
Emerging and Biological Concepts in Pediatric High-Grade Gliomas


 and
and
Abstract
:1. Introduction
2. High-Grade Gliomas in a Nutshell
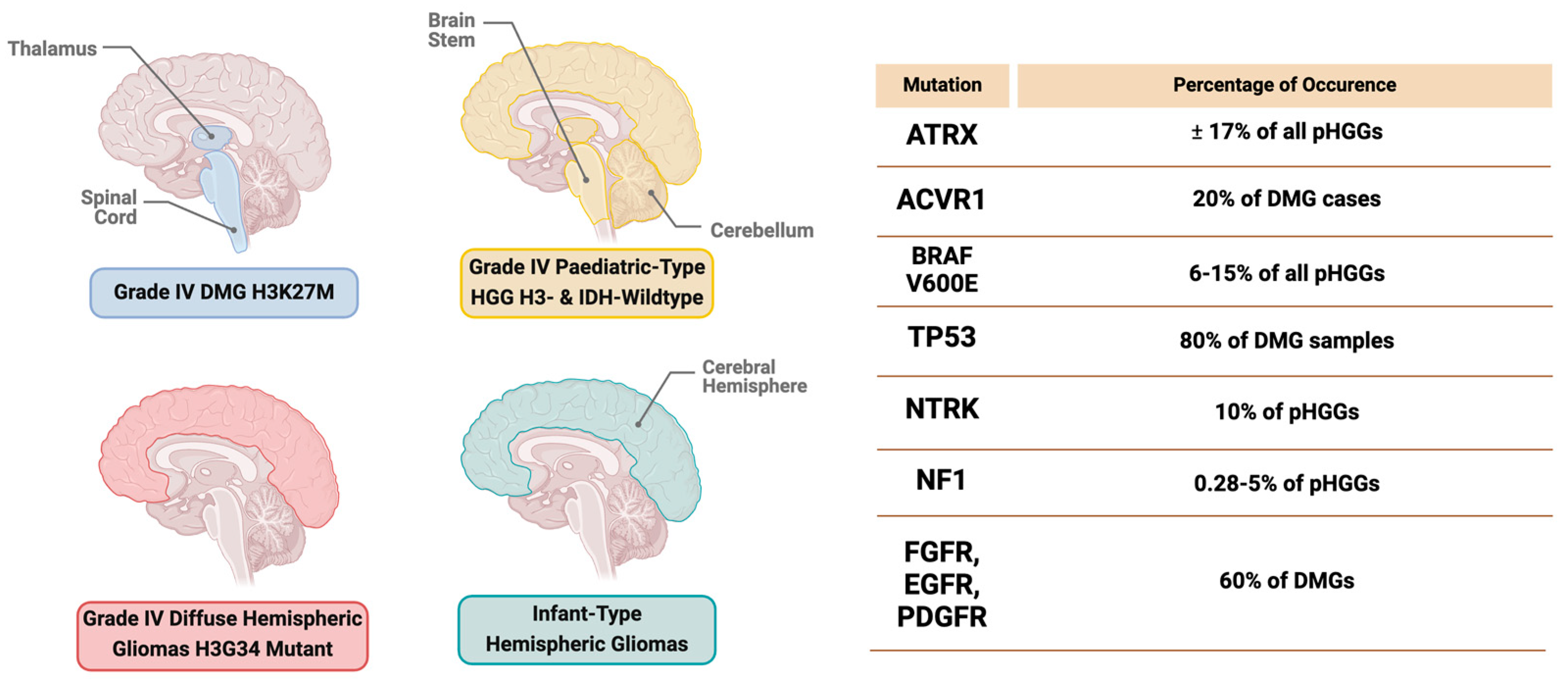
2.1. Diffuse Midline Glioma (DMG) H3K27-Altered Subtype
2.2. Grade 4 Diffuse Hemispheric Glioma, H3G34-Mutant Subtype
2.3. Grade 4 Pediatric High-Grade Glioma H3-Wildtype and IDH-Wildtype
2.4. Infant-Type Hemispheric Gliomas
3. Clinical Diagnosis of pHGGs
3.1. Imaging Techniques
3.2. Histopathological and Immunohistochemistry Diagnosis
3.3. Liquid Biopsy
4. The Molecular Landscape of pHGGs
4.1. Genetic Mutations in pHGGs
4.1.1. Receptor Tyrosine Kinase (RTK)
4.1.2. Tumor Protein p53 (TP53)
4.1.3. Activin A Receptor, Type 1 (ACVR1)
4.1.4. ATRX
4.1.5. BRAF V600E Mutation
4.1.6. Neurofibromatosis Type 1 (NF-1)
4.1.7. Neurotrophic Tyrosine Receptor Kinase (NTRK) Fusion
5. The Tumor Microenvironment in pHGGs
6. Current Standard of Care for pHGG Patients
6.1. Surgery
6.2. Radiotherapy (RT)
6.3. Chemotherapy
7. Emerging Therapies for pHGGs
7.1. Immunotherapy
Checkpoint Blockade Inhibitors
7.2. Chimeric Antigen Receptor T Cell Therapy
7.3. Cancer Vaccines
8. New Therapeutic Strategies for Treating pHGGs
8.1. Precision Medicine and Targeted Therapy
8.2. RTK Inhibitors
8.3. Epigenetic Therapies
8.4. Functional Drug and Genetic Screens
9. Conclusions and Future Directions
Funding
Acknowledgments
Conflicts of Interest
References
- Shirazi, N.; Gupta, M.; Bhat, N.K.; Kalra, B.P.; Kumar, R.; Saini, M. Profile of Primary Pediatric Brain and Spinal Cord Tumors from North India. Indian J. Med. Paediatr. Oncol. 2017, 38, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.J.; Cullen, J.; Barnholtz-Sloan, J.S.; Ostrom, Q.T.; Langer, C.E.; Turner, M.C.; McKean-Cowdin, R.; Fisher, J.L.; Lupo, P.J.; Partap, S. Childhood brain tumor epidemiology: A brain tumor epidemiology consortium review. Cancer Epidemiol. Biomark. Prev. 2014, 23, 2716–2736. [Google Scholar] [CrossRef] [PubMed]
- Hanif, F.; Muzaffar, K.; Perveen, K.; Malhi, S.M.; Simjee, S.U. Glioblastoma multiforme: A review of its epidemiology and pathogenesis through clinical presentation and treatment. Asian Pac. J. Cancer Prev. APJCP 2017, 18, 3. [Google Scholar] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; Von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization classification of tumors of the central nervous system: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO classification of tumors of the central nervous system: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Mackay, A.; Burford, A.; Carvalho, D.; Izquierdo, E.; Fazal-Salom, J.; Taylor, K.R.; Bjerke, L.; Clarke, M.; Vinci, M.; Nandhabalan, M. Integrated molecular meta-analysis of 1000 pediatric high-grade and diffuse intrinsic pontine glioma. Cancer Cell 2017, 32, 520–537. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS statistical report: Primary brain and other central nervous system tumors diagnosed in the United States in 2014–2018. Neuro-Oncology 2021, 23 (Suppl. 3), iii1–iii105. [Google Scholar] [CrossRef]
- Aggarwal, P.; Luo, W.; Pehlivan, K.C.; Hoang, H.; Rajappa, P.; Cripe, T.P.; Cassady, K.A.; Lee, D.A.; Cairo, M.S. Pediatric versus adult high grade glioma: Immunotherapeutic and genomic considerations. Front. Immunol. 2022, 13, 1038096. [Google Scholar] [CrossRef]
- Davis, M.E. Glioblastoma: Overview of disease and treatment. Clin. J. Oncol. Nurs. 2016, 20, S2. [Google Scholar] [CrossRef]
- Cacciotti, C.; Fleming, A.; Ramaswamy, V. Advances in the molecular classification of pediatric brain tumors: A guide to the galaxy. J. Pathol. 2020, 251, 249–261. [Google Scholar] [CrossRef]
- Ryall, S.; Tabori, U.; Hawkins, C. Pediatric low-grade glioma in the era of molecular diagnostics. Acta Neuropathol. Commun. 2020, 8, 30. [Google Scholar] [CrossRef] [PubMed]
- Hauser, P. Classification and treatment of pediatric gliomas in the molecular era. Children 2021, 8, 739. [Google Scholar] [CrossRef] [PubMed]
- Kline, C.; Felton, E.; Allen, I.E.; Tahir, P.; Mueller, S. Survival outcomes in pediatric recurrent high-grade glioma: Results of a 20-year systematic review and meta-analysis. J. Neuro-Oncology 2018, 137, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Lam, S.; Lin, Y.; Auffinger, B.; Melkonian, S. Analysis of survival in pediatric high-grade brainstem gliomas: A population-based study. J. Pediatr. Neurosci. 2015, 10, 199–206. [Google Scholar] [CrossRef]
- Fangusaro, J. Pediatric high grade glioma: A review and update on tumor clinical characteristics and biology. Front. Oncol. 2012, 2, 105. [Google Scholar] [CrossRef]
- Jones, C.; Perryman, L.; Hargrave, D. Paediatric and adult malignant glioma: Close relatives or distant cousins? Nat. Rev. Clin. Oncol. 2012, 9, 400–413. [Google Scholar] [CrossRef]
- Ceccarelli, M.; Barthel, F.P.; Malta, T.M.; Sabedot, T.S.; Salama, S.R.; Murray, B.A.; Morozova, O.; Newton, Y.; Radenbaugh, A.; Pagnotta, S.M. Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell 2016, 164, 550–563. [Google Scholar] [CrossRef]
- Sun, C.X.; Daniel, P.; Bradshaw, G.; Shi, H.; Loi, M.; Chew, N.; Parackal, S.; Tsui, V.; Liang, Y.; Koptyra, M. Generation and multi-dimensional profiling of a childhood cancer cell line atlas defines new therapeutic opportunities. Cancer Cell 2023, 41, 660–677. [Google Scholar] [CrossRef]
- Fabbri, V.P.; Caporalini, C.; Asioli, S.; Buccoliero, A. Paediatric-type diffuse low-grade gliomas: A clinically and biologically distinct group of tumours with a favourable outcome. Pathologica 2022, 114, 410. [Google Scholar] [CrossRef]
- Gianno, F.; Giovannoni, I.; Cafferata, B.; Diomedi-Camassei, F.; Minasi, S.; Barresi, S.; Buttarelli, F.R.; Alesi, V.; Cardoni, A.; Antonelli, M. Paediatric-type diffuse high-grade gliomas in the 5th CNS WHO Classification. Pathologica 2022, 114, 422. [Google Scholar] [CrossRef]
- Park, Y.W.; Vollmuth, P.; Foltyn-Dumitru, M.; Sahm, F.; Ahn, S.S.; Chang, J.H.; Kim, S.H. The 2021 WHO Classification for Gliomas and Implications on Imaging Diagnosis: Part 2—Summary of Imaging Findings on Pediatric-Type Diffuse High-Grade Gliomas, Pediatric-Type Diffuse Low-Grade Gliomas, and Circumscribed Astrocytic Gliomas. J. Magn. Reson. Imaging 2023, 58, 690–708. [Google Scholar] [CrossRef]
- McNamara, C.; Mankad, K.; Thust, S.; Dixon, L.; Limback-Stanic, C.; D’Arco, F.; Jacques, T.S.; Löbel, U. 2021 WHO classification of tumours of the central nervous system: A review for the neuroradiologist. Neuroradiology 2022, 64, 1919–1950. [Google Scholar] [CrossRef] [PubMed]
- Sejda, A.; Grajkowska, W.; Trubicka, J.; Szutowicz, E.; Wojdacz, T.; Kloc, W.; Iżycka-Świeszewska, E. WHO CNS5 2021 classification of gliomas: A practical review and road signs for diagnosing pathologists and proper patho-clinical and neuro-oncological cooperation. Folia Neuropathol. 2022, 60, 137–152. [Google Scholar] [CrossRef] [PubMed]
- Castel, D.; Philippe, C.; Calmon, R.; Le Dret, L.; Truffaux, N.; Boddaert, N.; Pagès, M.; Taylor, K.R.; Saulnier, P.; Lacroix, L. Histone H3F3A and HIST1H3B K27M mutations define two subgroups of diffuse intrinsic pontine gliomas with different prognosis and phenotypes. Acta Neuropathol. 2015, 130, 815–827. [Google Scholar] [CrossRef] [PubMed]
- Cosnarovici, M.M.; Cosnarovici, R.V.; Piciu, D. Updates on the 2016 world health organization classification of pediatric tumors of the central nervous system-a systematic review. Med. Pharm. Rep. 2021, 94, 282. [Google Scholar] [CrossRef]
- Bender, S.; Tang, Y.; Lindroth, A.M.; Hovestadt, V.; Jones, D.T.; Kool, M.; Zapatka, M.; Northcott, P.A.; Sturm, D.; Wang, W. Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell 2013, 24, 660–672. [Google Scholar] [CrossRef]
- Mondal, G.; Lee, J.C.; Ravindranathan, A.; Villanueva-Meyer, J.E.; Tran, Q.T.; Allen, S.J.; Barreto, J.; Gupta, R.; Doo, P.; Van Ziffle, J. Pediatric bithalamic gliomas have a distinct epigenetic signature and frequent EGFR exon 20 insertions resulting in potential sensitivity to targeted kinase inhibition. Acta Neuropathol. 2020, 139, 1071–1088. [Google Scholar] [CrossRef]
- Solomon, D.A.; Wood, M.D.; Tihan, T.; Bollen, A.W.; Gupta, N.; Phillips, J.J.; Perry, A. Diffuse midline gliomas with histone H3-K27M mutation: A series of 47 cases assessing the spectrum of morphologic variation and associated genetic alterations. Brain Pathol. 2016, 26, 569–580. [Google Scholar] [CrossRef]
- Fontebasso, A.M.; Papillon-Cavanagh, S.; Schwartzentruber, J.; Nikbakht, H.; Gerges, N.; Fiset, P.-O.; Bechet, D.; Faury, D.; De Jay, N.; Ramkissoon, L.A. Recurrent somatic mutations in ACVR1 in pediatric midline high-grade astrocytoma. Nat. Genet. 2014, 46, 462–466. [Google Scholar] [CrossRef]
- Taylor, K.R.; Mackay, A.; Truffaux, N.; Butterfield, Y.S.; Morozova, O.; Philippe, C.; Castel, D.; Grasso, C.S.; Vinci, M.; Carvalho, D. Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nat. Genet. 2014, 46, 457–461. [Google Scholar] [CrossRef]
- Chen, C.C.; Deshmukh, S.; Jessa, S.; Hadjadj, D.; Lisi, V.; Andrade, A.F.; Faury, D.; Jawhar, W.; Dali, R.; Suzuki, H. Histone H3. 3G34-mutant interneuron progenitors co-opt PDGFRA for gliomagenesis. Cell 2020, 183, 1617–1633. [Google Scholar] [CrossRef] [PubMed]
- Lowe, B.R.; Maxham, L.A.; Hamey, J.J.; Wilkins, M.R.; Partridge, J.F. Histone H3 mutations: An updated view of their role in chromatin deregulation and cancer. Cancers 2019, 11, 660. [Google Scholar] [CrossRef] [PubMed]
- Shao, H.; Gong, J.; Su, X.; Chen, N.; Li, S.; Yang, X.; Zhang, S.; Huang, Z.; Hu, W.; Gong, Q. MRI characteristics of H3 G34–mutant diffuse hemispheric gliomas and possible differentiation from IDH–wild-type glioblastomas in adolescents and young adults. J. Neurosurg. Pediatr. 2023, 33, 236–244. [Google Scholar] [CrossRef]
- Crowell, C.; Mata-Mbemba, D.; Bennett, J.; Matheson, K.; Mackley, M.; Perreault, S.; Erker, C. Systematic review of diffuse hemispheric glioma, H3 G34-mutant: Outcomes and associated clinical factors. Neurooncol. Adv. 2022, 4, vdac133. [Google Scholar] [CrossRef]
- Bozkurt, S.U.; Dagcinar, A.; Tanrikulu, B.; Comunoglu, N.; Meydan, B.C.; Ozek, M.; Oz, B. Significance of H3K27M mutation with specific histomorphological features and associated molecular alterations in pediatric high-grade glial tumors. Childs Nerv. Syst. 2018, 34, 107–116. [Google Scholar] [CrossRef]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.-Y.; Jones, D.T.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.-A.K.; Tönjes, M. Driver mutations in histone H3. 3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef]
- Korshunov, A.; Capper, D.; Reuss, D.; Schrimpf, D.; Ryzhova, M.; Hovestadt, V.; Sturm, D.; Meyer, J.; Jones, C.; Zheludkova, O. Histologically distinct neuroepithelial tumors with histone 3 G34 mutation are molecularly similar and comprise a single nosologic entity. Acta Neuropathol. 2016, 131, 137–146. [Google Scholar] [CrossRef]
- Jain, S.U.; Khazaei, S.; Marchione, D.M.; Lundgren, S.M.; Wang, X.; Weinberg, D.N.; Deshmukh, S.; Juretic, N.; Lu, C.; Allis, C.D. Histone H3. 3 G34 mutations promote aberrant PRC2 activity and drive tumor progression. Proc. Natl. Acad. Sci. USA 2020, 117, 27354–27364. [Google Scholar] [CrossRef]
- Cheng, Z.; Cheung, P.; Kuo, A.J.; Yukl, E.T.; Wilmot, C.M.; Gozani, O.; Patel, D.J. A molecular threading mechanism underlies Jumonji lysine demethylase KDM2A regulation of methylated H3K36. Genes Dev. 2014, 28, 1758–1771. [Google Scholar] [CrossRef] [PubMed]
- Korshunov, A.; Schrimpf, D.; Ryzhova, M.; Sturm, D.; Chavez, L.; Hovestadt, V.; Sharma, T.; Habel, A.; Burford, A.; Jones, C. H3-/IDH-wild type pediatric glioblastoma is comprised of molecularly and prognostically distinct subtypes with associated oncogenic drivers. Acta Neuropathol. 2017, 134, 507–516. [Google Scholar] [CrossRef]
- Bender, K.; Kahn, J.; Perez, E.; Ehret, F.; Roohani, S.; Capper, D.; Schmid, S.; Kaul, D. Diffuse paediatric-type high-grade glioma, H3-wildtype and IDH-wildtype: Case series of a new entity. Brain Tumor Pathol. 2023, 40, 204–214. [Google Scholar] [CrossRef]
- Korshunov, A.; Ryzhova, M.; Hovestadt, V.; Bender, S.; Sturm, D.; Capper, D.; Meyer, J.; Schrimpf, D.; Kool, M.; Northcott, P.A. Integrated analysis of pediatric glioblastoma reveals a subset of biologically favorable tumors with associated molecular prognostic markers. Acta Neuropathol. 2015, 129, 669–678. [Google Scholar] [CrossRef] [PubMed]
- Tauziède-Espariat, A.; Debily, M.; Castel, D.; Grill, J.; Puget, S.; Roux, A.; Saffroy, R.; Pagès, M.; Gareton, A.; Chrétien, F. The pediatric supratentorial MYCN-amplified high-grade gliomas methylation class presents the same radiological, histopathological and molecular features as their pontine counterparts. Acta Neuropathol. Commun. 2020, 8, 104. [Google Scholar] [CrossRef] [PubMed]
- Fernando, D.; Ahmed, A.U.; Williams, B.R. Therapeutically targeting the unique disease landscape of pediatric high-grade gliomas. Front. Oncol. 2024, 14, 1347694. [Google Scholar] [CrossRef]
- Clarke, M.; Mackay, A.; Ismer, B.; Pickles, J.C.; Tatevossian, R.G.; Newman, S.; Bale, T.A.; Stoler, I.; Izquierdo, E.; Temelso, S. Infant high-grade gliomas comprise multiple subgroups characterized by novel targetable gene fusions and favorable outcomes. Cancer Discov. 2020, 10, 942–963. [Google Scholar] [CrossRef]
- Ceglie, G.; Vinci, M.; Carai, A.; Rossi, S.; Colafati, G.S.; Cacchione, A.; Tornesello, A.; Miele, E.; Locatelli, F.; Mastronuzzi, A. Infantile/congenital high-grade gliomas: Molecular features and therapeutic perspectives. Diagnostics 2020, 10, 648. [Google Scholar] [CrossRef]
- Guerreiro Stucklin, A.S.; Ryall, S.; Fukuoka, K.; Zapotocky, M.; Lassaletta, A.; Li, C.; Bridge, T.; Kim, B.; Arnoldo, A.; Kowalski, P.E.; et al. Alterations in ALK/ROS1/NTRK/MET drive a group of infantile hemispheric gliomas. Nat. Commun. 2019, 10, 4343. [Google Scholar] [CrossRef]
- Vecht, C.J.; Kerkhof, M.; Duran-Pena, A. Seizure prognosis in brain tumors: New insights and evidence-based management. Oncologist 2014, 19, 751–759. [Google Scholar] [CrossRef]
- Gattringer, T.; Enzinger, C.; Ropele, S.; Fazekas, F. Brain imaging (ct/mri). In Ischaemic Stroke in the Young; Oxford University Press: Oxford, UK, 2018; Volume 113. [Google Scholar]
- Uduma Felix, U.; Fokam Pius, G. Magnetic Resonance Imaging (Mri), The Prefered Evaluation Tool in Soft Tissue Sarcoma: Literature Review, Demonstrated with a Case Report. J. Asian Sci. Res. 2012, 2, 87–99. [Google Scholar]
- Maheshwari, M. Pediatric presurgical functional MRI. Top. Magn. Reson. Imaging 2019, 28, 197–204. [Google Scholar] [CrossRef]
- Grudzień, K.; Klimeczek-Chrapusta, M.; Kwiatkowski, S.; Milczarek, O. Predicting the WHO Grading of Pediatric Brain Tumors Based on Their MRI Appearance: A Retrospective Study. Cureus 2023, 15, e47333. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Ahlawat, S.; Panda, A.K.; Sarangi, J.; Jain, P.; Gupta, R.K.; Vaishya, S.; Patir, R. Pediatric high grade gliomas: A comprehensive histopathological, immunohistochemical and molecular integrated approach in routine practice. Pathol. Res. Pract. 2024, 258, 155347. [Google Scholar] [CrossRef]
- Blasco-Santana, L.; Colmenero, I. Molecular and Pathological Features of Paediatric High-Grade Gliomas. Int. J. Mol. Sci. 2024, 25, 8498. [Google Scholar] [CrossRef]
- El-Ayadi, M.; Ansari, M.; Sturm, D.; Gielen, G.H.; Warmuth-Metz, M.; Kramm, C.M.; von Bueren, A.O. High-grade glioma in very young children: A rare and particular patient population. Oncotarget 2017, 8, 64564. [Google Scholar] [CrossRef]
- Sareen, H.; Garrett, C.; Lynch, D.; Powter, B.; Brungs, D.; Cooper, A.; Po, J.; Koh, E.-S.; Vessey, J.Y.; McKechnie, S. The role of liquid biopsies in detecting molecular tumor biomarkers in brain cancer patients. Cancers 2020, 12, 1831. [Google Scholar] [CrossRef]
- Tan, J.Y.; Wijesinghe, I.V.S.; Alfarizal Kamarudin, M.N.; Parhar, I. Paediatric gliomas: BRAF and histone H3 as biomarkers, therapy and perspective of liquid biopsies. Cancers 2021, 13, 607. [Google Scholar] [CrossRef]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M. Detection of circulating tumor DNA in early-and late-stage human malignancies. Sci. Transl. Med. 2014, 6, 224ra24. [Google Scholar] [CrossRef]
- Atallah, O.; Krauss, J.K.; Hermann, E.J. External ventricular drainage in pediatric patients: Indications, management, and shunt conversion rates. Childs Nerv. Syst. 2024, 40, 2071–2079. [Google Scholar] [CrossRef]
- Laconi, E.; Marongiu, F.; DeGregori, J. Cancer as a disease of old age: Changing mutational and microenvironmental landscapes. Br. J. Cancer 2020, 122, 943–952. [Google Scholar] [CrossRef] [PubMed]
- Buczkowicz, P.; Hoeman, C.; Rakopoulos, P.; Pajovic, S.; Letourneau, L.; Dzamba, M.; Morrison, A.; Lewis, P.; Bouffet, E.; Bartels, U. Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat. Genet. 2014, 46, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Puget, S.; Philippe, C.; Bax, D.A.; Job, B.; Varlet, P.; Junier, M.-P.; Andreiuolo, F.; Carvalho, D.; Reis, R.; Guerrini-Rousseau, L. Mesenchymal transition and PDGFRA amplification/mutation are key distinct oncogenic events in pediatric diffuse intrinsic pontine gliomas. PLoS ONE 2012, 7, e30313. [Google Scholar] [CrossRef]
- Jones, C.; Baker, S.J. Unique genetic and epigenetic mechanisms driving paediatric diffuse high-grade glioma. Nat. Rev. Cancer 2014, 14, 651–661. [Google Scholar] [CrossRef]
- Ferguson, H.R.; Smith, M.P.; Francavilla, C. Fibroblast growth factor receptors (FGFRs) and noncanonical partners in cancer signaling. Cells 2021, 10, 1201. [Google Scholar] [CrossRef]
- Damodharan, S.; Lara-Velazquez, M.; Williamsen, B.C.; Helgager, J.; Dey, M. Diffuse intrinsic pontine glioma: Molecular landscape, evolving treatment strategies and emerging clinical trials. J. Pers. Med. 2022, 12, 840. [Google Scholar] [CrossRef] [PubMed]
- Pollack, I.F.; Hamilton, R.L.; James, C.D.; Finkelstein, S.D.; Burnham, J.; Yates, A.J.; Holmes, E.J.; Zhou, T.; Finlay, J.L. Rarity of PTEN deletions and EGFR amplification in malignant gliomas of childhood: Results from the Children’s Cancer Group 945 cohort. J. Neurosurg. Pediatr. 2006, 105, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Bax, D.A.; Gaspar, N.; Little, S.E.; Marshall, L.; Perryman, L.; Regairaz, M.; Viana-Pereira, M.; Vuononvirta, R.; Sharp, S.Y.; Reis-Filho, J.S. EGFRvIII deletion mutations in pediatric high-grade glioma and response to targeted therapy in pediatric glioma cell lines. Clin. Cancer Res. 2009, 15, 5753–5761. [Google Scholar] [CrossRef] [PubMed]
- Parrales, A.; Iwakuma, T. Targeting oncogenic mutant p53 for cancer therapy. Front. Oncol. 2015, 5, 288. [Google Scholar] [CrossRef]
- Borrero, L.J.H.; El-Deiry, W.S. Tumor suppressor p53: Biology, signaling pathways, and therapeutic targeting. Biochim. Biophys. Acta (BBA) Rev. Cancer 2021, 1876, 188556. [Google Scholar]
- Humpton, T.J.; Vousden, K.H. Regulation of cellular metabolism and hypoxia by p53. Cold Spring Harb. Perspect. Med. 2016, 6, a026146. [Google Scholar] [CrossRef]
- Zhang, Y.; Dube, C.; Gibert Jr, M.; Cruickshanks, N.; Wang, B.; Coughlan, M.; Yang, Y.; Setiady, I.; Deveau, C.; Saoud, K. The p53 pathway in glioblastoma. Cancers 2018, 10, 297. [Google Scholar] [CrossRef]
- Khuong-Quang, D.-A.; Buczkowicz, P.; Rakopoulos, P.; Liu, X.-Y.; Fontebasso, A.M.; Bouffet, E.; Bartels, U.; Albrecht, S.; Schwartzentruber, J.; Letourneau, L. K27M mutation in histone H3. 3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol. 2012, 124, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Werbrouck, C.; Evangelista, C.C.; Lobón-Iglesias, M.-J.; Barret, E.; Le Teuff, G.; Merlevede, J.; Brusini, R.; Kergrohen, T.; Mondini, M.; Bolle, S. TP53 pathway alterations drive radioresistance in diffuse intrinsic pontine gliomas (DIPG). Clin. Cancer Res. 2019, 25, 6788–6800. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Rigueur, D.; Lyons, K.M. TGFβ as a gatekeeper of BMP action in the developing growth plate. Bone 2020, 137, 115439. [Google Scholar] [CrossRef]
- Heldin, C.-H.; Moustakas, A. Signaling receptors for TGF-β family members. Cold Spring Harb. Perspect. Biol. 2016, 8, a022053. [Google Scholar] [CrossRef]
- Taylor, K.R.; Vinci, M.; Bullock, A.N.; Jones, C. ACVR1 mutations in DIPG: Lessons learned from FOP. Cancer Res. 2014, 74, 4565–4570. [Google Scholar] [CrossRef]
- Bentayebi, K.; El Aked, R.; Ezzahidi, O.; Alami, A.B.; Louati, S.; Ouadghiri, M.; Aanniz, T.; Amzazi, S.; Belyamani, L.; Ibrahimi, A. Targeting molecular mechanisms underlying treatment efficacy and resistance in DIPG: A review of current and future strategies. Brain Disord. 2024, 14, 100132. [Google Scholar] [CrossRef]
- Carvalho, D.; Taylor, K.R.; Olaciregui, N.G.; Molinari, V.; Clarke, M.; Mackay, A.; Ruddle, R.; Henley, A.; Valenti, M.; Hayes, A. ALK2 inhibitors display beneficial effects in preclinical models of ACVR1 mutant diffuse intrinsic pontine glioma. Commun. Biol. 2019, 2, 156. [Google Scholar] [CrossRef] [PubMed]
- Haase, S.; Nuñez, F.M.; Gauss, J.C.; Thompson, S.; Brumley, E.; Lowenstein, P.; Castro, M.G. Hemispherical pediatric high-grade glioma: Molecular basis and therapeutic opportunities. Int. J. Mol. Sci. 2020, 21, 9654. [Google Scholar] [CrossRef]
- Haase, S.; Garcia-Fabiani, M.B.; Carney, S.; Altshuler, D.; Núñez, F.J.; Méndez, F.M.; Núñez, F.; Lowenstein, P.R.; Castro, M.G. Mutant ATRX: Uncovering a new therapeutic target for glioma. Expert Opin. Ther. Targets 2018, 22, 599–613. [Google Scholar] [CrossRef]
- Voon, H.P.J.; Wong, L.H. Chromatin Mutations in Pediatric High Grade Gliomas. Front. Oncol. 2023, 12, 1104129. Available online: https://www.frontiersin.org/journals/oncology/articles/10.3389/fonc.2022.1104129/full (accessed on 9 August 2024). [CrossRef]
- Schreck, K.C.; Grossman, S.A.; Pratilas, C.A. BRAF mutations and the utility of RAF and MEK inhibitors in primary brain tumors. Cancers 2019, 11, 1262. [Google Scholar] [CrossRef]
- Di Nunno, V.; Gatto, L.; Tosoni, A.; Bartolini, S.; Franceschi, E. Implications of BRAF V600E mutation in gliomas: Molecular considerations, prognostic value and treatment evolution. Front. Oncol. 2023, 12, 1067252. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Yaeger, R.; Rodrik-Outmezguine, V.S.; Tao, A.; Torres, N.M.; Chang, M.T.; Drosten, M.; Zhao, H.; Cecchi, F.; Hembrough, T. Tumours with class 3 BRAF mutants are sensitive to the inhibition of activated RAS. Nature 2017, 548, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Behling, F.; Schittenhelm, J. Oncogenic BRAF alterations and their role in brain tumors. Cancers 2019, 11, 794. [Google Scholar] [CrossRef]
- Nobre, L.; Bouffet, E. BRAF inhibitors in BRAFV600E-mutated pediatric high-grade gliomas: Upfront or at recurrence? Neuro-Oncology 2022, 24, 1976–1977. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, T.; Yeo, K.K.; Mauguen, A.; Alexandrescu, S.; Prabhu, S.P.; Tsai, J.W.; Malinowski, S.; Joshirao, M.; Parikh, K.; Farouk Sait, S.; et al. Upfront molecular targeted therapy for the treatment of BRAF-mutant pediatric high-grade glioma. Neuro-Oncology 2022, 24, 1964–1975. [Google Scholar] [CrossRef]
- Spyris, C.; Castellino, R.; Schniederjan, M.; Kadom, N. High-grade gliomas in children with neurofibromatosis type 1: Literature review and illustrative cases. Am. J. Neuroradiol. 2019, 40, 366–369. [Google Scholar] [CrossRef]
- Byrne, S.; Connor, S.; Lascelles, K.; Siddiqui, A.; Hargrave, D.; Ferner, R.E. Clinical presentation and prognostic indicators in 100 adults and children with neurofibromatosis 1 associated non-optic pathway brain gliomas. J. Neuro-Oncology 2017, 133, 609–614. [Google Scholar] [CrossRef]
- Lobbous, M.; Bernstock, J.D.; Coffee, E.; Friedman, G.K.; Metrock, L.K.; Chagoya, G.; Elsayed, G.; Nakano, I.; Hackney, J.R.; Korf, B.R. An update on neurofibromatosis type 1-associated gliomas. Cancers 2020, 12, 114. [Google Scholar] [CrossRef]
- Costa, A.D.A.; Gutmann, D.H. Brain tumors in neurofibromatosis type 1. Neuro-Oncol. Adv. 2020, 2 (Suppl. S1), i85–i97. [Google Scholar] [CrossRef]
- D’Angelo, F.; Ceccarelli, M.; Tala; Garofano, L.; Zhang, J.; Frattini, V.; Caruso, F.P.; Lewis, G.; Alfaro, K.D.; Bauchet, L.; et al. The molecular landscape of glioma in patients with Neurofibromatosis 1. Nat. Med. 2019, 25, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.R.; Bell, L.; Miller, C.; Segal, D. A case of infant-type hemispheric glioma with NTRK1 fusion. Child Neurol. Open 2022, 9, 2329048X221146982. [Google Scholar] [CrossRef] [PubMed]
- Torre, M.; Vasudevaraja, V.; Serrano, J.; DeLorenzo, M.; Malinowski, S.; Blandin, A.-F.; Pages, M.; Ligon, A.H.; Dong, F.; Meredith, D.M. Molecular and clinicopathologic features of gliomas harboring NTRK fusions. Acta Neuropathol. Commun. 2020, 8, 107. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.C.; Ellison, D.W. Molecular pathology of paediatric central nervous system tumours. J. Pathol. 2017, 241, 159–172. [Google Scholar] [CrossRef] [PubMed]
- Amatu, A.; Sartore-Bianchi, A.; Bencardino, K.; Pizzutilo, E.; Tosi, F.; Siena, S. Tropomyosin receptor kinase (TRK) biology and the role of NTRK gene fusions in cancer. Ann. Oncol. 2019, 30, viii5–viii15. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.E.; Park, C.K.; Kim, S.K.; Phi, J.H.; Paek, S.H.; Choi, J.Y.; Kang, H.J.; Lee, J.H.; Won, J.K.; Yun, H.; et al. NTRK-Fused Central Nervous System Tumours: Clinicopathological and Genetic Insights and Response to TRK Inhibitors. Acta Neuropathol. Commun. 2024, 12, 118. Available online: https://link.springer.com/article/10.1186/s40478-024-01798-9 (accessed on 9 August 2024). [CrossRef]
- Antonucci, L.; Canciani, G.; Mastronuzzi, A.; Carai, A.; Del Baldo, G.; Del Bufalo, F. CAR-T Therapy for Pediatric High-Grade Gliomas: Peculiarities, Current Investigations and Future Strategies. Front. Immunol. 2022, 13, 867154. Available online: https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2022.867154/full (accessed on 9 August 2024). [CrossRef]
- Sharma, P.; Aaroe, A.; Liang, J.; Puduvalli, V.K. Tumor microenvironment in glioblastoma: Current and emerging concepts. Neuro-Oncology Adv. 2023, 5, vdad009. [Google Scholar] [CrossRef]
- Lieberman, N.A.; DeGolier, K.; Kovar, H.M.; Davis, A.; Hoglund, V.; Stevens, J.; Winter, C.; Deutsch, G.; Furlan, S.N.; Vitanza, N.A. Characterization of the immune microenvironment of diffuse intrinsic pontine glioma: Implications for development of immunotherapy. Neuro-Oncology 2019, 21, 83–94. [Google Scholar] [CrossRef]
- Lin, G.L.; Nagaraja, S.; Filbin, M.G.; Suvà, M.L.; Vogel, H.; Monje, M. Non-inflammatory tumor microenvironment of diffuse intrinsic pontine glioma. Acta Neuropathol. Commun. 2018, 6, 51. [Google Scholar] [CrossRef]
- Mangani, D.; Weller, M.; Roth, P. The network of immunosuppressive pathways in glioblastoma. Biochem. Pharmacol. 2017, 130, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Njonkou, R.; Jackson, C.M.; Woodworth, G.F.; Hersh, D.S. Pediatric glioblastoma: Mechanisms of immune evasion and potential therapeutic opportunities. Cancer Immunol. Immunother. 2022, 71, 1813–1822. [Google Scholar] [CrossRef] [PubMed]
- du Chatinier, A.; Velilla, I.Q.; Meel, M.H.; Hoving, E.W.; Hulleman, E.; Metselaar, D.S. Microglia in pediatric brain tumors: The missing link to successful immunotherapy. CR Med. 2023, 4, 101246. [Google Scholar] [CrossRef]
- Klemm, F.; Maas, R.R.; Bowman, R.L.; Kornete, M.; Soukup, K.; Nassiri, S.; Brouland, J.-P.; Iacobuzio-Donahue, C.A.; Brennan, C.; Tabar, V.; et al. Interrogation of the Microenvironmental Landscape in Brain Tumors Reveals Disease-Specific Alterations of Immune Cells. Cell 2020, 181, 1643–1660.e17. [Google Scholar] [CrossRef] [PubMed]
- Rozowsky, J.S.; Meesters-Ensing, J.I.; Lammers, J.A.S.; Belle, M.L.; Nierkens, S.; Kranendonk, M.E.G.; Kester, L.A.; Calkoen, F.G.; van der Lugt, J. A Toolkit for Profiling the Immune Landscape of Pediatric Central Nervous System Malignancies. Front. Immunol. 2022, 13, 864423. [Google Scholar] [CrossRef] [PubMed]
- Thon, N.; Tonn, J.-C.; Kreth, F.-W. The surgical perspective in precision treatment of diffuse gliomas. OncoTargets Ther. 2019, 12, 1497–1508. [Google Scholar] [CrossRef]
- Pawloski, J.A.; Awan, O.; Ziu, M.; Robin, A.M. Indications and Techniques for Surgical Intervention in Patients with Metastatic Brain Tumors. In Cancer Metastasis through the Lymphovascular System; Springer: Berlin/Heidelberg, Germany, 2022; pp. 547–558. [Google Scholar]
- Foo, C.Y.; Munir, N.; Kumaria, A.; Akhtar, Q.; Bullock, C.J.; Narayanan, A.; Fu, R.Z. Medical device advances in the treatment of glioblastoma. Cancers 2022, 14, 5341. [Google Scholar] [CrossRef]
- Hirano, Y.; Shinya, Y.; Aono, T.; Hasegawa, H.; Kawashima, M.; Shin, M.; Takami, H.; Takayanagi, S.; Umekawa, M.; Ikemura, M. The role of stereotactic frame-based biopsy for brainstem tumors in the era of molecular-based diagnosis and treatment decisions. Curr. Oncol. 2022, 29, 4558–4565. [Google Scholar] [CrossRef]
- Yang, T.; Temkin, N.; Barber, J.; Geyer, J.R.; Leary, S.; Browd, S.; Ojemann, J.G.; Ellenbogen, R.G. Gross total resection correlates with long-term survival in pediatric patients with glioblastoma. World Neurosurg. 2013, 79, 537–544. [Google Scholar] [CrossRef]
- Han, Q.; Liang, H.; Cheng, P.; Yang, H.; Zhao, P. Gross total vs. subtotal resection on survival outcomes in elderly patients with high-grade glioma: A systematic review and meta-analysis. Front. Oncol. 2020, 10, 151. [Google Scholar] [CrossRef]
- Hatoum, R.; Chen, J.-S.; Lavergne, P.; Shlobin, N.A.; Wang, A.; Elkaim, L.M.; Dodin, P.; Couturier, C.P.; Ibrahim, G.M.; Fallah, A. Extent of tumor resection and survival in pediatric patients with high-grade gliomas: A systematic review and meta-analysis. JAMA Netw. Open 2022, 5, e2226551. [Google Scholar] [CrossRef] [PubMed]
- Löfgren, D.; Valachis, A.; Olivecrona, M. Risk for morbidity and mortality after neurosurgery in older patients with high grade gliomas–a retrospective population based study. BMC Geriatr. 2022, 22, 805. [Google Scholar] [CrossRef]
- Kessler, A.T.; Bhatt, A.A. Brain tumour post-treatment imaging and treatment-related complications. Insights Imaging 2018, 9, 1057–1075. [Google Scholar] [CrossRef] [PubMed]
- De Simone, M.; Conti, V.; Palermo, G.; De Maria, L.; Iaconetta, G. Advancements in Glioma Care: Focus on Emerging Neurosurgical Techniques. Biomedicines 2023, 12, 8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, C.; Zeng, X. Risk factors for early hydrocephalus on post unilateral thalamic tumor resection. Front. Surg. 2022, 9, 814308. [Google Scholar] [CrossRef] [PubMed]
- Skliarenko, J.; Barry, A. Clinical and practical applications of radiation therapy: When should radiation therapy be considered for my patient? Medicine 2020, 48, 84–89. [Google Scholar] [CrossRef]
- Baskar, R.; Lee, K.A.; Yeo, R.; Yeoh, K.-W. Cancer and radiation therapy: Current advances and future directions. Int. J. Med. Sci. 2012, 9, 193. [Google Scholar] [CrossRef]
- Hu, X.; Fang, Y.; Hui, X.; Jv, Y.; You, C. Radiotherapy for diffuse brainstem glioma in children and young adults. Cochrane Database Syst. Rev. 2016, 2016, CD010439. [Google Scholar] [CrossRef]
- Major, N.; Patel, N.A.; Bennett, J.; Novakovic, E.; Poloni, D.; Abraham, M.; Brown, N.J.; Gendreau, J.L.; Sahyouni, R.; Loya, J. The Current state of radiotherapy for pediatric brain tumors: An overview of post-radiotherapy neurocognitive decline and outcomes. J. Pers. Med. 2022, 12, 1050. [Google Scholar] [CrossRef]
- Kaur, S.; Mayanglambam, P.; Bajwan, D.; Thakur, N. Chemotherapy and its adverse effects-A systematic review. Int. J. Nurs. Educ. Res. 2022, 10, 399–402. [Google Scholar] [CrossRef]
- Tamburini, E.; Tassinari, D.; Ramundo, M.; De Stefano, A.; Viola, M.G.; Romano, C.; Elia, M.T.; Zanaletti, N.; Rudnas, B.; Casadei-Gardini, A. Adjuvant chemotherapy after neoadjuvant chemo-radiotherapy and surgery in locally advanced rectal cancer. A systematic review of literature with a meta-analysis of randomized clinical trials. Crit. Rev. Oncol. Hematol. 2022, 172, 103627. [Google Scholar] [CrossRef] [PubMed]
- Wimmer, K.; Bolliger, M.; Bago-Horvath, Z.; Steger, G.; Kauer-Dorner, D.; Helfgott, R.; Gruber, C.; Moinfar, F.; Mittlböck, M.; Fitzal, F. Impact of surgical margins in breast cancer after preoperative systemic chemotherapy on local recurrence and survival. Ann. Surg. Oncol. 2020, 27, 1700–1707. [Google Scholar] [CrossRef]
- Bhowmik, A.; Khan, R.; Ghosh, M.K. Blood brain barrier: A challenge for effectual therapy of brain tumors. BioMed Res. Int. 2015, 2015, 320941. [Google Scholar] [CrossRef] [PubMed]
- Frosina, G. Advances in drug delivery to high grade gliomas. Brain Pathol. 2016, 26, 689–700. [Google Scholar] [CrossRef]
- Bhaskaran, D.; Savage, J.; Patel, A.; Collinson, F.; Mant, R.; Boele, F.; Brazil, L.; Meade, S.; Buckle, P.; Lax, S. A randomised phase II trial of temozolomide with or without cannabinoids in patients with recurrent glioblastoma (ARISTOCRAT): Protocol for a multi-centre, double-blind, placebo-controlled trial. BMC Cancer 2024, 24, 83. [Google Scholar] [CrossRef] [PubMed]
- Asano, K.; Fumoto, T.; Matsuzaka, M.; Hasegawa, S.; Suzuki, N.; Akasaka, K.; Katayama, K.; Kamataki, A.; Kurose, A.; Ohkuma, H. Combination chemoradiotherapy with temozolomide, vincristine, and interferon-β might improve outcomes regardless of O6-methyl-guanine-DNA-methyltransferase (MGMT) promoter methylation status in newly glioblastoma. BMC Cancer 2021, 21, 867. [Google Scholar] [CrossRef] [PubMed]
- Lwin, Z.; MacFadden, D.; Al-Zahrani, A.; Atenafu, E.; Miller, B.A.; Sahgal, A.; Menard, C.; Laperriere, N.; Mason, W.P. Glioblastoma management in the temozolomide era: Have we improved outcome? J. Neuro-Oncology 2013, 115, 303–310. [Google Scholar] [CrossRef]
- Cohen, K.J.; Heideman, R.L.; Zhou, T.; Holmes, E.J.; Lavey, R.S.; Bouffet, E.; Pollack, I.F. Temozolomide in the treatment of children with newly diagnosed diffuse intrinsic pontine gliomas: A report from the Children’s Oncology Group. Neuro-Oncology 2011, 13, 410–416. [Google Scholar] [CrossRef]
- Jakacki, R.I.; Cohen, K.J.; Buxton, A.; Krailo, M.D.; Burger, P.C.; Rosenblum, M.K.; Brat, D.J.; Hamilton, R.L.; Eckel, S.P.; Zhou, T. Phase 2 study of concurrent radiotherapy and temozolomide followed by temozolomide and lomustine in the treatment of children with high-grade glioma: A report of the Children’s Oncology Group ACNS0423 study. Neuro-Oncology 2016, 18, 1442–1450. [Google Scholar] [CrossRef]
- Gupta, T.; Talukdar, R.; Kannan, S.; Dasgupta, A.; Chatterjee, A.; Patil, V. Efficacy and safety of extended adjuvant temozolomide compared to standard adjuvant temozolomide in glioblastoma: Updated systematic review and meta-analysis. Neuro-Oncology Pract. 2022, 9, 354–363. [Google Scholar] [CrossRef]
- Mansouri, A.; Hachem, L.D.; Mansouri, S.; Nassiri, F.; Laperriere, N.J.; Xia, D.; Lindeman, N.I.; Wen, P.Y.; Chakravarti, A.; Mehta, M.P. MGMT promoter methylation status testing to guide therapy for glioblastoma: Refining the approach based on emerging evidence and current challenges. Neuro-Oncology 2019, 21, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Perry, J.R.; Laperriere, N.; O’Callaghan, C.J.; Brandes, A.A.; Menten, J.; Phillips, C.; Fay, M.; Nishikawa, R.; Cairncross, J.G.; Roa, W. Short-course radiation plus temozolomide in elderly patients with glioblastoma. N. Engl. J. Med. 2017, 376, 1027–1037. [Google Scholar] [CrossRef] [PubMed]
- Egaña, L.; Auzmendi-Iriarte, J.; Andermatten, J.; Villanua, J.; Ruiz, I.; Elua-Pinin, A.; Aldaz, P.; Querejeta, A.; Sarasqueta, C.; Zubia, F. Methylation of MGMT promoter does not predict response to temozolomide in patients with glioblastoma in Donostia Hospital. Sci. Rep. 2020, 10, 18445. [Google Scholar] [CrossRef] [PubMed]
- Friedman, G.K.; Johnston, J.M.; Bag, A.K.; Bernstock, J.D.; Li, R.; Aban, I.; Kachurak, K.; Nan, L.; Kang, K.-D.; Totsch, S. Oncolytic HSV-1 G207 immunovirotherapy for pediatric high-grade gliomas. N. Engl. J. Med. 2021, 384, 1613–1622. [Google Scholar] [CrossRef]
- Dunn, D.B. Larotrectinib and entrectinib: TRK inhibitors for the treatment of pediatric and adult patients with NTRK gene fusion. J. Adv. Pract. Oncol. 2020, 11, 418. [Google Scholar]
- Benitez-Ribas, D.; Cabezón, R.; Flórez-Grau, G.; Molero, M.C.; Puerta, P.; Guillen, A.; González-Navarro, E.A.; Paco, S.; Carcaboso, A.M.; Santa-Maria Lopez, V. Immune response generated with the administration of autologous dendritic cells pulsed with an allogenic tumoral cell-lines lysate in patients with newly diagnosed diffuse intrinsic pontine glioma. Front. Oncol. 2018, 8, 127. [Google Scholar] [CrossRef]
- Lin, F.Y.; Stuckert, A.; Tat, C.; White, M.; Ruggieri, L.; Zhang, H.; Mehta, B.; Lapteva, N.; Mei, Z.; Major, A. Phase I Trial of GD2. CART Cells Augmented with Constitutive Interleukin-7 Receptor for Treatment of High-Grade Pediatric CNS Tumors. J. Clin. Oncol. 2024, 42, 2769–2779. [Google Scholar] [CrossRef]
- Boland, L.K.; Wang, S.; Fourati, S.; DeLay, S.; Chia, T.-Y.; Billingham, L.; Katz, J.; Wei, C.; Geng, Y.; Sipila, P. DIPG-32. neoantigen heat shock protein vaccine, rhsc-dipgvax, is associated with increased vaccine-induced b cells and bcr/tcr repertoire diversity in pediatric pa-tients with diffuse midline gliomas. Neuro-Oncology 2024, 26 (Suppl. 4). [Google Scholar] [CrossRef]
- Aldape, K.; Brindle, K.M.; Chesler, L.; Chopra, R.; Gajjar, A.; Gilbert, M.R.; Gottardo, N.; Gutmann, D.H.; Hargrave, D.; Holland, E.C. Challenges to curing primary brain tumours. Nat. Rev. Clin. Oncol. 2019, 16, 509–520. [Google Scholar] [CrossRef]
- Brinkman, T.M.; Krasin, M.J.; Liu, W.; Armstrong, G.T.; Ojha, R.P.; Sadighi, Z.S.; Gupta, P.; Kimberg, C.; Srivastava, D.; Merchant, T.E. Long-term neurocognitive functioning and social attainment in adult survivors of pediatric CNS tumors: Results from the St Jude Lifetime Cohort Study. J. Clin. Oncol. 2016, 34, 1358. [Google Scholar] [CrossRef]
- Farkona, S.; Diamandis, E.P.; Blasutig, I.M. Cancer immunotherapy: The beginning of the end of cancer? BMC Med. 2016, 14, 73. [Google Scholar] [CrossRef] [PubMed]
- Mahoney, K.M.; Rennert, P.D.; Freeman, G.J. Combination cancer immunotherapy and new immunomodulatory targets. Nat. Rev. Drug Discov. 2015, 14, 561–584. [Google Scholar] [CrossRef]
- Coventry, B.J.; Henneberg, M. The immune system and responses to cancer: Coordinated evolution. F1000Research 2015, 4, 552. [Google Scholar] [CrossRef]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef]
- Zhang, J.; Shi, Z.; Xu, X.; Yu, Z.; Mi, J. The influence of microenvironment on tumor immunotherapy. FEBS J. 2019, 286, 4160–4175. [Google Scholar] [CrossRef]
- Ross, J.L.; Velazquez Vega, J.; Plant, A.; MacDonald, T.J.; Becher, O.J.; Hambardzumyan, D. Tumour immune landscape of paediatric high-grade gliomas. Brain 2021, 144, 2594–2609. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef]
- Bouffet, E.; Larouche, V.; Campbell, B.B.; Merico, D.; De Borja, R.; Aronson, M.; Durno, C.; Krueger, J.; Cabric, V.; Ramaswamy, V. Immune checkpoint inhibition for hypermutant glioblastoma multiforme resulting from germline biallelic mismatch repair deficiency. J. Clin. Oncol. 2016, 34, 2206–2211. [Google Scholar] [CrossRef] [PubMed]
- Guerra-García, P.; Marshall, L.V.; Cockle, J.V.; Ramachandran, P.V.; Saran, F.H.; Jones, C.; Carceller, F. Challenging the indiscriminate use of temozolomide in pediatric high-grade gliomas: A review of past, current, and emerging therapies. Pediatr. Blood Cancer 2020, 67, e28011. [Google Scholar] [CrossRef]
- Schuster, S.J.; Svoboda, J.; Chong, E.A.; Nasta, S.D.; Mato, A.R.; Anak, Ö.; Brogdon, J.L.; Pruteanu-Malinici, I.; Bhoj, V.; Landsburg, D. Chimeric antigen receptor T cells in refractory B-cell lymphomas. N. Engl. J. Med. 2017, 377, 2545–2554. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef] [PubMed]
- Foster, J.B.; Madsen, P.J.; Hegde, M.; Ahmed, N.; Cole, K.A.; Maris, J.M.; Resnick, A.C.; Storm, P.B.; Waanders, A.J. Immunotherapy for pediatric brain tumors: Past and present. Neuro-Oncology 2019, 21, 1226–1238. [Google Scholar] [CrossRef]
- Majzner, R.G.; Theruvath, J.L.; Nellan, A.; Heitzeneder, S.; Cui, Y.; Mount, C.W.; Rietberg, S.P.; Linde, M.H.; Xu, P.; Rota, C. CAR T cells targeting B7-H3, a pan-cancer antigen, demonstrate potent preclinical activity against pediatric solid tumors and brain tumors. Clin. Cancer Res. 2019, 25, 2560–2574. [Google Scholar] [CrossRef]
- Tang, X.; Zhao, S.; Zhang, Y.; Wang, Y.; Zhang, Z.; Yang, M.; Zhu, Y.; Zhang, G.; Guo, G.; Tong, A. B7-H3 as a novel CAR-T therapeutic target for glioblastoma. Mol. Ther. Oncolytics 2019, 14, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Mount, C.W.; Majzner, R.G.; Sundaresh, S.; Arnold, E.P.; Kadapakkam, M.; Haile, S.; Labanieh, L.; Hulleman, E.; Woo, P.J.; Rietberg, S.P. Potent antitumor efficacy of anti-GD2 CAR T cells in H3-K27M+ diffuse midline gliomas. Nat. Med. 2018, 24, 572–579. [Google Scholar] [CrossRef] [PubMed]
- Nazha, B.; Inal, C.; Owonikoko, T.K. Disialoganglioside GD2 expression in solid tumors and role as a target for cancer therapy. Front. Oncol. 2020, 10, 1000. [Google Scholar] [CrossRef]
- Mougel, A.; Terme, M.; Tanchot, C. Therapeutic cancer vaccine and combinations with antiangiogenic therapies and immune checkpoint blockade. Front. Immunol. 2019, 10, 432525. [Google Scholar] [CrossRef]
- Hollingsworth, R.E.; Jansen, K. Turning the corner on therapeutic cancer vaccines. NPJ Vaccines 2019, 4, 7. [Google Scholar] [CrossRef]
- Sellars, M.C.; Wu, C.J.; Fritsch, E.F. Cancer vaccines: Building a bridge over troubled waters. Cell 2022, 185, 2770–2788. [Google Scholar] [CrossRef]
- Olsen, H.E.; Lynn, G.M.; Valdes, P.A.; Cerecedo Lopez, C.D.; Ishizuka, A.S.; Arnaout, O.; Bi, W.L.; Peruzzi, P.P.; Chiocca, E.A.; Friedman, G.K. Therapeutic cancer vaccines for pediatric malignancies: Advances, challenges, and emerging technologies. Neuro-Oncology Adv. 2021, 3, vdab027. [Google Scholar] [CrossRef] [PubMed]
- Sampson, J.H.; Mitchell, D.A. Vaccination strategies for neuro-oncology. Neuro-Oncology 2015, 17 (Suppl. S7), vii15–vii25. [Google Scholar] [CrossRef] [PubMed]
- Gambardella, V.; Tarazona, N.; Cejalvo, J.M.; Lombardi, P.; Huerta, M.; Roselló, S.; Fleitas, T.; Roda, D.; Cervantes, A. Personalized medicine: Recent progress in cancer therapy. Cancers 2020, 12, 1009. [Google Scholar] [CrossRef] [PubMed]
- Krzyszczyk, P.; Acevedo, A.; Davidoff, E.J.; Timmins, L.M.; Marrero-Berrios, I.; Patel, M.; White, C.; Lowe, C.; Sherba, J.J.; Hartmanshenn, C. The growing role of precision and personalized medicine for cancer treatment. Technology 2018, 6, 79–100. [Google Scholar] [CrossRef]
- Dewdney, B.; Jenkins, M.R.; Best, S.A.; Freytag, S.; Prasad, K.; Holst, J.; Endersby, R.; Johns, T.G. From signalling pathways to targeted therapies: Unravelling glioblastoma’s secrets and harnessing two decades of progress. Signal Transduct. Target. Ther. 2023, 8, 400. [Google Scholar] [CrossRef]
- Bolcaen, J.; Nair, S.; Driver, C.H.; Boshomane, T.M.; Ebenhan, T.; Vandevoorde, C. Novel receptor tyrosine kinase pathway inhibitors for targeted radionuclide therapy of glioblastoma. Pharmaceuticals 2021, 14, 626. [Google Scholar] [CrossRef]
- Yesilkanal, A.E.; Johnson, G.L.; Ramos, A.F.; Rosner, M.R. New strategies for targeting kinase networks in cancer. J. Biol. Chem. 2021, 297, 101128. [Google Scholar] [CrossRef]
- Huchedé, P.; Leblond, P.; Castets, M. The Intricate Epigenetic and Transcriptional Alterations in Pediatric High-Grade Gliomas: Targeting the Crosstalk as the Oncogenic Achilles’ Heel. Biomedicines 2022, 10, 1311. [Google Scholar] [CrossRef]
- Dalpatraj, N.; Naik, A.; Thakur, N. GSK-J4: An H3K27 histone demethylase inhibitor, as a potential anti-cancer agent. Int. J. Cancer 2023, 153, 1130–1138. [Google Scholar] [CrossRef]
- Williams, M.J.; Singleton, W.G.; Lowis, S.P.; Malik, K.; Kurian, K.M. Therapeutic targeting of histone modifications in adult and pediatric high-grade glioma. Front. Oncol. 2017, 7, 45. [Google Scholar] [CrossRef]
- Lee, E.Q.; Reardon, D.A.; Schiff, D.; Drappatz, J.; Muzikansky, A.; Grimm, S.A.; Norden, A.D.; Nayak, L.; Beroukhim, R.; Rinne, M.L. Phase II study of panobinostat in combination with bevacizumab for recurrent glioblastoma and anaplastic glioma. Neuro-Oncology 2015, 17, 862–867. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Palmer, J.D.; Werner-Wasik, M.; Andrews, D.W.; Evans, J.J.; Glass, J.; Kim, L.; Bar-Ad, V.; Judy, K.; Farrell, C. Phase I trial of panobinostat and fractionated stereotactic re-irradiation therapy for recurrent high grade gliomas. J. Neuro-Oncology 2016, 127, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Peters, K.B.; Vredenburgh, J.J.; Desjardins, A.; Friedman, H.S.; Herndon, J.E.; Coan, A.D.; McSherry, F.; Lipp, E.S.; Brickhouse, A.; Massey, W.C. Vorinostat, temozolomide, and bevacizumab for patients with recurrent glioblastoma: A phase I/II trial. J. Clin. Oncol. 2012, 30, 2027. [Google Scholar] [CrossRef]
- Tran, A.N.; Lai, A.; Li, S.; Pope, W.B.; Teixeira, S.; Harris, R.J.; Woodworth, D.C.; Nghiemphu, P.L.; Cloughesy, T.F.; Ellingson, B.M. Increased sensitivity to radiochemotherapy in IDH1 mutant glioblastoma as demonstrated by serial quantitative MR volumetry. Neuro-Oncology 2014, 16, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Cairncross, J.G.; Wang, M.; Jenkins, R.B.; Shaw, E.G.; Giannini, C.; Brachman, D.G.; Buckner, J.C.; Fink, K.L.; Souhami, L.; Laperriere, N.J. Benefit from procarbazine, lomustine, and vincristine in oligodendroglial tumors is associated with mutation of IDH. J. Clin. Oncol. 2014, 32, 783. [Google Scholar] [CrossRef]
- Ray Chaudhuri, A.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef]
- Wang, Y.; Wild, A.T.; Turcan, S.; Wu, W.H.; Sigel, C.; Klimstra, D.S.; Ma, X.; Gong, Y.; Holland, E.C.; Huse, J.T. Targeting therapeutic vulnerabilities with PARP inhibition and radiation in IDH-mutant gliomas and cholangiocarcinomas. Sci. Adv. 2020, 6, eaaz3221. [Google Scholar] [CrossRef]
- Mayoh, C.; Mao, J.; Xie, J.; Tax, G.; Chow, S.-O.; Cadiz, R.; Pazaky, K.; Barahona, P.; Ajuyah, P.; Trebilcock, P. High-Throughput Drug Screening of Primary Tumor Cells Identifies Therapeutic Strategies for Treating Children with High-Risk Cancer. Cancer Res. 2023, 83, 2716–2732. [Google Scholar] [CrossRef]
- Liu, R.; Li, X.; Xiao, W.; Lam, K.S. Tumor-targeting peptides from combinatorial libraries. Adv. Drug Deliv. Rev. 2017, 110, 13–37. [Google Scholar] [CrossRef]
- Gröbner, S.N.; Worst, B.C.; Weischenfeldt, J.; Buchhalter, I.; Kleinheinz, K.; Rudneva, V.A.; Johann, P.D.; Balasubramanian, G.P.; Segura-Wang, M.; Brabetz, S. The landscape of genomic alterations across childhood cancers. Nature 2018, 555, 321–327. [Google Scholar] [CrossRef]
- Dharia, N.V.; Kugener, G.; Guenther, L.M.; Malone, C.F.; Durbin, A.D.; Hong, A.L.; Howard, T.P.; Bandopadhayay, P.; Wechsler, C.S.; Fung, I. A first-generation pediatric cancer dependency map. Nat. Genet. 2021, 53, 529–538. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Inhibitors, Immunotherapy or Others | NCT # | Phase of Clinical Trial | Target Gene/s | Cohort |
|---|---|---|---|---|
| NK cells injection | NCT04254419 | 1 | MGMT, BRAFV600E, ACVR1, ATRx, TP53, H3G34, H3.3/H3.1K27, IDH1, CDKN2A, PDGFR | Recurrent HGG |
| Intratumoral injection of G207 + RT [136] | NCT04482933 | 2 | - | Neoplasms, HGG, astrocytoma |
| Lutathera (177Lu-DOTATATE) | NCT05278208 | 1 and 2 | Type-2A somatostatin receptors (SST2A) | HGG, medulloblastoma |
| iC9-GD2-CAR-T cells | NCT05298995 | 1 | GD2 | Medulloblastoma, HGG, DMG |
| Loc3CART: Locoregional Delivery of B7-H3-CAR T cells | NCT05835687 | 1 | - | HGG (B7-H3-positive) |
| CLR 131 | NCT03478462 | 1 | Breakdown of dsDNA | DMG, HGG |
| Berubicin | NCT05082493 | 1 | Topoisomerase II (Topo II) | Progressive, refractory, or recurrent HGG |
| NEO100 | NCT06357377 | 1 | Rad/Raf pathway | HGG, DMG |
| AsiDNA (etidaligide) + RT | NCT05394558 | 1 and 2 | DNA-PK | HGG, DMG |
| Larotrectinib [137] | NCT04655404 | 1 | TRK | HGG with NTRK fusion, DMG |
| LAM561 acid | NCT04299191 | 1 and 2 | MAPK, CDK, PI3K inhibitor | HGG |
| Selinezor + RT | NCT05099003 | 1 and 2 | CRM1 | HGG (H3K27M-mutant or H3K27-WT), DMG |
| Fimepinostat | NCT03893487 | 1 | PI3K, HDAC | DIPG, HGG, medulloblastoma |
| DC vaccine + TMZ [134,138] | NCT04911621 | 1 and 2 | Anti-tumor defense mechanisms | HGG, DIPG |
| Trametinib + Everolimus | NCT04485559 | 1 | MEK, mTOR | Recurrent HGG, grade 2 glioma, LGG |
| Novel peptide vaccine (PEP-CMV) + TMZ + Tetanus–Diphtheria vaccine | NCT05096481 | 2 | - | Medulloblastoma, HGG, DIPG |
| Lorlatinib + BABYPOG/HIT-SKK | NCT06333899 | 1 | ALK, TRK receptors | HGG with ROS1 or ALK fusion |
| Abemaciclib + TMZ + RT | NCT06413706 | 2 | CKD4/6, Rb phosphorylation | HGG |
| C7R-GD2 CAR-T cells + cyclophosphamide and fludarabine [139] | NCT04099797 | 2 | GD2 | GAIL-B in DMG, HGG, DIPG, medulloblastoma |
| Ribociclib + Everolimus | NCT05843253 | 2 | CDK4/6, growth-driven transduction signals in T cell response | HGG and DIPG with PI3K/mTOR mutations |
| Dabrafenib, Trametinib and Hydroxychloroquine | NCT04201457 | 1 and 2 | BRAFV600E, MEK | HGG with BRAFV600E, BRAF fusion/duplication positive, or NF1-associated mutations |
| Olutasidenib + TMZ | NCT06161974 | 2 | IDH1 | HGG with IDH1 mutations |
| Panobinostat + Everolimus | NCT03632317 | 2 | Histone deacetylase | H3.1 or H3.3 K27M DIPG |
| Neo-antigen HSP vaccine (rHSC-DIPGVax) + Balstilimab/Zalifrelimab [140] | NCT04943848 | 1 | 16 peptides on DIPG and DMG | DIPG, DMG |
| Dabrafenib + Trametinib | NCT03975829 | 4 | BRAF, MEK1/2 | HGG |
| Topotecan + TMZ + Ribociclib | NCT05429502 | 1 and 2 | Topoisomerase I, CDK4/6 | R/R neuroblastoma, medulloblastoma, HGG |
| Abemaciclib + Irinotecan + TMZ + Dinutuximab + GM-CSF | NCT04238819 | 1 and 2 | CDK4/6, topoisomerase I, GD2 | HGG |
| Nivolumab | NCT04323046 | 1 | PD-1 receptor | Malignant, recurrent HGG |
| CD200 Activation Receptor Ligand (CD200AR-L) and Allogeneic tumor lysate vaccine + adjuvant re-irradiation | NCT06305910 | 1 | CD200R1 | DMG, H3K27M HGG |
| INCB7839 | NCT04295759 | 1 | ADAM | HGG, DIPG, DMG |
| SurVaxM + Montanide ISA 51 | NCT04978727 | 1 | IAP | HGG, DIPG |
| NKTR-214 + Nivolumab | NCT04730349 | 1 and 2 | CD8T | HGG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoel, A.; Adjumain, S.; Liang, Y.; Daniel, P.; Firestein, R.; Tsui, V. Emerging and Biological Concepts in Pediatric High-Grade Gliomas. Cells 2024, 13, 1492. https://doi.org/10.3390/cells13171492
Yoel A, Adjumain S, Liang Y, Daniel P, Firestein R, Tsui V. Emerging and Biological Concepts in Pediatric High-Grade Gliomas. Cells. 2024; 13(17):1492. https://doi.org/10.3390/cells13171492
Chicago/Turabian StyleYoel, Abigail, Shazia Adjumain, Yuqing Liang, Paul Daniel, Ron Firestein, and Vanessa Tsui. 2024. "Emerging and Biological Concepts in Pediatric High-Grade Gliomas" Cells 13, no. 17: 1492. https://doi.org/10.3390/cells13171492