

Importance of Transcript Variants in Transcriptome Analyses

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Model
2.2. RNA Sequencing Data
2.3. RNA Sequencing Analysis
2.4. Analysis of the Transcript Variants
2.5. Statistical Analyses
3. Results
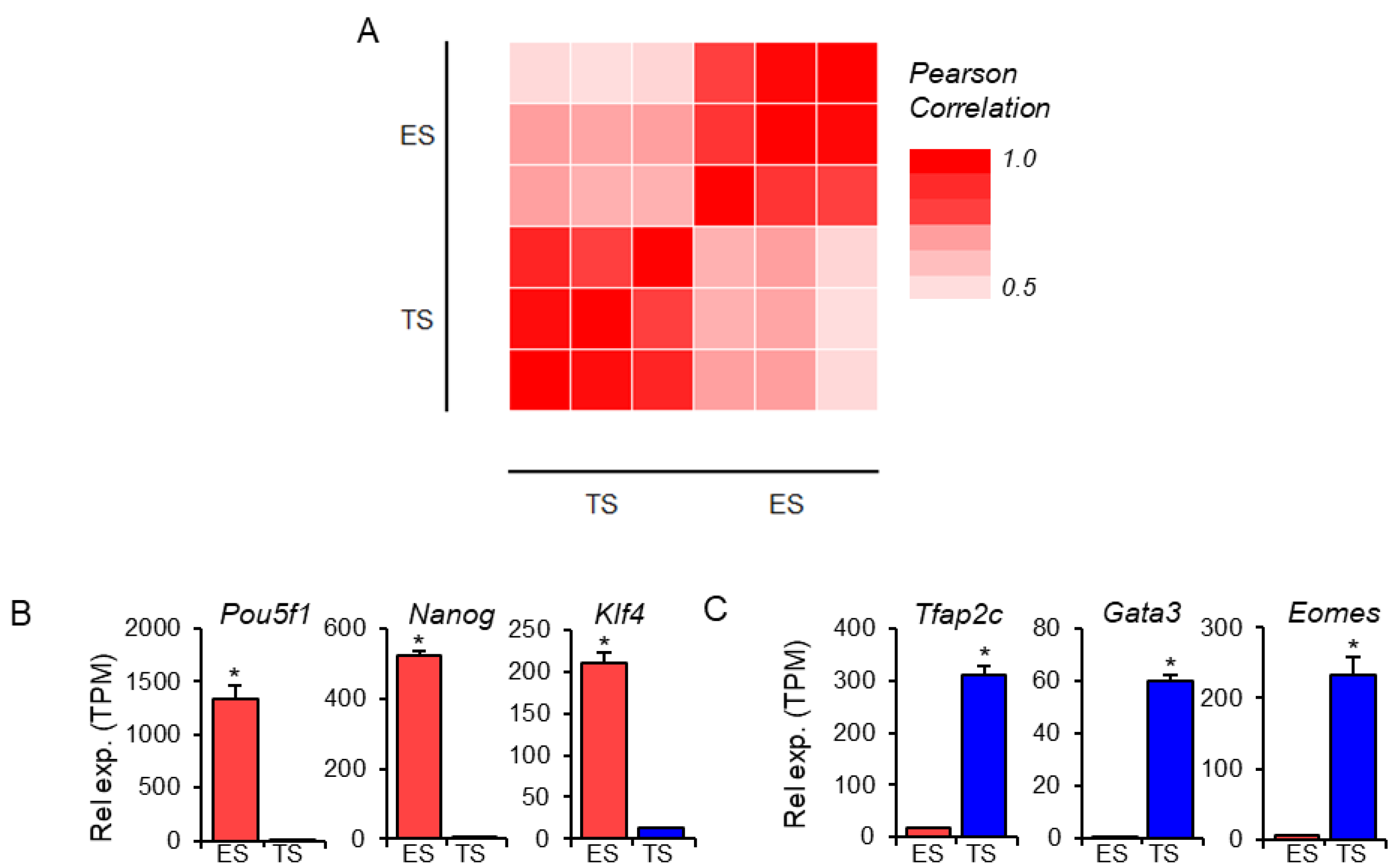
3.1. Lineage-Specific Expression of Transcription Factors
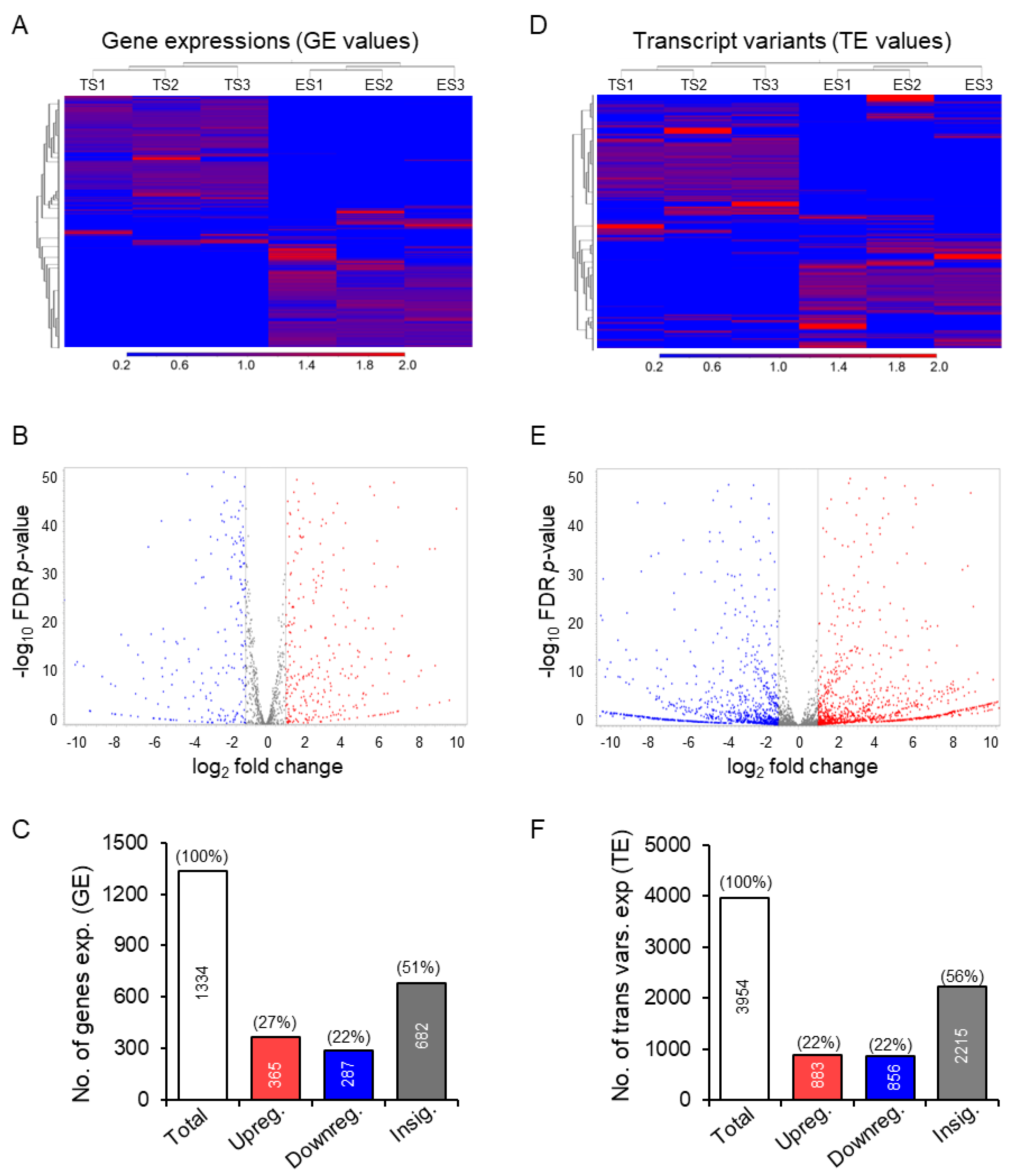
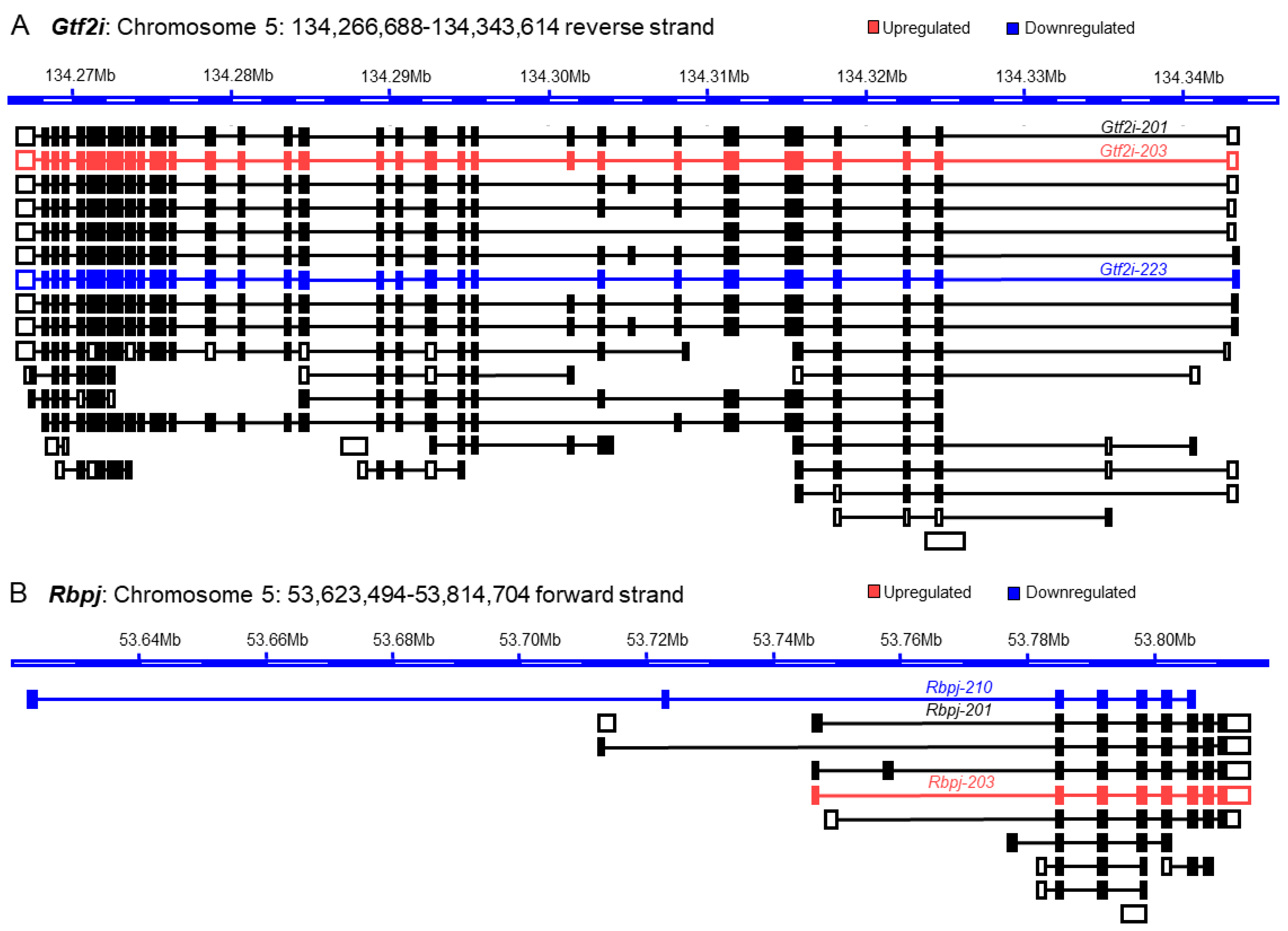
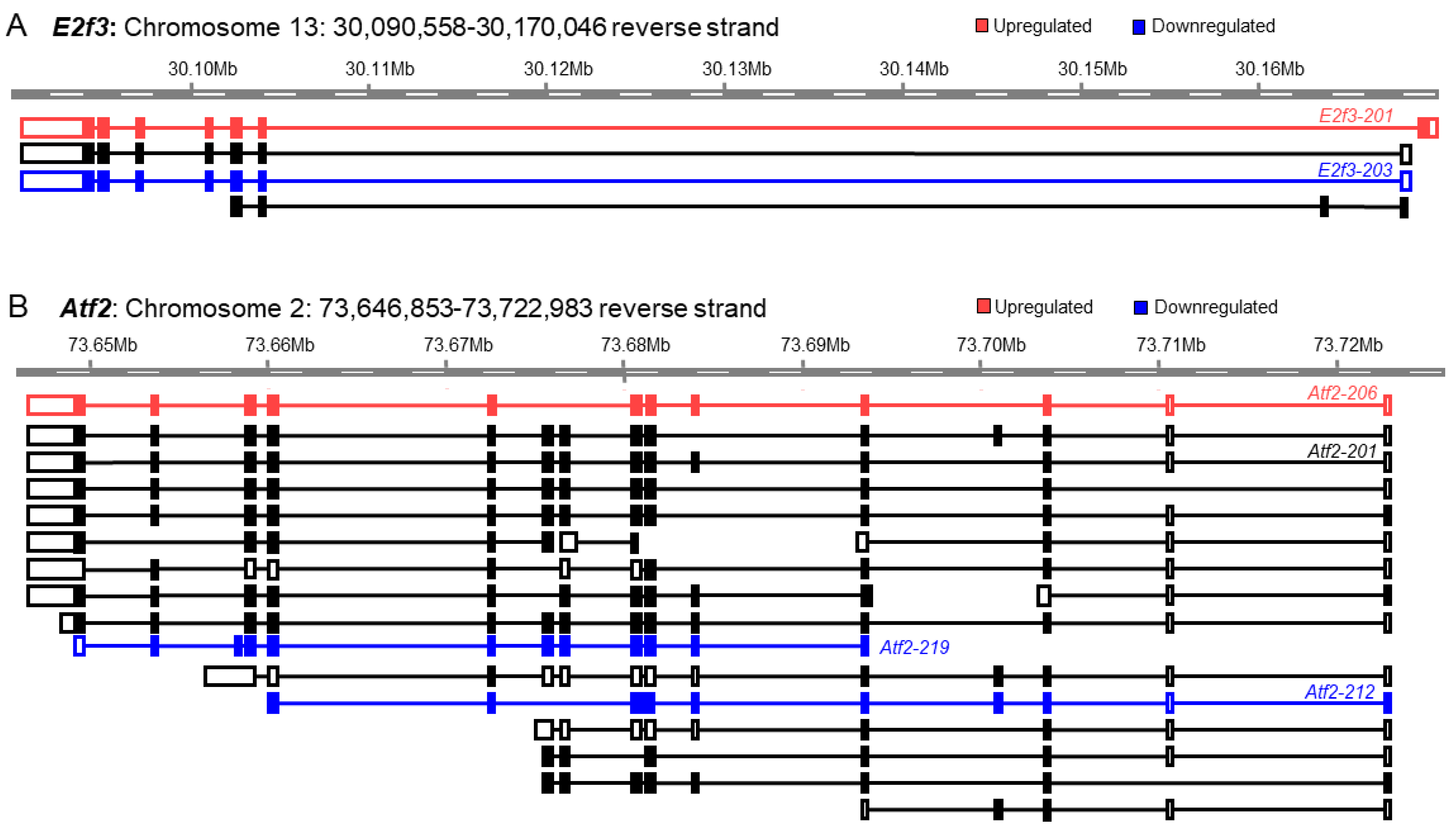
3.2. Differential Expression of the Transcription Factor Genes and Transcript Variants
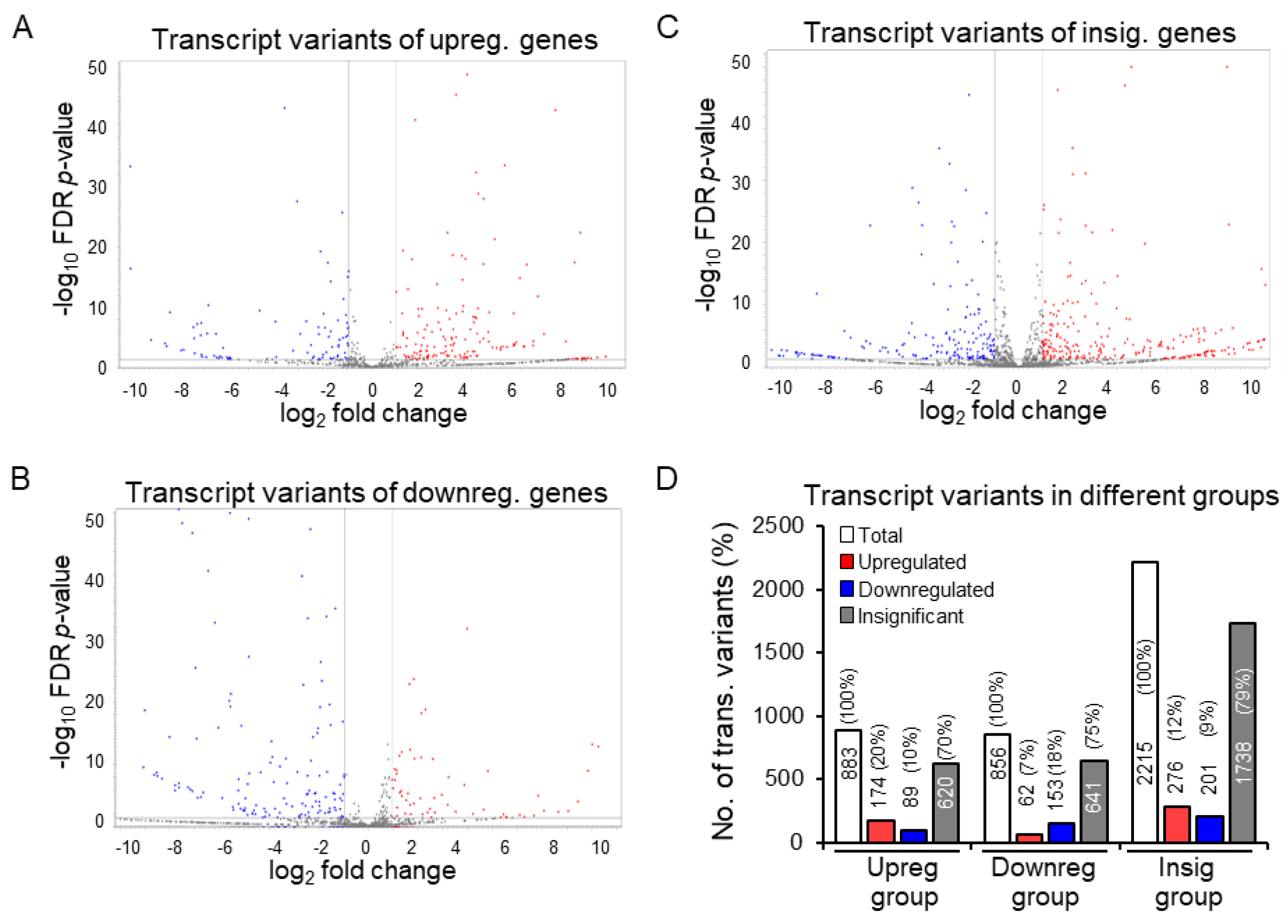
3.3. Discrepancy between Gene Expression and Transcript Variants
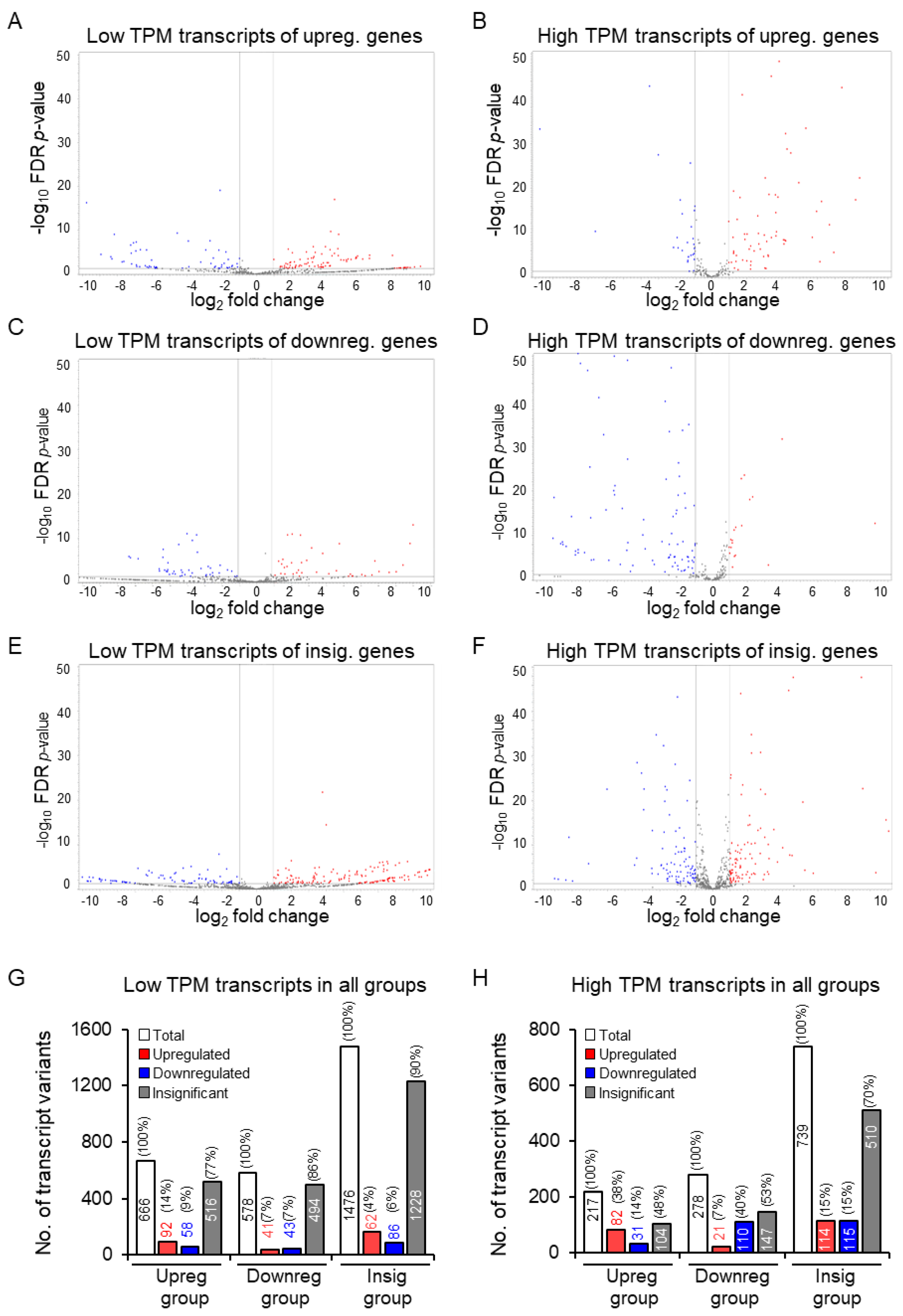
3.4. Increased Discrepancy among the Low-Abundance Transcript Variants
3.5. The Basis of Discrepancy between Gene Expression and Transcript Variant Analyses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, S.; Pyne, S.; Pietrzak, S.; Halberg, S.; McCalla, S.G.; Siahpirani, A.F.; Sridharan, R.; Roy, S. Inference of cell type-specific gene regulatory networks on cell lineages from single cell omic datasets. Nat. Commun. 2023, 14, 3064. [Google Scholar] [CrossRef]
- Lowe, R.; Shirley, N.; Bleackley, M.; Dolan, S.; Shafee, T. Transcriptomics technologies. PLoS Comput. Biol. 2017, 13, e1005457. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Corey, D.R. RNA sequencing: Platform selection, experimental design, and data interpretation. Nucleic Acid 2012, 22, 271–274. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Ballard, J.; Zhao, Y.; Long, Q. Knowledge-guided learning methods for integrative analysis of multi-omics data. Comput. Struct. Biotechnol. J. 2024, 23, 1945–1950. [Google Scholar] [CrossRef] [PubMed]
- Limbu, M.S.; Xiong, T.; Wang, S. A review of Ribosome profiling and tools used in Ribo-seq data analysis. Comput. Struct. Biotechnol. J. 2024, 23, 1912–1918. [Google Scholar] [CrossRef]
- Samuels, D.S.; Lybecker, M.C.; Yang, X.F.; Ouyang, Z.; Bourret, T.J.; Boyle, W.K.; Stevenson, B.; Drecktrah, D.; Caimano, M.J. Gene Regulation and Transcriptomics. Curr. Issues Mol. Biol. 2021, 42, 223–266. [Google Scholar] [CrossRef]
- Pal, S.; Gupta, R.; Kim, H.; Wickramasinghe, P.; Baubet, V.; Showe, L.C.; Dahmane, N.; Davuluri, R.V. Alternative transcription exceeds alternative splicing in generating the transcriptome diversity of cerebellar development. Genome Res. 2011, 21, 1260–1272. [Google Scholar] [CrossRef]
- Reyes, A.; Huber, W. Alternative start and termination sites of transcription drive most transcript isoform differences across human tissues. Nucleic Acids Res. 2018, 46, 582–592. [Google Scholar] [CrossRef]
- Alfonso-Gonzalez, C.; Hilgers, V. (Alternative) transcription start sites as regulators of RNA processing. Trends Cell Biol. 2024. [Google Scholar] [CrossRef]
- Xin, D.; Hu, L.; Kong, X. Alternative promoters influence alternative splicing at the genomic level. PLoS ONE 2008, 3, e2377. [Google Scholar] [CrossRef] [PubMed]
- Kelemen, O.; Convertini, P.; Zhang, Z.; Wen, Y.; Shen, M.; Falaleeva, M.; Stamm, S. Function of alternative splicing. Gene 2013, 514, 1–30. [Google Scholar] [CrossRef]
- Piazzi, M.; Bavelloni, A.; Salucci, S.; Faenza, I.; Blalock, W.L. Alternative splicing, RNA editing, and the current limits of next generation sequencing. Genes 2023, 14, 1386. [Google Scholar] [CrossRef] [PubMed]
- Ha, I.; Roberts, S.; Maldonado, E.; Sun, X.; Kim, L.U.; Green, M.; Reinberg, D. Multiple functional domains of human transcription factor IIB: Distinct interactions with two general transcription factors and RNA polymerase II. Genes Dev 1993, 7, 1021–1032. [Google Scholar] [CrossRef] [PubMed]
- Sonam, D.; Manoj, B.M. Non-coding transcript variants of protein-coding genes—What are they good for? RNA Biol. 2018, 15, 1025–1031. [Google Scholar] [CrossRef]
- Johnson, K.A.; Krishnan, A. Robust normalization and transformation techniques for constructing gene coexpression networks from RNA-seq data. Genome Biol. 2022, 23, 1–26. [Google Scholar] [CrossRef]
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-Cabrero, D.; Cervera, A.; McPherson, A.; Szcześniak, M.W.; Gaffney, D.J.; Elo, L.L.; Zhang, X. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016, 17, 1–19. [Google Scholar] [CrossRef]
- Jiang, Z.; Zhou, X.; Li, R.; Michal, J.J.; Zhang, S.; Dodson, M.V.; Zhang, Z.; Harland, R.M. Whole transcriptome analysis with sequencing: Methods, challenges and potential solutions. Cell Mol. Life Sci. 2015, 72, 3425–3439. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. A decade of transcription factor-mediated reprogramming to pluripotency. Nat. Rev. Mol. Cell Biol. 2016, 17, 183–193. [Google Scholar] [CrossRef]
- Kubaczka, C.; Senner, C.E.; Cierlitza, M.; Araúzo-Bravo, M.J.; Kuckenberg, P.; Peitz, M.; Hemberger, M.; Schorle, H. Direct Induction of Trophoblast Stem Cells from Murine Fibroblasts. Cell Stem Cell 2015, 17, 557–568. [Google Scholar] [CrossRef]
- Johnston, A.D.; Simões-Pires, C.A.; Thompson, T.V.; Suzuki, M.; Greally, J.M. Functional genetic variants can mediate their regulatory effects through alteration of transcription factor binding. Nat. Commun. 2019, 10, 3472. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets--update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Kunath, T.; Hadjantonakis, A.K.; Nagy, A.; Rossant, J. Promotion of trophoblast stem cell proliferation by FGF4. Science 1998, 282, 2072–2075. [Google Scholar] [CrossRef] [PubMed]
- Chakravarthi, V.P.; Ratri, A.; Masumi, S.; Borosha, S.; Ghosh, S.; Christenson, L.K.; Roby, K.F.; Wolfe, M.W.; Rumi, M.A.K. Granulosa cell genes that regulate ovarian follicle development beyond the antral stage: The role of estrogen receptor β. Mol. Cell Endocrinol 2021, 528, 111212. [Google Scholar] [CrossRef]
- Khristi, V.; Chakravarthi, V.P.; Singh, P.; Ghosh, S.; Pramanik, A.; Ratri, A.; Borosha, S.; Roby, K.F.; Wolfe, M.W.; Rumi, M.A.K. ESR2 regulates granulosa cell genes essential for follicle maturation and ovulation. Mol. Cell Endocrinol 2018, 474, 214–226. [Google Scholar] [CrossRef]
- Khristi, V.; Ratri, A.; Ghosh, S.; Pathak, D.; Borosha, S.; Dai, E.; Roy, R.; Chakravarthi, V.P.; Wolfe, M.W.; Karim Rumi, M.A. Disruption of ESR1 alters the expression of genes regulating hepatic lipid and carbohydrate metabolism in male rats. Mol. Cell Endocrinol 2019, 490, 47–56. [Google Scholar] [CrossRef]
- Lambert, S.A.; Jolma, A.; Campitelli, L.F.; Das, P.K.; Yin, Y.; Albu, M.; Chen, X.; Taipale, J.; Hughes, T.R.; Weirauch, M.T. The Human Transcription Factors. Cell 2018, 172, 650–665. [Google Scholar] [CrossRef]
- Nelder, J.A.; Wedderburn, R.W. Generalized linear models. J. R. Stat. Soc. Ser. A: Stat. Soc. 1972, 135, 370–384. [Google Scholar] [CrossRef]
- Lin, J.; Khan, M.; Zapiec, B.; Mombaerts, P. Efficient derivation of extraembryonic endoderm stem cell lines from mouse postimplantation embryos. Sci. Rep. 2016, 6, 39457. [Google Scholar] [CrossRef]
- Ralston, A.; Cox, B.J.; Nishioka, N.; Sasaki, H.; Chea, E.; Rugg-Gunn, P.; Guo, G.; Robson, P.; Draper, J.S.; Rossant, J. Gata3 regulates trophoblast development downstream of Tead4 and in parallel to Cdx2. Development 2010, 137, 395–403. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Soneson, C.; Love, M.I.; Robinson, M.D. Differential analyses for RNA-seq: Transcript-level estimates improve gene-level inferences. F1000Research 2015, 4, 1521–1538. [Google Scholar] [CrossRef] [PubMed]
- Stamm, S.; Ben-Ari, S.; Rafalska, I.; Tang, Y.; Zhang, Z.; Toiber, D.; Thanaraj, T.; Soreq, H. Function of alternative splicing. Gene 2005, 344, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Ashkenas, J. Gene regulation by mRNA editing. Am. J. Hum. Genet. 1997, 60, 278. [Google Scholar]
- Ray, T.A.; Cochran, K.; Kozlowski, C.; Wang, J.; Alexander, G.; Cady, M.A.; Spencer, W.J.; Ruzycki, P.A.; Clark, B.S.; Laeremans, A. Comprehensive identification of mRNA isoforms reveals the diversity of neural cell-surface molecules with roles in retinal development and disease. Nat. Commun. 2020, 11, 3328. [Google Scholar] [CrossRef]
- Sun, B.; Chen, L. Mapping genetic variants for nonsense-mediated mRNA decay regulation across human tissues. Genome Biol. 2023, 24, 164. [Google Scholar] [CrossRef]
- Marchese, F.P.; Raimondi, I.; Huarte, M. The multidimensional mechanisms of long noncoding RNA function. Genome Biol. 2017, 18, 1–13. [Google Scholar] [CrossRef]
- Okonechnikov, K.; Golosova, O.; Fursov, M. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef]
- Golosova, O.; Henderson, R.; Vaskin, Y.; Gabrielian, A.; Grekhov, G.; Nagarajan, V.; Oler, A.J.; Quiñones, M.; Hurt, D.; Fursov, M.; et al. Unipro UGENE NGS pipelines and components for variant calling, RNA-seq and ChIP-seq data analyses. PeerJ 2014, 2, e644. [Google Scholar] [CrossRef]
- Rose, R.; Golosova, O.; Sukhomlinov, D.; Tiunov, A.; Prosperi, M. Flexible design of multiple metagenomics classification pipelines with UGENE. Bioinformatics 2019, 35, 1963–1965. [Google Scholar] [CrossRef]
- Lee, G.Y.; Ham, S.; Lee, S.V. Brief guide to RNA sequencing analysis for nonexperts in bioinformatics. Mol. Cells 2024, 47, 100060. [Google Scholar] [CrossRef] [PubMed]
- Dillies, M.A.; Rau, A.; Aubert, J.; Hennequet-Antier, C.; Jeanmougin, M.; Servant, N.; Keime, C.; Marot, G.; Castel, D.; Estelle, J.; et al. A comprehensive evaluation of normalization methods for Illumina high-throughput RNA sequencing data analysis. Brief Bioinform 2013, 14, 671–683. [Google Scholar] [CrossRef] [PubMed]
- Yi, L.; Pimentel, H.; Bray, N.L.; Pachter, L. Gene-level differential analysis at transcript-level resolution. Genome Biol. 2018, 19, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Baruzzo, G.; Hayer, K.E.; Kim, E.J.; Di Camillo, B.; FitzGerald, G.A.; Grant, G.R. Simulation-based comprehensive benchmarking of RNA-seq aligners. Nat. Methods 2017, 14, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Ju, W.; Greene, C.S.; Eichinger, F.; Nair, V.; Hodgin, J.B.; Bitzer, M.; Lee, Y.-s.; Zhu, Q.; Kehata, M.; Li, M. Defining cell-type specificity at the transcriptional level in human disease. Genome Res. 2013, 23, 1862–1873. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vo, K.; Sharma, Y.; Paul, A.; Mohamadi, R.; Mohamadi, A.; Fields, P.E.; Rumi, M.A.K. Importance of Transcript Variants in Transcriptome Analyses. Cells 2024, 13, 1502. https://doi.org/10.3390/cells13171502
Vo K, Sharma Y, Paul A, Mohamadi R, Mohamadi A, Fields PE, Rumi MAK. Importance of Transcript Variants in Transcriptome Analyses. Cells. 2024; 13(17):1502. https://doi.org/10.3390/cells13171502
Chicago/Turabian StyleVo, Kevin, Yashica Sharma, Anohita Paul, Ryan Mohamadi, Amelia Mohamadi, Patrick E. Fields, and M. A. Karim Rumi. 2024. "Importance of Transcript Variants in Transcriptome Analyses" Cells 13, no. 17: 1502. https://doi.org/10.3390/cells13171502
APA StyleVo, K., Sharma, Y., Paul, A., Mohamadi, R., Mohamadi, A., Fields, P. E., & Rumi, M. A. K. (2024). Importance of Transcript Variants in Transcriptome Analyses. Cells, 13(17), 1502. https://doi.org/10.3390/cells13171502