, which lack T cells; (2) non-obese diabetic severe combined immunodeficiency (NOD-SCID) mice, which lack T and B cells; and (3) NOD-SCID IL2R-γ null (NSG or NOG) mice, which lack T, B or NK cell activity [75,76]. Disadvantages of xenograft models include the inability to study the effects and impact of the endogenous immune system.
By engrafting patient-derived GBM cells into immunocompromised mice, the histopathologic, genomic and phenotypic characteristics of the primary tumor are preserved. The wide variety of human gliomas that have been tested in these models have been catalogued elsewhere [77,78,79]. In general, xenograft models offer the advantage of using human cancer cells, providing more accurate insights into aspects of tumor development and therapeutic response to therapies that can be used in patients. However, these models lack an endogenous immune system.
4. Overview of In Vitro Models
4.1. Cell Culture Assays
CAR T-cell efficacy is often correlated with the ability of a particular approach to induce T-cell activation, persistence, proliferation, inhibition and exhaustion. Perhaps the most straightforward method of evaluating CAR T cells in an in vitro system is a cytotoxicity assay. In these assays, effector response is measured against cells expressing tumor-associated antigens (TAAs). The killing of target cell lines can be quantified and analyzed using live cell imaging, bioluminescence assays or flow cytometry analysis [80]. The chromium release assay, developed in the 1968, provides an alternative approach [81]. The co-culturing of tumor target cells with CAR T cells at different densities can also be used to assess CAR T-cell proliferation, activation and degranulation. With the addition of certain agents, such as 3H-thymidine or presto blue, fluorescence intensity can be measured after several days as a proxy for proliferation and expansion. T-cell activation, degranulation and differentiation can also be evaluated by flow cytometry. These assays offer efficient, inexpensive and valuable methods of characterizing CAR T-cell therapies.
4.2. Patient-Derived Organoids
Cerebral organoid models represent an attempt to better recapitulate the environment of the brain. They are three-dimensional cell cultures derived from a patient’s tumor and used to model the TME, although earlier versions were generated from normal tissue organoids by introducing pro-tumorigenic mutations [82,83]. By utilizing induced pluripotent stem cells or embryonic stem cells and a specialized growth process, a primitive organoid resembling certain structures in the brain can be cultured in vitro [84]. These organoid models retain key features of the patient’s tumor, such as its heterogeneity and microenvironment [85]. They serve as an intermediary between in vitro and in vivo models.
4.3. Spheroid Model
Spheroids are 3D cell aggregates that are not attached to any surface, providing a unique environment that more accurately mimics solid tumors compared to 2D cell cultures. Spheroid cells are often cultured with a hydrogel matrix, which functions as an extracellular matrix [86,87]. Multiple spheroid models have been constructed for GBM to study immunological approaches [86,88,89]. Spheroids are either matrix-based or matrix-free (i.e., suspension cells). Matrix-based models provide the advantage of demonstrating the invasion of tumor cells [90]. Generally, these scaffolds are made up of natural or synthetic hydrogels in an effort to mimic the brain ECM.
4.4. Organ-on-a-Chip Model
Organ-on-a-chip (OoC) models also seek to incorporate critical features of the human TME [91]. Inside each chip is a co-culture of primary human cells with a tissue–vascular interface. Media flowing in the vascular channel provide shear stress, which is required to model human immune cell recruitment with specificity [92]. For instance, CAR T cells may flow through these channels and migrate through the endothelium in response to local inflammatory conditions. Limitations of OoC models include the fact that the chip materials are artificially engineered and may not exactly replicate the TME. The absence or imbalance of specific cellular components can also lead to significant differences between models [93].
5. Overview of In Vivo Models
Prior to translation to clinical trials, in vivo models are essential in the evaluation of the efficacy and safety of CAR T-cell therapies for brain tumors. To date, the most commonly used models are syngeneic and xenograft murine models. Both have unique advantages and limitations, and no single approach provides a perfect pre-clinical framework. Recent advances and new models such as transgenic or humanized mice, canine models and non-human primate models provide novel ways to evaluate CAR T-cell therapies (Figure 2).
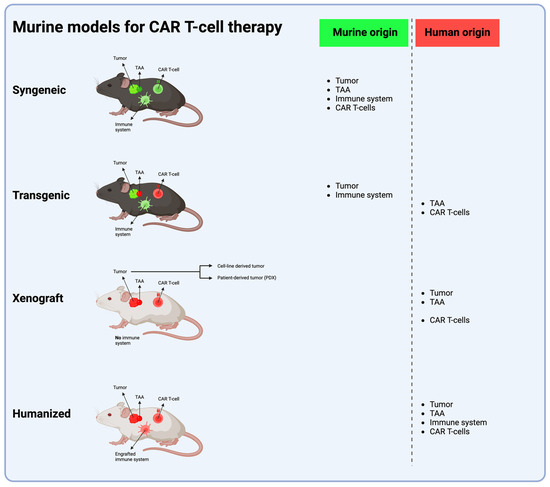
Figure 2.
The four murine models most frequently used in CAR T-cell therapy development. The syngeneic model uses an immunocompetent mouse with a functional murine immune system implanted with a tumor of murine origin; this tumor holds a murine TAA, which is targeted with murine CAR T cells. The transgenic model utilizes an immunocompetent mouse genetically engineered to express a human TAA, which is targeted with human CAR T cells. Xenograft and humanized models both use immunoincompetent mice, with NSG mice being the most widely used. In the xenograft model, in vitro cultured human tumor cells or patient-derived human cells are implanted into mice lacking a functional immune system, and the mice are, thereafter, treated with human CAR T cells. In the humanized model, (a part of) the functional human immune system is engrafted into the mouse, human tumor cells are implanted and the mouse is treated with human CAR T cells. (Abbreviations: TAA = tumor-associated antigen) (Figure created with BioRender.com (accessed on 1 April 2024)).
5.1. Syngeneic Mouse Model
The syngeneic mouse model is the oldest and most utilized model for immunotherapy [94]. A syngeneic mouse model uses immunocompetent mice in which histocompatible tumor cells are implanted [95]. These tumor cells are derived from a genetically identical murine strain. Orthotopic inoculation provides a more realistic TME [96]. The site of injection can trigger variable antitumor responses [97].
The advantages of a syngeneic model includes a functional immune system; and TME and, in certain cases, the ability to study pre-conditioning therapy prior to adoptive transfer [69,98]. This model is also particularly well-suited for combination regimens of CAR T-cell therapy and other immunotherapies, such as oncolytic viruses (OV), checkpoint inhibitors or small-molecule inhibitors [99,100]. However, syngeneic models have not been especially useful in other settings, such as in the recapitulation of cytokine release syndrome (CRS), the most commonly found toxicity in patients treated with CAR T-cell therapy. Furthermore, syngeneic tumor cells, similar to all cell lines cultured in vitro, are subject to mutations and genetic drift over time.
5.2. Xenograft Mouse Model
Xenografts from cell lines and patient-derived xenografts (PDXs) for GBM have been extensively utilized to recapitulate molecular and genomic characteristics of human tumors and to assess therapeutic efficacy in the setting of challenges such as tumor heterogeneity [101].
5.2.1. Xenografts from Cell Lines
Cell line-derived xenografts, particularly from the U87 and U251 glioblastoma cell lines, have been a mainstay in brain tumor research for decades [102]. These models involve the transplantation of human tumor cell lines into immunocompromised mice, either in the brain (orthotopic) or in other tissues (heterotopic), achieved by subcutaneously injecting cells into the flank, for example. Typically, severe combined immunodeficient (SCID) [103], non-obese diabetic (NOD) [104] and NOD-SCID IL2rγnull (NSG) mice are used to establish xenografts [105,106]. As a caveat to the use of these models, one study found that the U87 cell line had a drastically different DNA profile from that of the original tumor it was generated from [107]. Additionally, many of these cell lines have demonstrated failure to infiltrate the brain parenchyma when orthotopically implanted [108].
5.2.2. Patient-Derived Xenografts (PDXs)
Patient-derived xenograft models represent a significant advancement over cell line-derived xenografts, as they may maintain the cellular heterogeneity and architecture of the original patient tumors. PDX models are established by transplanting fresh tumor tissue or cells obtained from patient tissue directly into immunocompromised mice [101].
While cells can also be grown in adherent culture, and studies have shown superior tumor engraftment of neurospheres injected into the brain [109,110]. PDXs generated by these methods have been used in GBM CAR T-cell experiments [28,111]. One PDX, namely BT74, was used to accurately recapitulate glioma heterogeneity and the physiological expression of two GBM antigens, namely EGFRv3 and IL13Rα2, in pre-clinical studies of CAR T-cell approaches [111]. Despite these translational advantages, PDXs are laborious to generate and maintain, and engraftment rates for GBM range from 25 to 75 percent [78,102,112,113,114].
5.3. Transgenic Mouse Model
Transgenic (Tg) mouse models or genetically engineered mouse models (GEMMs) are unique models for the study of the effects of CAR T-cell therapy. In these models, the phenotype of a mouse can be engineered, for example, by stimulating the expression of oncogenes or inactivating tumor-suppressor genes [115]. Transgenic models insert oncogenes into tumor precursor cells to obtain carcinogenesis in mice, and genetically engineered mouse models broadly refer to these and other strategies used for genetic perturbation [116]. Genetically engineered mouse models can be obtained through either viral delivery, the Cre-LoxP system, the RCAS-TVA system, CRISPR-Cas9 or transposons [88]. This means tumor growth in the murine model occurs more naturally than that of artificially implanted cancer cells. As these tumors would grow in situ and are not implanted, which provides a unique environment to study tumor progression [117]. Additionally, specific tumor mutations and TAAs can be incorporated into the tumor generation process to allow for more specific murine models [118]. Numerous GEMM glioma cell lines have been created [116,119]. Key published GEMMs are summarized in Table 2.
Table 2.
GEMMs of glioma cell lines.
5.4. Humanized Mouse Model
A humanized mouse has no murine immune system but is engrafted with a human immune system. The most commonly used model is NSG (NOD SCID) mice transplanted with human CD34+ HSPCs or PBMCs [131,132]. HSPCs can regenerate lymphoid and myeloid compartments, but T-cell development in these animals is not optimal due to the lack of a thymus [133]. It is also possible to inject human CD34+ cells without thymectomizing the mice [134]. Since these mice lack T, B, NK and functional DCs, they are highly receptive to engraftment of human cells—both healthy and tumor tissues [135]. Another approach to this model requires the implantation of bone marrow, liver and thymus (BLT) into immunodeficient mice, typically NSG. This favors the lymphoid compartment and results in improved T-cell reconstitution, maturation and selection [136].
5.5. Canine Model
One infrequently used approach for CAR T-cell therapy for gliomas is the use of canine models, which typically arise in genetically outbred animals with intact immune systems [137]. Canine tumors are quite similar to human tumors and have shown comparable responses to various treatments [138]. Many of the adversities seen in clinical trials have also been observed in canine trials, reinforcing the similarity between humans and dogs [139]. However, canine trials require unique expertise, funding and specific infrastructure.
5.6. Primate Model
A few groups have recently tested CAR T-cell therapies using non-human primate (NHP) models. One relative strength of NHP models is their ability to provide greater fidelity when recapitulating immune system characteristics and genetic heterogeneity. These similarities give primate models an advantage when testing the safety of CAR T-cell therapies, specifically for on-target/off-tumor toxicity and cytokine release syndrome (CRS) [133].
A limitation of NHP systems is the dearth of tumor models available for meaningful therapeutic evaluation. Primary tumors in NHP models are rare and have limited practicality. The use of tumor cell lines from other animals would also quickly lead to rejection of the tumor [140]. As such, NHP models may be better suited for the modeling of the toxicity and safety of CAR T-cell therapies.
The first study evaluating CAR T cells in NHP models was conducted in 2015 [141]. Researchers tested CAR T cells targeting orphan tyrosine kinase receptor ROR1, which is frequently expressed in many hematological and solid malignancies. They observed no symptoms of toxicity after treating two macaques with autologous CAR T cells, even at high dosages [142]. As of April 2024, there are five ongoing clinical trials investigating the safety of ROR1-targeting CAR T cells in humans [143]. Another group performed a study on non-human primates using an anti-CD20 CAR T cell therapy. They found signs of significant cytotoxicity correlated with pro-inflammatory CSF cytokines and pan-T-cell encephalitis. Evidence of CRS included levels of IL-6 and IL-8 that were recapitulated in clinical trials [144]. Ephrin type B receptor 4 (EPHB4) CAR T cells were also tested in a lymphodepleted NHP model. Researchers found a slight elevation in IL-6 but no symptoms of CRS. This might be attributed to this certain model having no tumor and, therefore, no TAA to fully activate the CAR T cells. This could prevent CRS or immune effector cell-associated neurotoxicity syndrome (ICANS) [145]. To date, no CAR T-cell therapies targeting brain tumors in NHP models have been described, but previous studies show their promising utility in modeling the safety of these therapies.
6. Recommendations
The authors recommend considering both xenograft and syngeneic in vivo murine models for the pre-clinical validation of CAR T-cell therapies for glioma in order to enhance the evaluation of both specificity and toxicity. Clinical trials with CAR T-cell therapies for solid tumors have highlighted the importance of the TME, underscoring the value of models that can recapitulate the TME. There should be some consideration given to the planning of experiments that match in vitro and in vivo systems. These models will become essential in the future for the establishment of more reliable CAR T-cell therapies. We recommend that researchers critically evaluate all models and consider the specific goals of investigation of their therapies.
7. Conclusions
The integration of different pre-clinical models is essential for the advancement of CAR T-cell therapy for brain tumors towards clinical success. While each model contributes valuable insights, none is able to fully replicate the complexities of human brain tumors. Researchers must carefully consider the benefits and limitations of each model for pre-clinical evaluation of their therapies. Although imperfect, in vitro and in vivo models have proven vital in the discovery and development of new treatment opportunities for brain tumors. Future efforts should focus on refining existing models, developing novel systems and, most, importantly maintaining collaboration between pre-clinical and clinical programs.
Author Contributions
Conceptualization: G.V., R.R., L.G.R., B.D.C. and E.P.G.; investigation: G.V., R.R., S.J.S., M.T., A.K., E.P.G., J.S., B.D.C. and W.T.C.; methodology: G.V., R.R., L.G.R., S.J.S., A.K., E.P.G., B.D.C. and W.T.C.; formal analysis: G.V., R.R. and B.D.C.; resources: G.V., L.G.R., R.R., B.D.C. and W.T.C.; data curation: G.V. and R.R.; writing—original draft preparation: G.V., R.R., M.G., A.K. and E.P.G.; writing—review and editing: G.V., R.R., M.G., L.G.R., M.T., A.K., S.J.S., E.P.G., J.S., W.T.C. and B.D.C.; visualization: G.V.; supervision: W.T.C. and B.D.C.; funding acquisition: G.V. and B.D.C. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by The Jenny Fund (B.D.C. and W.T.C.), A Shot for Life (B.D.C. and W.T.C.), Swim Across America (B.D.C.), the Marcus Foundation (B.D.C. and W.T.C.), the Brian D. Silber Memorial Fund (B.D.C. and W.T.C.) and HHS/National Institutes of Health (NIH) K12CA090354 and R25NS065743 (B.D.C.). G.V. is supported by the Belgian American Eduational Foundation.
Conflicts of Interest
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests. B.D.C. is an inventor on patents and patent applications relating to T-cell engineering approaches. B.D.C. serves as a consultant for Third Rock Ventures on CAR T-cell strategies for glioma.
References
- Choi, B.D.; Gerstner, E.R.; Frigault, M.J.; Leick, M.B.; Mount, C.W.; Balaj, L.; Nikiforow, S.; Carter, B.S.; Curry, W.T.; Gallagher, K.; et al. Intraventricular CARv3-TEAM-E T Cells in Recurrent Glioblastoma. N. Engl. J. Med. 2024, 390, 1290–1298. [Google Scholar] [CrossRef] [PubMed]
- Davila, M.L.; Riviere, I.; Wang, X.; Bartido, S.; Park, J.; Curran, K.; Chung, S.S.; Stefanski, J.; Borquez-Ojeda, O.; Olszewska, M.; et al. Efficacy and Toxicity Management of 19-28z CAR T Cell Therapy in B Cell Acute Lymphoblastic Leukemia. Sci. Transl. Med. 2014, 6, 224ra25. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.W.; Kochenderfer, J.N.; Stetler-Stevenson, M.; Cui, Y.K.; Delbrook, C.; Feldman, S.A.; Fry, T.J.; Orentas, R.; Sabatino, M.; Shah, N.N.; et al. T Cells Expressing CD19 Chimeric Antigen Receptors for Acute Lymphoblastic Leukaemia in Children and Young Adults: A Phase 1 Dose-Escalation Trial. Lancet 2015, 385, 517–528. [Google Scholar] [CrossRef]
- Kalos, M.; Levine, B.L.; Porter, D.L.; Katz, S.; Grupp, S.A.; Bagg, A.; June, C.H. T Cells with Chimeric Antigen Receptors Have Potent Antitumor Effects and Can Establish Memory in Patients with Advanced Leukemia. Sci. Transl. Med. 2011, 3, 95ra73. [Google Scholar] [CrossRef]
- Eshhar, Z.; Waks, T.; Gross, G.; Schindler, D.G. Specific Activation and Targeting of Cytotoxic Lymphocytes through Chimeric Single Chains Consisting of Antibody-Binding Domains and the Gamma or Zeta Subunits of the Immunoglobulin and T-Cell Receptors. Proc. Natl. Acad. Sci. USA 1993, 90, 720–724. [Google Scholar] [CrossRef] [PubMed]
- Dagar, G.; Gupta, A.; Masoodi, T.; Nisar, S.; Merhi, M.; Hashem, S.; Chauhan, R.; Dagar, M.; Mirza, S.; Bagga, P.; et al. Harnessing the Potential of CAR-T Cell Therapy: Progress, Challenges, and Future Directions in Hematological and Solid Tumor Treatments. J. Transl. Med. 2023, 21, 449. [Google Scholar] [CrossRef] [PubMed]
- Thirumalaisamy, R.; Vasuki, S.; Sindhu, S.M.; Mothilal, T.M.; Srimathi, V.; Poornima, B.; Bhuvaneswari, M.; Hariharan, M. FDA-Approved Chimeric Antigen Receptor (CAR)-T Cell Therapy for Different Cancers-A Recent Perspective. Mol. Biotechnol. 2024, 1–15. [Google Scholar] [CrossRef]
- Albelda, S.M. CAR T Cell Therapy for Patients with Solid Tumours: Key Lessons to Learn and Unlearn. Nat. Rev. Clin. Oncol. 2024, 21, 47–66. [Google Scholar] [CrossRef]
- Grewal, E.P.; Richardson, L.G.K.; Sun, J.; Ramapriyan, R.; Martinez-Lage, M.; Miller, J.J.; Cahill, D.P.; Choi, B.D.; Curry, W.T. Suppression of Antitumor Immune Signatures and Upregulation of VEGFA as IDH-Mutant Gliomas Progress to Higher Grade. Neurosurg. Focus 2024, 56, E2. [Google Scholar] [CrossRef]
- Richardson, L.G.; Nieman, L.T.; Stemmer-Rachamimov, A.O.; Zheng, X.S.; Stafford, K.; Nagashima, H.; Miller, J.J.; Kiyokawa, J.; Ting, D.T.; Wakimoto, H.; et al. IDH-Mutant Gliomas Harbor Fewer Regulatory T Cells in Humans and Mice. Oncoimmunology 2020, 9, 1806662. [Google Scholar] [CrossRef]
- Keam, S.J. Lifileucel: First Approval. Mol. Diagn. Ther. 2024, 28, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A Summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Choi, B.D.; Curry, W.T.; Carter, B.S.; Maus, M.V. Chimeric Antigen Receptor T-Cell Immunotherapy for Glioblastoma: Practical Insights for Neurosurgeons. Neurosurg. Focus 2018, 44, E13. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.D.; O’Rourke, D.M.; Maus, M.V. Engineering Chimeric Antigen Receptor T Cells to Treat Glioblastoma. J. Target. Ther. Cancer 2017, 6, 22–25. [Google Scholar] [PubMed]
- Suryadevara, C.M.; Desai, R.; Abel, M.L.; Riccione, K.A.; Batich, K.A.; Shen, S.H.; Chongsathidkiet, P.; Gedeon, P.C.; Elsamadicy, A.A.; Snyder, D.J.; et al. Temozolomide Lymphodepletion Enhances CAR Abundance and Correlates with Antitumor Efficacy against Established Glioblastoma. Oncoimmunology 2018, 7, e1434464. [Google Scholar] [CrossRef] [PubMed]
- Del Baldo, G.; Del Bufalo, F.; Pinacchio, C.; Carai, A.; Quintarelli, C.; De Angelis, B.; Merli, P.; Cacchione, A.; Locatelli, F.; Mastronuzzi, A. The Peculiar Challenge of Bringing CAR-T Cells into the Brain: Perspectives in the Clinical Application to the Treatment of Pediatric Central Nervous System Tumors. Front. Immunol. 2023, 14, 1142597. [Google Scholar]
- Zhao, T.; Li, C.; Ge, H.; Lin, Y.; Kang, D. Glioblastoma Vaccine Tumor Therapy Research Progress. Chin. Neurosurg. J. 2022, 8, 2. [Google Scholar] [CrossRef]
- Luksik, A.S.; Yazigi, E.; Shah, P.; Jackson, C.M. CAR T Cell Therapy in Glioblastoma: Overcoming Challenges Related to Antigen Expression. Cancers 2023, 15, 1414. [Google Scholar] [CrossRef]
- Suryadevara, C.M.; Desai, R.; Farber, S.H.; Choi, B.D.; Swartz, A.M.; Shen, S.H.; Gedeon, P.C.; Snyder, D.J.; Herndon, J.E.; Healy, P.; et al. Preventing Lck Activation in CAR T Cells Confers Treg Resistance but Requires 4-1BB Signaling for Them to Persist and Treat Solid Tumors in Nonlymphodepleted Hosts. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 358–368. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Gatto, L.; Ricciotti, I.; Tosoni, A.; Di Nunno, V.; Bartolini, S.; Ranieri, L.; Franceschi, E. CAR-T Cells Neurotoxicity from Consolidated Practice in Hematological Malignancies to Fledgling Experience in CNS Tumors: Fill the Gap. Front. Oncol. 2023, 13, 1206983. [Google Scholar] [CrossRef]
- Ramapriyan, R.; Vykunta, V.S.; Vandecandelaere, G.; Richardson, L.G.K.; Sun, J.; Curry, W.T.; Choi, B.D. Altered Cancer Metabolism and Implications for Next-Generation CAR T-Cell Therapies. Pharmacol. Ther. 2024, 259, 108667. [Google Scholar] [CrossRef] [PubMed]
- Majzner, R.G.; Ramakrishna, S.; Yeom, K.W.; Patel, S.; Chinnasamy, H.; Schultz, L.M.; Richards, R.M.; Jiang, L.; Barsan, V.; Mancusi, R.; et al. GD2-CAR T Cell Therapy for H3K27M-Mutated Diffuse Midline Gliomas. Nature 2022, 603, 934–941. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Liu, F.; Liu, Z.; Cao, Y.; Zhang, Z.; Wang, Y.; Huang, J.; Fan, S.; Zhao, S.; Chen, Y.; et al. Bioactivity and Safety of B7-H3-targeted Chimeric Antigen Receptor T Cells against Anaplastic Meningioma. Clin. Transl. Immunol. 2020, 9, e1137. [Google Scholar] [CrossRef]
- Choi, B.D.; Maus, M.V.; June, C.H.; Sampson, J.H. Immunotherapy for Glioblastoma: Adoptive T-Cell Strategies. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 2042–2048. [Google Scholar] [CrossRef]
- Miao, H.; Choi, B.D.; Suryadevara, C.M.; Sanchez-Perez, L.; Yang, S.; De Leon, G.; Sayour, E.J.; McLendon, R.; Herndon, J.E.; Healy, P.; et al. EGFRvIII-Specific Chimeric Antigen Receptor T Cells Migrate to and Kill Tumor Deposits Infiltrating the Brain Parenchyma in an Invasive Xenograft Model of Glioblastoma. PLoS ONE 2014, 9, e94281. [Google Scholar] [CrossRef]
- Choi, B.D.; Suryadevara, C.M.; Gedeon, P.C.; Herndon, J.E., II; Sanchez-Perez, L.; Bigner, D.D.; Sampson, J.H. Intracerebral Delivery of a Third Generation EGFRvIII-Specific Chimeric Antigen Receptor Is Efficacious against Human Glioma. J. Clin. Neurosci. Off. J. Neurosurg. Soc. Australas. 2014, 21, 189–190. [Google Scholar] [CrossRef]
- Choi, B.D.; Yu, X.; Castano, A.P.; Bouffard, A.A.; Schmidts, A.; Larson, R.C.; Bailey, S.R.; Boroughs, A.C.; Frigault, M.J.; Leick, M.B.; et al. CAR-T Cells Secreting BiTEs Circumvent Antigen Escape without Detectable Toxicity. Nat. Biotechnol. 2019, 37, 1049–1058. [Google Scholar] [CrossRef]
- Jain, M.D.; Smith, M.; Shah, N.N. How I Treat Refractory CRS and ICANS after CAR T-Cell Therapy. Blood 2023, 141, 2430–2442. [Google Scholar]
- Morris, E.C.; Neelapu, S.S.; Giavridis, T.; Sadelain, M. Cytokine Release Syndrome and Associated Neurotoxicity in Cancer Immunotherapy. Nat. Rev. Immunol. 2022, 22, 85–96. [Google Scholar] [CrossRef]
- Kalaitsidou, M.; Kueberuwa, G.; Schütt, A.; Gilham, D.E. CAR T-Cell Therapy: Toxicity and the Relevance of Preclinical Models. Immunotherapy 2015, 7, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Fyfe, I. CAR T Cells Offer Hope in Glioblastoma. Nat. Rev. Neurol. 2024, 20, 315. [Google Scholar] [CrossRef]
- Stock, S.; Schmitt, M.; Sellner, L. Optimizing Manufacturing Protocols of Chimeric Antigen Receptor T Cells for Improved Anticancer Immunotherapy. Int. J. Mol. Sci. 2019, 20, 6223. [Google Scholar] [CrossRef]
- Riccione, K.; Suryadevara, C.M.; Snyder, D.; Cui, X.; Sampson, J.H.; Sanchez-Perez, L. Generation of CAR T Cells for Adoptive Therapy in the Context of Glioblastoma Standard of Care. J. Vis. Exp. 2015, 96, 52397. [Google Scholar] [CrossRef]
- Sadelain, M.; Brentjens, R.; Riviere, I. The Basic Principles of Chimeric Antigen Receptor Design. Cancer Discov. 2013, 3, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Dejenie, T.A.; G/Medhin, M.T.; Terefe, G.D.; Admasu, F.T.; Tesega, W.W.; Abebe, E.C. Current Updates on Generations, Approvals, and Clinical Trials of CAR T-Cell Therapy. Hum. Vaccines Immunother. 2022, 18, 2114254. [Google Scholar] [CrossRef]
- McGarrity, G.J.; Hoyah, G.; Winemiller, A.; Andre, K.; Stein, D.; Blick, G.; Greenberg, R.N.; Kinder, C.; Zolopa, A.; Binder-Scholl, G.; et al. Patient Monitoring and Follow-Up in Lentiviral Clinical Trials. J. Gene Med. 2013, 15, 78–82. [Google Scholar] [CrossRef]
- Atsavapranee, E.S.; Billingsley, M.M.; Mitchell, M.J. Delivery Technologies for T Cell Gene Editing: Applications in Cancer Immunotherapy. eBioMedicine 2021, 67, 103354. [Google Scholar] [CrossRef]
- Pinto, I.S.; Cordeiro, R.A.; Faneca, H. Polymer- and Lipid-Based Gene Delivery Technology for CAR T Cell Therapy. J. Control. Release 2023, 353, 196–215. [Google Scholar] [CrossRef] [PubMed]
- Balke-Want, H.; Keerthi, V.; Cadinanos-Garai, A.; Fowler, C.; Gkitsas, N.; Brown, A.K.; Tunuguntla, R.; Abou-el-Enein, M.; Feldman, S.A. Non-Viral Chimeric Antigen Receptor (CAR) T Cells Going Viral. Immuno-Oncol. Technol. 2023, 18, 100375. [Google Scholar] [CrossRef]
- Zhang, J.; Hu, Y.; Yang, J.; Li, W.; Zhang, M.; Wang, Q.; Zhang, L.; Wei, G.; Tian, Y.; Zhao, K.; et al. Non-Viral, Specifically Targeted CAR-T Cells Achieve High Safety and Efficacy in B-NHL. Nature 2022, 609, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Magnani, C.F.; Gaipa, G.; Lussana, F.; Belotti, D.; Gritti, G.; Napolitano, S.; Matera, G.; Cabiati, B.; Buracchi, C.; Borleri, G.; et al. Sleeping Beauty–Engineered CAR T Cells Achieve Antileukemic Activity without Severe Toxicities. J. Clin. Investig. 2020, 130, 6021–6033. [Google Scholar] [CrossRef] [PubMed]
- Haydar, D.; Ibañez-Vega, J.; Crawford, J.C.; Chou, C.-H.; Guy, C.; Meehl, M.; Yi, Z.; Langfitt, D.; Vogel, P.; DeRenzo, C.; et al. CAR T-Cell Design Dependent Remodeling of the Brain Tumor Immune Microenvironment Identify Macrophages as Key Players That Inhibit or Promote Anti-Tumor Activity. Res. Sq. 2023, 12, 2430–2446. [Google Scholar] [CrossRef]
- Chuntova, P.; Hou, Y.; Naka, R.; Yamamichi, A.; Chen, T.; Goretsky, Y.; Hatae, R.; Nejo, T.; Kohanbash, G.; Mende, A.L.; et al. Novel EGFRvIII-CAR Transgenic Mice for Rigorous Preclinical Studies in Syngeneic Mice. Neuro-Oncology 2021, 24, 259–272. [Google Scholar] [CrossRef]
- Yong, C.S.; John, L.B.; Devaud, C.; Prince, M.H.; Johnstone, R.W.; Trapani, J.A.; Darcy, P.K.; Kershaw, M.H. A Role for Multiple Chimeric Antigen Receptor-Expressing Leukocytes in Antigen-Specific Responses to Cancer. Oncotarget 2016, 7, 34582–34598. [Google Scholar]
- Seligman, A.; Shear, M.J.; Alexander, L. Studies in Carcinogenesis: VIII. Experimental Production of Brain Tumors in Mice with Methylcholanthrene. Am. J. Cancer 1939, 37, 364–395. [Google Scholar]
- Haddad, A.F.; Young, J.S.; Amara, D.; Berger, M.S.; Raleigh, D.R.; Aghi, M.K.; Butowski, N.A. Mouse Models of Glioblastoma for the Evaluation of Novel Therapeutic Strategies. Neuro-Oncol. Adv. 2021, 3, vdab100. [Google Scholar] [CrossRef]
- Hewitt, H.B.; Blake, E.R.; Walder, A.S. A Critique of the Evidence for Active Host Defence against Cancer, Based on Personal Studies of 27 Murine Tumours of Spontaneous Origin. Br. J. Cancer 1976, 33, 241–259. [Google Scholar] [CrossRef]
- Uhrbom, L.; Hesselager, G.; Nistér, M.; Westermark, B. Induction of Brain Tumors in Mice Using a Recombinant Platelet-Derived Growth Factor B-Chain Retrovirus. Cancer Res. 1998, 58, 5275–5279. [Google Scholar]
- Peterson, D.L.; Sheridan, P.J.; Brown, W.E. Animal Models for Brain Tumors: Historical Perspectives and Future Directions. J. Neurosurg. 1994, 80, 865–876. [Google Scholar] [CrossRef]
- Shelton, L.M.; Mukherjee, P.; Huysentruyt, L.C.; Urits, I.; Rosenberg, J.A.; Seyfried, T.N. A Novel Pre-Clinical In Vivo Mouse Model for Malignant Brain Tumor Growth and Invasion. J. Neurooncol. 2010, 99, 165–176. [Google Scholar] [CrossRef]
- Fraser, H. Astrocytomas in an Inbred Mouse Strain. J. Pathol. 1971, 103, 266–270. [Google Scholar] [CrossRef]
- Garcia-Fabiani, M.B.; Kadiyala, P.; Lowenstein, P.R.; Castro, M.G. An Optimized Protocol for In Vivo Analysis of Tumor Cell Division in a Sleeping Beauty-Mediated Mouse Glioma Model. STAR Protoc. 2020, 1, 100044. [Google Scholar] [CrossRef] [PubMed]
- Wiesner, S.M.; Decker, S.A.; Larson, J.D.; Ericson, K.; Forster, C.; Gallardo, J.L.; Long, C.; Demorest, Z.L.; Zamora, E.A.; Low, W.C.; et al. De Novo Induction of Genetically Engineered Brain Tumors in Mice Using Plasmid DNA. Cancer Res. 2009, 69, 431–439. [Google Scholar] [CrossRef]
- Calinescu, A.-A.; Núñez, F.J.; Koschmann, C.; Kolb, B.L.; Lowenstein, P.R.; Castro, M.G. Transposon Mediated Integration of Plasmid DNA into the Subventricular Zone of Neonatal Mice to Generate Novel Models of Glioblastoma. J. Vis. Exp. 2015, 96, 52443. [Google Scholar] [CrossRef]
- Koschmann, C.; Calinescu, A.-A.; Nunez, F.J.; Mackay, A.; Fazal-Salom, J.; Thomas, D.; Mendez, F.; Kamran, N.; Dzaman, M.; Mulpuri, L.; et al. ATRX Loss Promotes Tumor Growth and Impairs Nonhomologous End Joining DNA Repair in Glioma. Sci. Transl. Med. 2016, 8, 328ra28. [Google Scholar] [CrossRef]
- Alghamri, M.S.; Núñez, F.J.; Kamran, N.; Carney, S.; Altshuler, D.; Lowenstein, P.R.; Castro, M.G. Chapter Six—Functional Characterization of Tumor Antigen-Specific T-Cells Isolated from the Tumor Microenvironment of Sleeping Beauty Induced Murine Glioma Models. In Methods in Enzymology; Tumor Immunology and Immunotherapy—Cellular Methods Part A; Galluzzi, L., Rudqvist, N.-P., Eds.; Academic Press: New York, NY, USA, 2020; Volume 631, pp. 91–106. [Google Scholar]
- Ausman, J.I.; Shapiro, W.R.; Rall, D.P. Studies on the Chemotherapy of Experimental Brain Tumors: Development of an Experimental Model. Cancer Res. 1970, 30, 2394–2400. [Google Scholar]
- Szatmári, T.; Lumniczky, K.; Désaknai, S.; Trajcevski, S.; Hídvégi, E.J.; Hamada, H.; Sáfrány, G. Detailed Characterization of the Mouse Glioma 261 Tumor Model for Experimental Glioblastoma Therapy. Cancer Sci. 2006, 97, 546–553. [Google Scholar] [CrossRef]
- Maes, W.; Van Gool, S.W. Experimental Immunotherapy for Malignant Glioma: Lessons from Two Decades of Research in the GL261 Model. Cancer Immunol. Immunother. 2011, 60, 153–160. [Google Scholar] [CrossRef]
- Johanns, T.M.; Ward, J.P.; Miller, C.A.; Wilson, C.; Kobayashi, D.K.; Bender, D.; Fu, Y.; Alexandrov, A.; Mardis, E.R.; Artyomov, M.N.; et al. Endogenous Neoantigen-Specific CD8 T Cells Identified in Two Glioblastoma Models Using a Cancer Immunogenomics Approach. Cancer Immunol. Res. 2016, 4, 1007–1015. [Google Scholar] [CrossRef]
- Genoud, V.; Marinari, E.; Nikolaev, S.I.; Castle, J.C.; Bukur, V.; Dietrich, P.-Y.; Okada, H.; Walker, P.R. Responsiveness to Anti-PD-1 and Anti-CTLA-4 Immune Checkpoint Blockade in SB28 and GL261 Mouse Glioma Models. OncoImmunology 2018, 7, e1501137. [Google Scholar] [CrossRef]
- Wainwright, D.A.; Chang, A.L.; Dey, M.; Balyasnikova, I.V.; Kim, C.K.; Tobias, A.; Cheng, Y.; Kim, J.W.; Qiao, J.; Zhang, L.; et al. Durable Therapeutic Efficacy Utilizing Combinatorial Blockade against IDO, CTLA-4 and PD-L1 in Mice with Brain Tumors. Clin. Cancer Res. 2014, 20, 5290–5301. [Google Scholar] [CrossRef] [PubMed]
- Seyfried, T.N.; El-Abbadi, M.; Roy, M.L. Ganglioside Distribution in Murine Neural Tumors. Mol. Chem. Neuropathol. 1992, 17, 147–167. [Google Scholar] [CrossRef]
- Noffsinger, B.; Witter, A.; Sheybani, N.; Xiao, A.; Manigat, L.; Zhong, Q.; Taori, S.; Harris, T.; Bullock, T.; Price, R.; et al. Technical Choices Significantly Alter the Adaptive Immune Response against Immunocompetent Murine Gliomas in a Model-Dependent Manner. J. Neurooncol. 2021, 154, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Martínez Murillo, R.; Martínez, A. Standardization of an Orthotopic Mouse Brain Tumor Model Following Transplantation of CT-2A Astrocytoma Cells. Histol. Histopathol. 2007, 22, 1309–1326. [Google Scholar]
- Serano, R.D.; Pegram, C.N.; Bigner, D.D. Tumorigenic Cell Culture Lines from a Spontaneous VM/Dk Murine Astrocytoma (SMA). Acta Neuropathol. 1980, 51, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Sampson, J.H.; Ashley, D.M.; Archer, G.E.; Fuchs, H.E.; Dranoff, G.; Hale, L.P.; Bigner, D.D. Characterization of a Spontaneous Murine Astrocytoma and Abrogation of Its Tumorigenicity by Cytokine Secretion. Neurosurgery 1997, 41, 1365–1372. [Google Scholar] [CrossRef]
- Sampson, J.H.; Choi, B.D.; Sanchez-Perez, L.; Suryadevara, C.M.; Snyder, D.J.; Flores, C.T.; Schmittling, R.J.; Nair, S.; Reap, E.A.; Norberg, P.K.; et al. EGFRvIII mCAR-Modified T-Cell Therapy Cures Mice with Established Intracerebral Glioma and Generates Host Immunity against Tumor-Antigen Loss. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 972–984. [Google Scholar] [CrossRef]
- Kosaka, A.; Ohkuri, T.; Okada, H. Combination of an Agonistic Anti-CD40 Monoclonal Antibody and the COX-2 Inhibitor Celecoxib Induces Anti-Glioma Effects by Promotion of Type-1 Immunity in Myeloid Cells and T-Cells. Cancer Immunol. Immunother. 2014, 63, 847–857. [Google Scholar] [CrossRef]
- Letchuman, V.; Ampie, L.; Shah, A.H.; Brown, D.A.; Heiss, J.D.; Chittiboina, P. Syngeneic Murine Glioblastoma Models: Reactionary Immune Changes and Immunotherapy Intervention Outcomes. Neurosurg. Focus 2022, 52, E5. [Google Scholar]
- Marumoto, T.; Tashiro, A.; Friedmann-Morvinski, D.; Scadeng, M.; Soda, Y.; Gage, F.H.; Verma, I.M. Development of a Novel Mouse Glioma Model Using Lentiviral Vectors. Nat. Med. 2009, 15, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Khalsa, J.K.; Cheng, N.; Keegan, J.; Chaudry, A.; Driver, J.; Bi, W.L.; Lederer, J.; Shah, K. Immune Phenotyping of Diverse Syngeneic Murine Brain Tumors Identifies Immunologically Distinct Types. Nat. Commun. 2020, 11, 3912. [Google Scholar] [CrossRef]
- Weiner, N.E.; Pyles, R.B.; Chalk, C.L.; Balko, M.G.; Miller, M.A.; Dyer, C.A.; Warnick, R.E.; Parysek, L.M. A Syngeneic Mouse Glioma Model for Study of Glioblastoma Therapy. J. Neuropathol. Exp. Neurol. 1999, 58, 54–60. [Google Scholar] [CrossRef][Green Version]
- Yoshida, G.J. Applications of Patient-Derived Tumor Xenograft Models and Tumor Organoids. J. Hematol. Oncol. 2020, 13, 4. [Google Scholar] [CrossRef]
- Gómez-Oliva, R.; Domínguez-García, S.; Carrascal, L.; Abalos-Martínez, J.; Pardillo-Díaz, R.; Verástegui, C.; Castro, C.; Nunez-Abades, P.; Geribaldi-Doldán, N. Evolution of Experimental Models in the Study of Glioblastoma: Toward Finding Efficient Treatments. Front. Oncol. 2021, 10, 614295. [Google Scholar] [CrossRef] [PubMed]
- Hicks, W.H.; Bird, C.E.; Traylor, J.I.; Shi, D.D.; El Ahmadieh, T.Y.; Richardson, T.E.; McBrayer, S.K.; Abdullah, K.G. Contemporary Mouse Models in Glioma Research. Cells 2021, 10, 712. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Qi, L.; Du, Y.; Huang, L.F.; Braun, F.K.; Kogiso, M.; Zhao, Y.; Li, C.; Lindsay, H.; Zhao, S.; et al. Patient-Derived Orthotopic Xenograft (PDOX) Mouse Models of Primary and Recurrent Meningioma. Cancers 2020, 12, 1478. [Google Scholar] [CrossRef]
- Zeng, W.; Tang, Z.; Li, Y.; Yin, G.; Liu, Z.; Gao, J.; Chen, Y.; Chen, F. Patient-Derived Xenografts of Different Grade Gliomas Retain the Heterogeneous Histological and Genetic Features of Human Gliomas. Cancer Cell Int. 2020, 20, 1. [Google Scholar] [CrossRef]
- Brakel, B.A.; Chokshi, C.R.; Salim, S.K.; Venugopal, C.; Singh, S. In Vitro Evaluation of CAR-T Cells in Patient-Derived Glioblastoma Models. STAR Protoc. 2021, 2, 100920. [Google Scholar] [CrossRef] [PubMed]
- Hickey, M.J.; Malone, C.C.; Erickson, K.E.; Gomez, G.G.; Young, E.L.; Liau, L.M.; Prins, R.M.; Kruse, C.A. Implementing Preclinical Study Findings to Protocol Design: Translational Studies with Alloreactive CTL for Gliomas. Am. J. Transl. Res. 2012, 4, 114–126. [Google Scholar]
- Eichmüller, O.L.; Knoblich, J.A. Human Cerebral Organoids—A New Tool for Clinical Neurology Research. Nat. Rev. Neurol. 2022, 18, 661–680. [Google Scholar] [CrossRef] [PubMed]
- Drost, J.; Van Jaarsveld, R.H.; Ponsioen, B.; Zimberlin, C.; Van Boxtel, R.; Buijs, A.; Sachs, N.; Overmeer, R.M.; Offerhaus, G.J.; Begthel, H.; et al. Sequential Cancer Mutations in Cultured Human Intestinal Stem Cells. Nature 2015, 521, 43–47. [Google Scholar] [CrossRef]
- Linkous, A.; Balamatsias, D.; Snuderl, M.; Edwards, L.; Miyaguchi, K.; Milner, T.; Reich, B.; Cohen-Gould, L.; Storaska, A.; Nakayama, Y.; et al. Modeling Patient-Derived Glioblastoma with Cerebral Organoids. Cell Rep. 2019, 26, 3203–3211.e5. [Google Scholar] [CrossRef] [PubMed]
- Khamis, Z.I.; Sarker, D.B.; Xue, Y.; Al-Akkary, N.; James, V.D.; Zeng, C.; Li, Y.; Sang, Q.-X.A. Modeling Human Brain Tumors and the Microenvironment Using Induced Pluripotent Stem Cells. Cancers 2023, 15, 1253. [Google Scholar] [CrossRef]
- Carrasco-Mantis, A.; Randelovic, T.; Castro-Abril, H.; Ochoa, I.; Doblaré, M.; Sanz-Herrera, J.A. A Mechanobiological Model for Tumor Spheroid Evolution with Application to Glioblastoma: A Continuum Multiphysics Approach. Comput. Biol. Med. 2023, 159, 106897. [Google Scholar] [CrossRef] [PubMed]
- Manikandan, C.; Jaiswal, A.K. Scaffold-Based Spheroid Models of Glioblastoma Multiforme and Its Use in Drug Screening. Biotechnol. Bioeng. 2023, 120, 2117–2132. [Google Scholar] [CrossRef]
- Pasupuleti, V.; Vora, L.; Prasad, R.; Nandakumar, D.N.; Khatri, D.K. Glioblastoma Preclinical Models: Strengths and Weaknesses. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2024, 1879, 189059. [Google Scholar] [CrossRef]
- Kong, D.; Kwon, D.; Moon, B.; Kim, D.-H.; Kim, M.-J.; Choi, J.; Kang, K.-S. CD19 CAR-Expressing iPSC-Derived NK Cells Effectively Enhance Migration and Cytotoxicity into Glioblastoma by Targeting to the Pericytes in Tumor Microenvironment. Biomed. Pharmacother. 2024, 174, 116436. [Google Scholar] [CrossRef]
- Abedin, M.J.; Michelhaugh, S.K.; Mittal, S.; Berdichevsky, Y. 3D Models of Glioblastoma Interaction with Cortical Cells. Front. Bioeng. Biotechnol. 2023, 11, 1150772. [Google Scholar] [CrossRef]
- Woodham, A.W.; Marianne, M.; Cespedes, A.V.; Heng, A.R.; Frischmann, A.S.; Fortes, G.M.; Kajula, V.J.; Ewart, L.; Carman, C.V. Chimeric Antigen Receptor-T Cell Efficacy Can Be Evaluated on an Organ-Chip Model System. J. Immunol. 2023, 210, 146.06. [Google Scholar] [CrossRef]
- Leung, C.M.; de Haan, P.; Ronaldson-Bouchard, K.; Kim, G.-A.; Ko, J.; Rho, H.S.; Chen, Z.; Habibovic, P.; Jeon, N.L.; Takayama, S.; et al. A Guide to the Organ-on-a-Chip. Nat. Rev. Methods Primers 2022, 2, 33. [Google Scholar] [CrossRef]
- Xie, Z.; Chen, M.; Lian, J.; Wang, H.; Ma, J. Glioblastoma-on-a-Chip Construction and Therapeutic Applications. Front. Oncol. 2023, 13, 1183059. [Google Scholar] [CrossRef]
- Olson, B.; Li, Y.; Lin, Y.; Liu, E.T.; Patnaik, A. Mouse Models for Cancer Immunotherapy Research. Cancer Discov. 2018, 8, 1358–1365. [Google Scholar] [CrossRef]
- Zeng, Z.; Wong, C.J.; Yang, L.; Ouardaoui, N.; Li, D.; Zhang, W.; Gu, S.; Zhang, Y.; Liu, Y.; Wang, X.; et al. TISMO: Syngeneic Mouse Tumor Database to Model Tumor Immunity and Immunotherapy Response. Nucleic Acids Res. 2022, 50, D1391–D1397. [Google Scholar] [CrossRef]
- Cook, D.P.; Galpin, K.J.C.; Rodriguez, G.M.; Shakfa, N.; Wilson-Sanchez, J.; Echaibi, M.; Pereira, M.; Matuszewska, K.; Haagsma, J.; Murshed, H.; et al. Comparative Analysis of Syngeneic Mouse Models of High-Grade Serous Ovarian Cancer. Commun. Biol. 2023, 6, 1152. [Google Scholar] [CrossRef] [PubMed]
- Carlson, P.M.; Mohan, M.; Rodriguez, M.; Subbotin, V.; Sun, C.X.; Patel, R.B.; Birstler, J.; Hank, J.A.; Rakhmilevich, A.L.; Morris, Z.S.; et al. Depth of Tumor Implantation Affects Response to in Situ Vaccination in a Syngeneic Murine Melanoma Model. J. Immunother. Cancer 2021, 9, e002107. [Google Scholar] [CrossRef]
- Kaminska, P.; Cyranowski, S.; Pilanc, P.; Malik, A.R. Syngeneic Mouse Model of Glioblastoma: Intracranial Implantation of GL261 Cells. In Neurobiology: Methods and Protocols; Methods in Molecular Biology; Dworkin, S., Ed.; Springer: New York, NY, USA, 2024; pp. 135–146. ISBN 978-1-07-163585-8. [Google Scholar]
- Al-Haideri, M.; Tondok, S.B.; Safa, S.H.; Maleki, A.H.; Rostami, S.; Jalil, A.T.; Al-Gazally, M.E.; Alsaikhan, F.; Rizaev, J.A.; Mohammad, T.A.M.; et al. CAR-T Cell Combination Therapy: The next Revolution in Cancer Treatment. Cancer Cell Int. 2022, 22, 365. [Google Scholar] [CrossRef] [PubMed]
- Grosser, R.; Cherkassky, L.; Chintala, N.; Adusumilli, P.S. Combination Immunotherapy with CAR T Cells and Checkpoint Blockade for the Treatment of Solid Tumors. Cancer Cell 2019, 36, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Akter, F.; Simon, B.; de Boer, N.L.; Redjal, N.; Wakimoto, H.; Shah, K. Pre-Clinical Tumor Models of Primary Brain Tumors: Challenges and Opportunities. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2021, 1875, 188458. [Google Scholar] [CrossRef]
- Patrizii, M.; Bartucci, M.; Pine, S.R.; Sabaawy, H.E. Utility of Glioblastoma Patient-Derived Orthotopic Xenografts in Drug Discovery and Personalized Therapy. Front. Oncol. 2018, 8, 23. [Google Scholar] [CrossRef]
- Bosma, G.C.; Custer, R.P.; Bosma, M.J. A Severe Combined Immunodeficiency Mutation in the Mouse. Nature 1983, 301, 527–530. [Google Scholar] [CrossRef]
- Makino, S.; Kunimoto, K.; Muraoka, Y.; Mizushima, Y.; Katagiri, K.; Tochino, Y. Breeding of a Non-Obese, Diabetic Strain of Mice. Exp. Anim. 1980, 29, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Racki, W.J.; Covassin, L.; Brehm, M.; Pino, S.; Ignotz, R.; Dunn, R.; Laning, J.; Graves, S.K.; Rossini, A.A.; Shultz, L.D.; et al. NOD-Scid IL2rγnull (NSG) Mouse Model of Human Skin Transplantation and Allograft Rejection. Transplantation 2010, 89, 527–536. [Google Scholar] [CrossRef]
- Choi, B.D.; Yu, X.; Castano, A.P.; Darr, H.; Henderson, D.B.; Bouffard, A.A.; Larson, R.C.; Scarfò, I.; Bailey, S.R.; Gerhard, G.M.; et al. CRISPR-Cas9 Disruption of PD-1 Enhances Activity of Universal EGFRvIII CAR T Cells in a Preclinical Model of Human Glioblastoma. J. Immunother. Cancer 2019, 7, 304. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.; Bjerke, M.; Edlund, H.; Nelander, S.; Westermark, B. Origin of the U87MG Glioma Cell Line: Good News and Bad News. Sci. Transl. Med. 2016, 8, 354re3. [Google Scholar] [CrossRef]
- Radaelli, E.; Ceruti, R.; Patton, V.; Russo, M.; Degrassi, A.; Croci, V.; Caprera, F.; Stortini, G.; Scanziani, E.; Pesenti, E.; et al. Immunohistopathological and Neuroimaging Characterization of Murine Orthotopic Xenograft Models of Glioblastoma Multiforme Recapitulating the Most Salient Features of Human Disease. Histol. Histopathol. 2009, 24, 879–891. [Google Scholar] [CrossRef] [PubMed]
- Nigim, F.; Esaki, S.; Hood, M.; Lelic, N.; James, M.F.; Ramesh, V.; Stemmer-Rachamimov, A.; Cahill, D.P.; Brastianos, P.K.; Rabkin, S.D.; et al. A New Patient-Derived Orthotopic Malignant Meningioma Model Treated with Oncolytic Herpes Simplex Virus. Neuro-Oncology 2016, 18, 1278–1287. [Google Scholar] [CrossRef]
- Wakimoto, H.; Kesari, S.; Farrell, C.J.; Curry, W.T.; Zaupa, C.; Aghi, M.; Kuroda, T.; Stemmer-Rachamimov, A.; Shah, K.; Liu, T.-C.; et al. Human Glioblastoma—Derived Cancer Stem Cells: Establishment of Invasive Glioma Models and Treatment with Oncolytic Herpes Simplex Virus Vectors. Cancer Res. 2009, 69, 3472–3481. [Google Scholar] [CrossRef]
- Schmidts, A.; Srivastava, A.A.; Ramapriyan, R.; Bailey, S.R.; Bouffard, A.A.; Cahill, D.P.; Carter, B.S.; Curry, W.T.; Dunn, G.P.; Frigault, M.J.; et al. Tandem Chimeric Antigen Receptor (CAR) T Cells Targeting EGFRvIII and IL-13Rα2 Are Effective against Heterogeneous Glioblastoma. Neuro-Oncol. Adv. 2023, 5, vdac185. [Google Scholar] [CrossRef]
- William, D.; Mullins, C.S.; Schneider, B.; Orthmann, A.; Lamp, N.; Krohn, M.; Hoffmann, A.; Classen, C.-F.; Linnebacher, M. Optimized Creation of Glioblastoma Patient Derived Xenografts for Use in Preclinical Studies. J. Transl. Med. 2017, 15, 27. [Google Scholar] [CrossRef]
- Kerstetter-Fogle, A.E.; Harris, P.L.R.; Brady-Kalnay, S.M.; Sloan, A.E. Generation of Glioblastoma Patient-Derived Intracranial Xenografts for Preclinical Studies. Int. J. Mol. Sci. 2020, 21, 5113. [Google Scholar] [CrossRef] [PubMed]
- Charbonneau, M.; Harper, K.; Brochu-Gaudreau, K.; Perreault, A.; Roy, L.-O.; Lucien, F.; Tian, S.; Fortin, D.; Dubois, C.M. The Development of a Rapid Patient-Derived Xenograft Model to Predict Chemotherapeutic Drug Sensitivity/Resistance in Malignant Glial Tumors. Neuro-Oncology 2023, 25, 1605–1616. [Google Scholar] [CrossRef] [PubMed]
- Lampreht Tratar, U.; Horvat, S.; Cemazar, M. Transgenic Mouse Models in Cancer Research. Front. Oncol. 2018, 8, 268. [Google Scholar] [CrossRef]
- Noorani, I. Genetically Engineered Mouse Models of Gliomas: Technological Developments for Translational Discoveries. Cancers 2019, 11, 1335. [Google Scholar] [CrossRef]
- Zitvogel, L.; Pitt, J.M.; Daillère, R.; Smyth, M.J.; Kroemer, G. Mouse Models in Oncoimmunology. Nat. Rev. Cancer 2016, 16, 759–773. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.B.; Pacheco, D.R.F.; Saxon, D.; Yang, A.; Sabet, S.; Dutra-Clarke, M.; Levy, R.; Watkins, A.; Park, H.; Akhtar, A.A.; et al. Rapid Generation of Somatic Mouse Mosaics with Locus-Specific, Stably-Integrated Transgenic Elements. Cell 2019, 179, 251–267.e24. [Google Scholar] [CrossRef]
- Huse, J.T.; Holland, E.C. Genetically Engineered Mouse Models of Brain Cancer and the Promise of Preclinical Testing. Brain Pathol. 2008, 19, 132–143. [Google Scholar] [CrossRef]
- Zuckermann, M.; Hovestadt, V.; Knobbe-Thomsen, C.B.; Zapatka, M.; Northcott, P.A.; Schramm, K.; Belic, J.; Jones, D.T.; Tschida, B.; Moriarity, B.; et al. Somatic CRISPR/Cas9-Mediated Tumour Suppressor Disruption Enables Versatile Brain Tumour Modelling. Nat. Commun. 2015, 6, 7391. [Google Scholar] [CrossRef]
- Pathania, M.; De Jay, N.; Maestro, N.; Harutyunyan, A.S.; Nitarska, J.; Pahlavan, P.; Henderson, S.; Mikael, L.G.; Richard-Londt, A.; Zhang, Y.; et al. H3.3K27M Cooperates with Trp53 Loss and PDGFRA Gain in Mouse Embryonic Neural Progenitor Cells to Induce Invasive High-Grade Gliomas. Cancer Cell 2017, 32, 684–700. [Google Scholar] [CrossRef]
- Ldrini, B.; Curiel-García, Á.; Marques, C.; Matia, V.; Uluçkan, Ö.; Graña-Castro, O.; Torres-Ruiz, R.; Rodriguez-Perales, S.; Huse, J.T.; Squatrito, M. Somatic Genome Editing with the RCAS-TVA-CRISPR-Cas9 System for Precision Tumor Modeling. Nat. Commun. 2018, 9, 1466. [Google Scholar] [CrossRef]
- Holland, E.C.; Hively, W.P.; DePinho, R.A.; Varmus, H.E. A Constitutively Active Epidermal Growth Factor Receptor Cooperates with Disruption of G1 Cell-Cycle Arrest Pathways to Induce Glioma-like Lesions in Mice. Genes Dev. 1998, 12, 3675–3685. [Google Scholar] [CrossRef]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.-M.; Gallia, G.L.; et al. An Integrated Genomic Analysis of Human Glioblastoma Multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef]
- Squatrito, M.; Brennan, C.W.; Helmy, K.; Huse, J.T.; Petrini, J.H.; Holland, E.C. Loss of ATM/Chk2/P53 Pathway Components Accelerates Tumor Development and Contributes to Radiation Resistance in Gliomas. Cancer Cell 2010, 18, 619–629. [Google Scholar] [CrossRef]
- Zheng, H.; Ying, H.; Yan, H.; Kimmelman, A.C.; Hiller, D.J.; Chen, A.J.; Perry, S.R.; Tonon, G.; Chu, G.C.; Ding, Z.; et al. P53 and Pten Control Neural and Glioma Stem/Progenitor Cell Renewal and Differentiation. Nature 2008, 455, 1129–1133. [Google Scholar] [CrossRef]
- Zhu, H.; Acquaviva, J.; Ramachandran, P.; Boskovitz, A.; Woolfenden, S.; Pfannl, R.; Bronson, R.T.; Chen, J.W.; Weissleder, R.; Housman, D.E.; et al. Oncogenic EGFR Signaling Cooperates with Loss of Tumor Suppressor Gene Functions in Gliomagenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 2712–2716. [Google Scholar] [CrossRef] [PubMed]
- Bardella, C.; Al-Dalahmah, O.; Krell, D.; Brazauskas, P.; Al-Qahtani, K.; Tomkova, M.; Adam, J.; Serres, S.; Lockstone, H.; Freeman-Mills, L.; et al. Expression of Idh1R132H in the Murine Subventricular Zone Stem Cell Niche Recapitulates Features of Early Gliomagenesis. Cancer Cell 2016, 30, 578–594. [Google Scholar] [CrossRef] [PubMed]
- Jacques, T.S.; Swales, A.; Brzozowski, M.J.; Henriquez, N.V.; Linehan, J.M.; Mirzadeh, Z.; O’Malley, C.; Naumann, H.; Alvarez-Buylla, A.; Brandner, S. Combinations of Genetic Mutations in the Adult Neural Stem Cell Compartment Determine Brain Tumour Phenotypes. EMBO J. 2010, 29, 222–235. [Google Scholar] [CrossRef] [PubMed]
- Friedmann-Morvinski, D.; Bushong, E.A.; Ke, E.; Soda, Y.; Marumoto, T.; Singer, O.; Ellisman, M.H.; Verma, I.M. Dedifferentiation of Neurons and Astrocytes by Oncogenes Can Induce Gliomas in Mice. Science 2012, 338, 1080–1084. [Google Scholar] [CrossRef]
- Mosier, D.E.; Gulizia, R.J.; Baird, S.M.; Wilson, D.B. Transfer of a Functional Human Immune System to Mice with Severe Combined Immunodeficiency. Nature 1988, 335, 256–259. [Google Scholar] [CrossRef]
- Traggiai, E.; Chicha, L.; Mazzucchelli, L.; Bronz, L.; Piffaretti, J.C.; Lanzavecchia, A.; Manz, M.G. Development of a Human Adaptive Immune System in Cord Blood Cell-Transplanted Mice. Science 2004, 304, 104–107. [Google Scholar] [CrossRef]
- Siegler, E.L.; Wang, P. Preclinical Models in Chimeric Antigen Receptor—Engineered T-Cell Therapy. Hum. Gene Ther. 2018, 29, 534–546. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Manner, K.; DeJesus, R.; White, K.; Gattis, C.; Ngo, P.; Bandoro, C.; Tham, E.; Chu, E.Y.; Young, C.; et al. Hypoimmune Anti-CD19 Chimeric Antigen Receptor T Cells Provide Lasting Tumor Control in Fully Immunocompetent Allogeneic Humanized Mice. Nat. Commun. 2023, 14, 2020. [Google Scholar] [CrossRef]
- Mhaidly, R.; Verhoeyen, E. Humanized Mice Are Precious Tools for Preclinical Evaluation of CAR T and CAR NK Cell Therapies. Cancers 2020, 12, 1915. [Google Scholar] [CrossRef] [PubMed]
- Karpel, M.E.; Boutwell, C.L.; Allen, T.M. BLT Humanized Mice as a Small Animal Model of HIV Infection. Curr. Opin. Virol. 2015, 13, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Atherton, M.J.; Morris, J.S.; McDermott, M.R.; Lichty, B.D. Cancer Immunology and Canine Malignant Melanoma: A Comparative Review. Vet. Immunol. Immunopathol. 2016, 169, 15–26. [Google Scholar] [CrossRef]
- Panjwani, M.K.; Atherton, M.J.; MaloneyHuss, M.A.; Haran, K.P.; Xiong, A.; Gupta, M.; Kulikovsaya, I.; Lacey, S.F.; Mason, N.J. Establishing a Model System for Evaluating CAR T Cell Therapy Using Dogs with Spontaneous Diffuse Large B Cell Lymphoma. Oncoimmunology 2019, 9, 1676615. [Google Scholar] [CrossRef]
- Migliorini, D.; Mason, N.J.; Posey, A.D., Jr. Keeping the Engine Running: The Relevance and Predictive Value of Preclinical Models for CAR-T Cell Development. ILAR J. 2018, 59, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Duncan, B.B.; Dunbar, C.E.; Ishii, K. Applying a Clinical Lens to Animal Models of CAR-T Cell Therapies. Mol. Ther.-Methods Clin. Dev. 2022, 27, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Berger, C.; Sommermeyer, D.; Hudecek, M.; Berger, M.; Balakrishnan, A.; Paszkiewicz, P.J.; Kosasih, P.L.; Rader, C.; Riddell, S.R. Safety of Targeting ROR1 in Primates with Chimeric Antigen Receptor—Modified T Cells. Cancer Immunol. Res. 2015, 3, 206–216. [Google Scholar] [CrossRef]
- Manni, S.; Del Bufalo, F.; Merli, P.; Silvestris, D.A.; Guercio, M.; Caruso, S.; Reddel, S.; Iaffaldano, L.; Pezzella, M.; Di Cecca, S.; et al. Neutralizing IFNγ Improves Safety without Compromising Efficacy of CAR-T Cell Therapy in B-Cell Malignancies. Nat. Commun. 2023, 14, 3423. [Google Scholar] [CrossRef]
- Lyell Immunopharma, Inc. A Phase 1 Study to Assess the Safety and Efficacy of LYL797, ROR1-Targeting CAR T Cells, in Adults with Relapsed and/or Refractory Solid-Tumor Malignancies; Clinicaltrials.gov: Bethesda, MD, USA, 2024.
- Taraseviciute, A.; Tkachev, V.; Ponce, R.; Turtle, C.J.; Snyder, J.M.; Liggitt, H.D.; Myerson, D.; Gonzalez-Cuyar, L.; Baldessari, A.; English, C.; et al. Chimeric Antigen Receptor T Cell—Mediated Neurotoxicity in Non-Human Primates. Cancer Discov. 2018, 8, 750–763. [Google Scholar] [CrossRef] [PubMed]
- Yagyu, S.; Mochizuki, H.; Yamashima, K.; Kubo, H.; Saito, S.; Tanaka, M.; Sakamoto, K.; Shimoi, A.; Nakazawa, Y. A Lymphodepleted Non-human Primate Model for the Assessment of Acute On-target and Off-tumor Toxicity of Human Chimeric Antigen receptor-T Cells. Clin. Transl. Immunol. 2021, 10, e1291. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).