
CCDC78: Unveiling the Function of a Novel Gene Associated with Hereditary Myopathy

 , ,
, ,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Standard Protocol Approvals and Patient Consents
2.2. Morphological Analysis of Muscle Biopsies and HeLa Cells
2.3. DNA Analysis
2.4. Cells Culture
2.5. RNA Extraction
2.6. Transcripts Analysis
2.7. Transcriptome Profiling
2.8. RT-qPCR
2.9. Western Blotting Study
2.10. Co-Immunoprecipitation (Co-Ip) Assay
2.11. Mass Spectroscopy Study
2.12. nLC-nESI-HRMS/MS
2.13. Filtering and Selecting Interactors Candidates for Validation
2.14. Three-Dimensional (3D) Modelling
3. Results
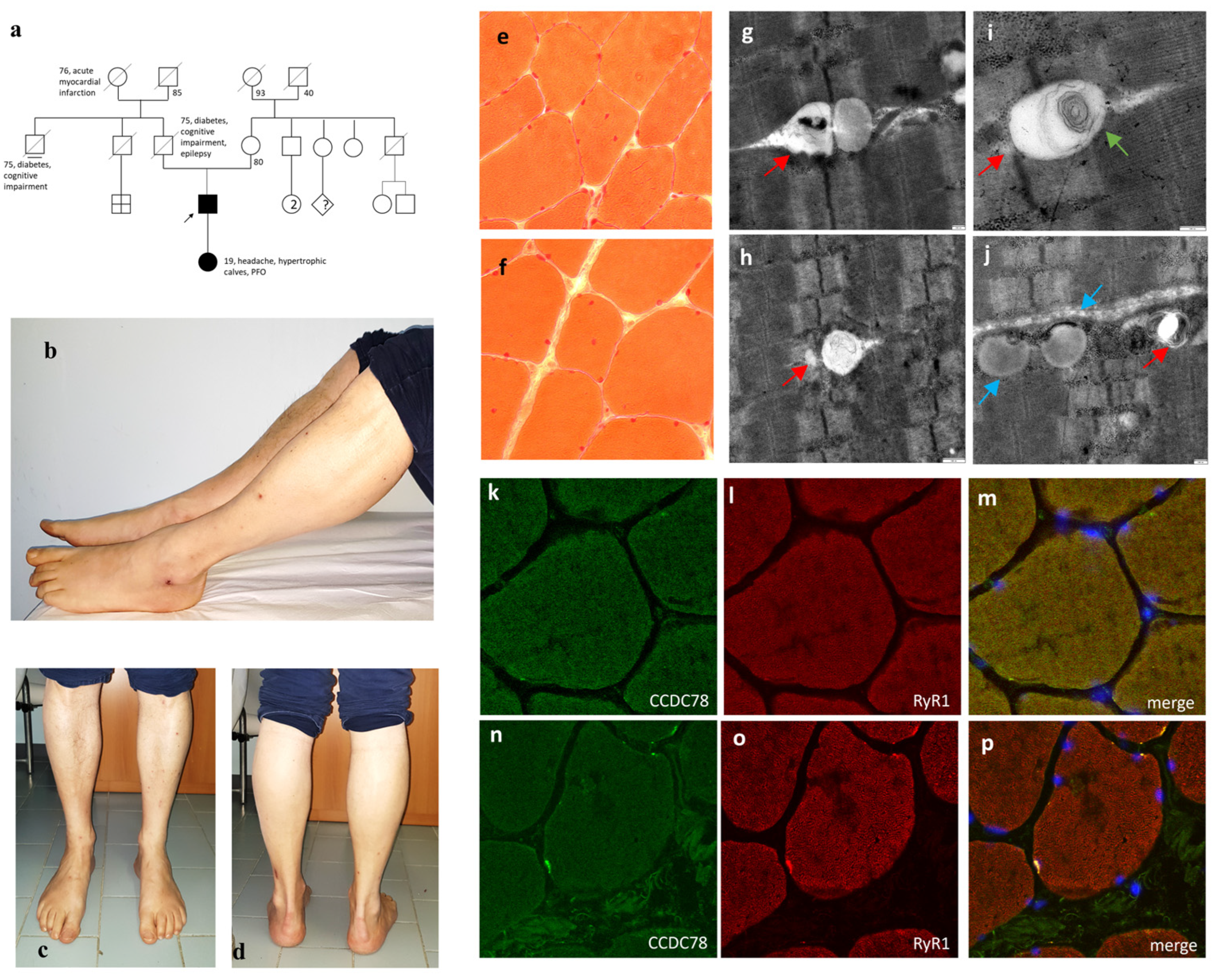
3.1. The Nonsense CCDC78 Mutation Leads to a Dystrophic Muscle Process Mainly Involving the SR
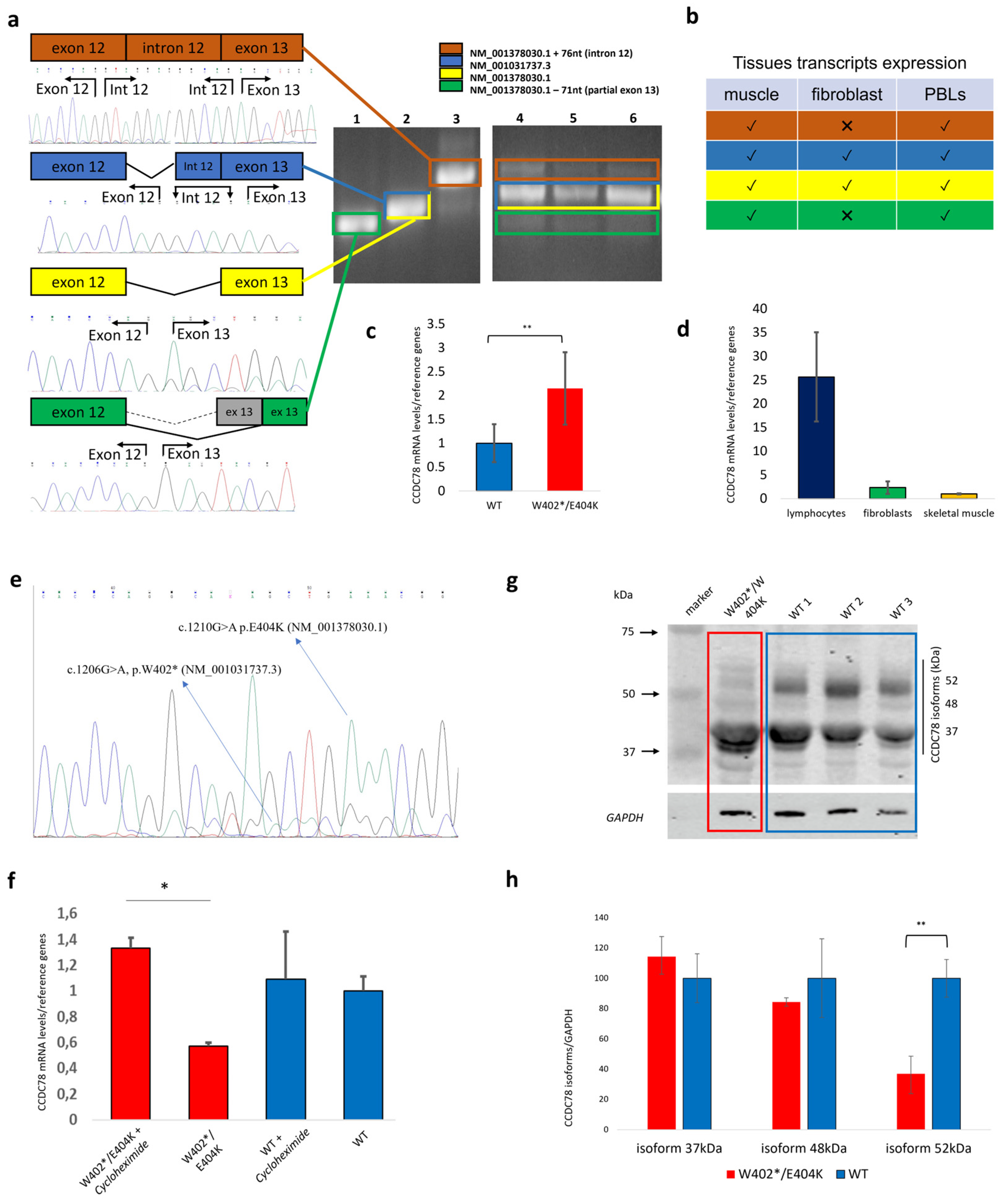
3.2. Human Tissues Express Multiple CCDC78 Isoforms, and the Nonsense CCDC78 Variant Leads to a Mutant Allele That Is Mostly Degraded by NMD
3.3. The Dysfunction of CCDC78 Leads to an Upregulation of Genes Involved in the SR
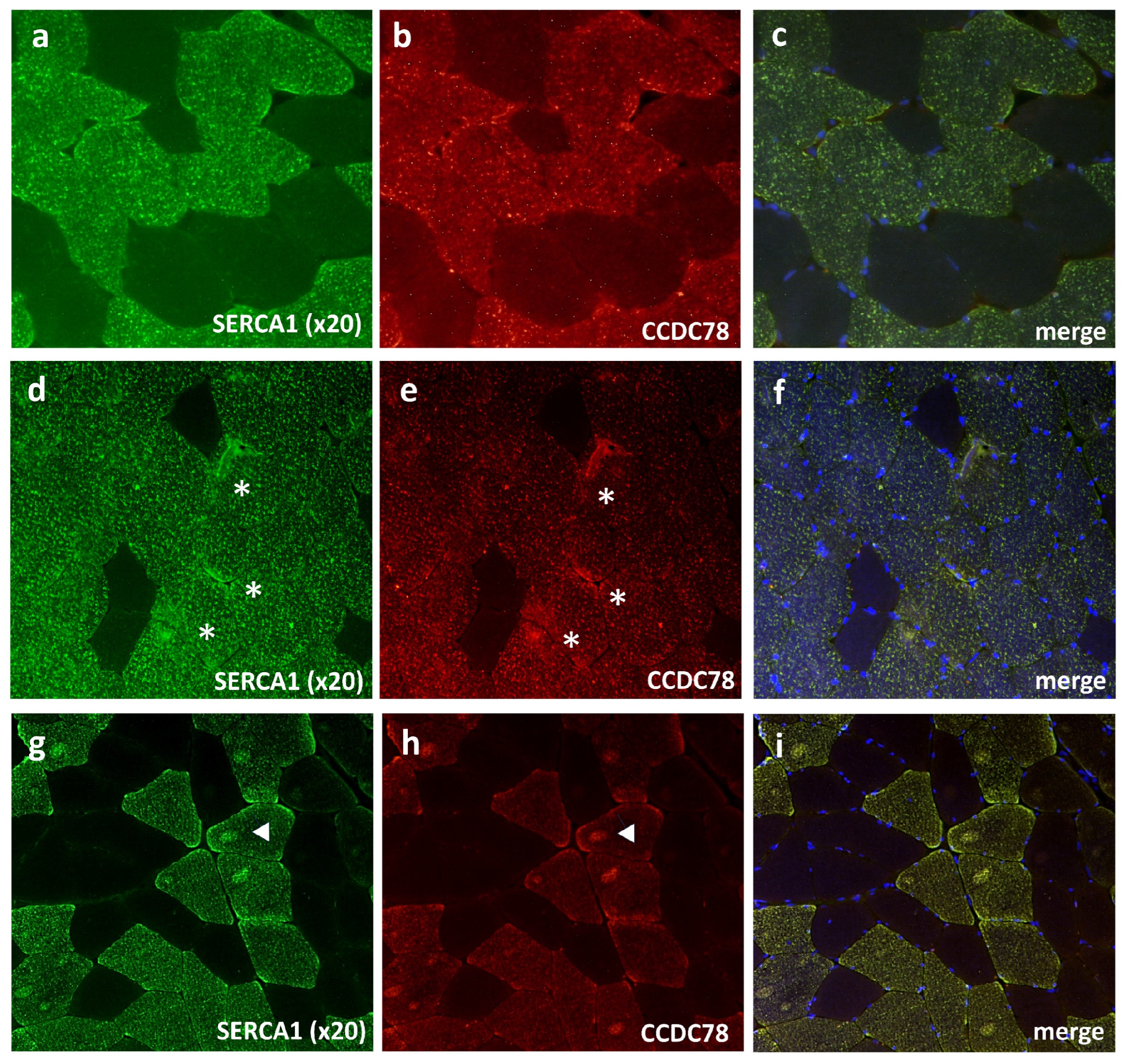
3.4. CCDC78 Interacts with Two Important SR Proteins: SERCA1 and CASQ1
3.5. CCDC78 Interacts with Other Proteins outside the SR: MYH1, ACTN2, and ACTA1
3.6. CCDC78 Does Not Directly Interact with RyR1
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cassandrini, D.; Trovato, R.; Rubegni, A.; Lenzi, S.; Fiorillo, C.; Baldacci, J.; Minetti, C.; Astrea, G.; Bruno, C.; Santorelli, F.M.; et al. Congenital Myopathies: Clinical Phenotypes and New Diagnostic Tools. Ital. J. Pediatr. 2017, 43, 1–16. [Google Scholar] [CrossRef] [PubMed]
- North, K.N.; Wang, C.H.; Clarke, N.; Jungbluth, H.; Vainzof, M.; Dowling, J.J.; Amburgey, K.; Quijano-Roy, S.; Beggs, A.H.; Sewry, C.; et al. Approach to the Diagnosis of Congenital Myopathies. Neuromuscul. Disord. 2014, 24, 97–116. [Google Scholar] [CrossRef]
- Papadimas, G.K.; Xirou, S.; Kararizou, E.; Papadopoulos, C. Update on Congenital Myopathies in Adulthood. Int. J. Mol. Sci. 2020, 21, 3694. [Google Scholar] [CrossRef]
- Volk, A.E.; Kubisch, C. The Rapid Evolution of Molecular Genetic Diagnostics in Neuromuscular Diseases. Curr. Opin. Neurol. 2017, 30, 523–528. [Google Scholar] [CrossRef]
- Boerkoel, C.F.; Takashima, H.; Garcia, C.A.; Olney, R.K.; Johnson, J.; Berry, K.; Russo, P.; Kennedy, S.; Teebi, A.S.; Scavina, M.; et al. Charcot-Marie-Tooth Disease and Related Neuropathies: Mutation Distribution and Genotype-Phenotype Correlation. Ann. Neurol. 2002, 51, 190–201. [Google Scholar] [CrossRef]
- Montenegro, R.M.; Costa-Riquetto, A.D.; Fernandes, V.O.; Montenegro, A.P.D.R.; de Santana, L.S.; de Jorge, A.A.L.; de Karbage, L.B.A.S.; Aguiar, L.B.; Carvalho, F.H.C.; Teles, M.G.; et al. Homozygous and Heterozygous Nuclear Lamin a p. R582C Mutation: Different Lipodystrophic Phenotypes in the Same Kindred. Front. Endocrinol. 2018, 9, 458. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.Z.; Bennett, C.L.; Huynh, H.M.; Blair, I.P.; Puls, I.; Irobi, J.; Dierick, I.; Abel, A.; Kennerson, M.L.; Rabin, B.A.; et al. DNA/RNA Helicase Gene Mutations in a Form of Juvenile Amyotrophic Lateral Sclerosis (ALS4). Am. J. Hum. Genet. 2004, 74, 458. [Google Scholar] [CrossRef] [PubMed]
- Münch, C.; Sedlmeier, R.; Meyer, T.; Homberg, V.; Sperfeld, A.D.; Kurt, A.; Prudlo, J.; Peraus, G.; Hanemann, C.O.; Stumm, G.; et al. Point Mutations of the P150 Subunit of Dynactin (DCTN1) Gene ALS. Neurology 2004, 63, 724–726. [Google Scholar] [CrossRef]
- Al-Saif, A.; Al-Mohanna, F.; Bohlega, S. A Mutation in Sigma-1 Receptor Causes Juvenile Amyotrophic Lateral Sclerosis. Ann. Neurol. 2011, 70, 913–919. [Google Scholar] [CrossRef]
- Salter, C.G.; Beijer, D.; Hardy, H.; Barwick, K.E.S.; Bower, M.; Mademan, I.; De Jonghe, P.; Deconinck, T.; Russell, M.A.; McEntagart, M.M.; et al. Truncating SLC5A7 Mutations Underlie a Spectrum of Dominant Hereditary Motor Neuropathies. Neurol. Genet. 2018, 4, e222. [Google Scholar] [CrossRef]
- Daniels, R.J.; Peden, J.F.; Lloyd, C.; Horsley, S.W.; Clark, K.; Tufarelli, C.; Kearney, L.; Buckle, V.J.; Doggett, N.A.; Flint, J.; et al. Sequence, Structure and Pathology of the Fully Annotated Terminal 2 Mb of the Short Arm of Human Chromosome 16. Hum. Mol. Genet. 2001, 10, 339–352. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.K.; Brotslaw, E.; Thome, V.; Mitchell, J.; Ventrella, R.; Collins, C.; Mitchell, B. A Role for Cep70 in Centriole Amplification in Multiciliated Cells. Dev. Biol. 2021, 471, 10–17. [Google Scholar] [CrossRef]
- Tang, T.K. Centriole Biogenesis in Multiciliated Cells. Nat. Cell Biol. 2013, 15, 1400–1402. [Google Scholar] [CrossRef]
- Majczenko, K.; Davidson, A.E.; Camelo-Piragua, S.; Agrawal, P.B.; Manfready, R.A.; Li, X.; Joshi, S.; Xu, J.; Peng, W.; Beggs, A.H.; et al. Dominant Mutation of CCDC78 in a Unique Congenital Myopathy with Prominent Internal Nuclei and Atypical Cores. Am. J. Hum. Genet. 2012, 91, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Tomelleri, G.; Palmucci, L.; Tonin, P.; Mongini, T.; Marini, M.; L’Erario, R.; Rizzuto, N.; Vattemi, G. SERCA1 and Calsequestrin Storage Myopathy: A New Surplus Protein Myopathy. Brain 2006, 129, 2085–2092. [Google Scholar] [CrossRef]
- Rossi, D.; Vezzani, B.; Galli, L.; Paolini, C.; Toniolo, L.; Pierantozzi, E.; Spinozzi, S.; Barone, V.; Pegoraro, E.; Bello, L.; et al. A Mutation in the CASQ1 Gene Causes a Vacuolar Myopathy with Accumulation of Sarcoplasmic Reticulum Protein Aggregates. Hum. Mutat. 2014, 35, 1163–1170. [Google Scholar] [CrossRef] [PubMed]
- Genovese, I.; Giamogante, F.; Barazzuol, L.; Battista, T.; Fiorillo, A.; Vicario, M.; D’Alessandro, G.; Cipriani, R.; Limatola, C.; Rossi, D.; et al. Sorcin Is an Early Marker of Neurodegeneration, Ca2+ Dysregulation and Endoplasmic Reticulum Stress Associated to Neurodegenerative Diseases. Cell Death Dis. 2020, 11, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Vorobjev, I.A.; Uzbekov, R.E.; Komarova, Y.A.; Alieva, I.B. γ-Tubulin Distribution in Interphase and Mitotic Cells upon Stabilization and Depolymerization of Microtubules. Membr. Cell Biol. 2000, 14, 219–235. [Google Scholar]
- Abràmoff, M.D.; Magalhães, P.J.; Ram, S.J. Image Processing with ImageJ. Biophotonics Int. 2004, 11, 36–42. [Google Scholar]
- Bøyum, A. [9] Separation of Lymphocytes, Granulocytes, and Monocytes from Human Blood Using Iodinated Density Gradient Media. Methods Enzymol. 1984, 108, 88–102. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and Quantifying Mammalian Transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential Expression Analysis for Sequence Count Data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; Van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript Assembly and Quantification by RNA-Seq Reveals Unannotated Transcripts and Isoform Switching during Cell Differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- des Georges, A.; Clarke, O.B.; Zalk, R.; Yuan, Q.; Condon, K.J.; Grassucci, R.A.; Hendrickson, W.A.; Marks, A.R.; Frank, J. Structural Basis for Gating and Activation of RyR1. Cell 2016, 167, 145–157.e17. [Google Scholar] [CrossRef] [PubMed]
- Rappsilber, J.; Mann, M.; Ishihama, Y. Protocol for Micro-Purification, Enrichment, Pre-Fractionation and Storage of Peptides for Proteomics Using StageTips. Nat. Protoc. 2007, 2, 1896–1906. [Google Scholar] [CrossRef]
- Dani, F.R.; Pieraccini, G. Proteomics of Arthropod Soluble Olfactory Proteins. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 2020; Volume 642. [Google Scholar]
- Cserzo, M.; Wallin, E.; Simon, I.; von Heijne, G.; Elofsson, A. Prediction of Transmembrane Alpha-Helices in Prokaryotic Membrane Proteins: The Dense Alignment Surface Method. Protein Eng. Des. Sel. 1997, 10, 673–676. [Google Scholar] [CrossRef]
- Venselaar, H.; te Beek, T.A.; Kuipers, R.K.; Hekkelman, M.L.; Vriend, G. Protein Structure Analysis of Mutations Causing Inheritable Diseases. An e-Science Approach with Life Scientist Friendly Interfaces. BMC Bioinform. 2010, 11, 548. [Google Scholar] [CrossRef]
- Gonorazky, H.D.; Naumenko, S.; Ramani, A.K.; Nelakuditi, V.; Mashouri, P.; Wang, P.; Kao, D.; Ohri, K.; Viththiyapaskaran, S.; Tarnopolsky, M.A.; et al. Expanding the Boundaries of RNA Sequencing as a Diagnostic Tool for Rare Mendelian Disease. Am. J. Hum. Genet. 2019, 104, 466–483. [Google Scholar] [CrossRef]
- Vasli, N.; Harris, E.; Karamchandani, J.; Bareke, E.; Majewski, J.; Romero, N.B.; Stojkovic, T.; Barresi, R.; Tasfaout, H.; Charlton, R.; et al. Recessive Mutations in the Kinase ZAK Cause a Congenital Myopathy with Fibre Type Disproportion. Brain 2017, 140, 466–483. [Google Scholar] [CrossRef]
- Rossi, D.; Pierantozzi, E.; Amadsun, D.O.; Buonocore, S.; Rubino, E.M.; Sorrentino, V. The Sarcoplasmic Reticulum of Skeletal Muscle Cells: A Labyrinth of Membrane Contact Sites. Biomolecules 2022, 12, 488. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kasai, M. Regulation of Calcium Channel in Sarcoplasmic Reticulum by Calsequestrin. Biochem. Biophys. Res. Commun. 1994, 199, 1120–1127. [Google Scholar] [CrossRef]
- Dulhunty, A.F.; Beard, N.A.; Casarotto, M.G. Recent Advances in Understanding the Ryanodine Receptor Calcium Release Channels and Their Role in Calcium Signalling [Version 1; Peer Review: 4 Approved]. F1000Research 2018, 7, F1000. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Bai, X.C.; Yan, C.; Wu, J.; Li, Z.; Xie, T.; Peng, W.; Yin, C.C.; Li, X.; Scheres, S.H.W.; et al. Structure of the Rabbit Ryanodine Receptor RyR1 at Near-Atomic Resolution. Nature 2015, 517, 50–55. [Google Scholar] [CrossRef]
- Lamboley, C.R.; Murphy, R.M.; Mckenna, M.J.; Lamb, G.D. Endogenous and Maximal Sarcoplasmic Reticulum Calcium Content and Calsequestrin Expression in Type I and Type II Human Skeletal Muscle Fibres. J. Physiol. 2013, 591, 6053–6068. [Google Scholar] [CrossRef]
- Lamboley, C.R.; Murphy, R.M.; Mckenna, M.J.; Lamb, G.D. Sarcoplasmic Reticulum Ca2+ Uptake and Leak Properties, and SERCA Isoform Expression, in Type I and Type II Fibres of Human Skeletal Muscle. J. Physiol. 2014, 592, 1381–1395. [Google Scholar] [CrossRef]
- Perni, S.; Beam, K.G. Voltage-Induced Ca2+ Release Is Supported by Junctophilins 1, 2 and 3, and Not by Junctophilin 4. J. Gen. Physiol. 2022, 154, e2021ecc22. [Google Scholar] [CrossRef]
- Rossi, D.; Scarcella, A.M.; Liguori, E.; Lorenzini, S.; Pierantozzi, E.; Kutchukian, C.; Jacquemond, V.; Messa, M.; De Camilli, P.; Sorrentino, V. Molecular Determinants of Homo- And Heteromeric Interactions of Junctophilin-1 at Triads in Adult Skeletal Muscle Fibers. Proc. Natl. Acad. Sci. USA 2019, 116, 15716–15724. [Google Scholar] [CrossRef] [PubMed]
- Srikanth, S.; Jew, M.; Kim, K.-D.; Yee, M.-K.; Abramson, J.; Gwack, Y. Junctate Is a Ca2+-Sensing Structural Component of Orai1 and Stromal Interaction Molecule 1 (STIM1). Proc. Natl. Acad. Sci. USA 2012, 109, 8682–8687. [Google Scholar] [CrossRef]
- Srikanth, S.; Jung, H.J.; Do Kim, K.; Souda, P.; Whitelegge, J.; Gwack, Y. A Novel EF-Hand Protein, CRACR2A, Is a Cytosolic Ca2+ Sensor That Stabilizes CRAC Channels in T Cells. Nat. Cell Biol. 2010, 12, 436–446. [Google Scholar] [CrossRef]
- Shen, X.; Franzini-Armstrong, C.; Lopez, J.R.; Jones, L.R.; Kobayashi, Y.M.; Wang, Y.; Kerrick, W.G.L.; Caswell, A.H.; Potter, J.D.; Miller, T.; et al. Triadins Modulate Intracellular Ca2+ Homeostasis but Are Not Essential for Excitation-Contraction Coupling in Skeletal Muscle. J. Biol. Chem. 2007, 282, 37864–37874. [Google Scholar] [CrossRef]
- Zhang, L.; Salgado-Somoza, A.; Vausort, M.; Leszek, P.; Devaux, Y. A Heart-Enriched Antisense Long Non-Coding RNA Regulates the Balance between Cardiac and Skeletal Muscle Triadin. Biochim. Biophys. Acta—Mol. Cell Res. 2018, 1865, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.; Lorenzini, S.; Pierantozzi, E.; Van Petegem, F.; Amadsun, D.O.; Sorrentino, V. Multiple Regions within Junctin Drive Its Interaction with Calsequestrin-1 and Its Localization to Triads in Skeletal Muscle. J. Cell Sci. 2022, 135, jcs259185. [Google Scholar] [CrossRef] [PubMed]
- Laing, N.G.; Dye, D.E.; Wallgren-Pettersson, C.; Richard, G.; Monnier, N.; Lillis, S.; Winder, T.L.; Lochmüller, H.; Graziano, C.; Mitrani-Rosenbaum, S.; et al. Mutations and Polymorphisms of the Skeletal Muscle α-Actin Gene (ACTA1). Hum. Mutat. 2009, 30, 1267–1277. [Google Scholar] [CrossRef] [PubMed]
- Sparrow, J.C.; Nowak, K.J.; Durling, H.J.; Beggs, A.H.; Wallgren-Pettersson, C.; Romero, N.; Nonaka, I.; Laing, N.G. Muscle Disease Caused by Mutations in the Skeletal Muscle Alpha-Actin Gene (ACTA1). Neuromuscul. Disord. 2003, 13, 519–531. [Google Scholar] [CrossRef]
- Beard, N.A.; Laver, D.R.; Dulhunty, A.F. Calsequestrin and the Calcium Release Channel of Skeletal and Cardiac Muscle. Prog. Biophys. Mol. Biol. 2004, 85, 33–69. [Google Scholar] [CrossRef]
- Gautel, M.; Djinović-Carugo, K. The Sarcomeric Cytoskeleton: From Molecules to Motion. J. Exp. Biol. 2016, 219, 135–145. [Google Scholar] [CrossRef]
- Wang, C.; Yue, F.; Kuang, S. Muscle Histology Characterization Using H&E Staining and Muscle Fiber Type Classification Using Immunofluorescence Staining. Bio-Protocol 2017, 7, e2279. [Google Scholar] [CrossRef]
- Valberg, S.J.; Henry, M.L.; Perumbakkam, S.; Gardner, K.L.; Finno, C.J. An E321G MYH1 Mutation Is Strongly Associated with Nonexertional Rhabdomyolysis in Quarter Horses. J. Vet. Intern. Med. 2018, 32, 1718–1725. [Google Scholar] [CrossRef]
- Alsaif, H.S.; Alshehri, A.; Sulaiman, R.A.; Al-Hindi, H.; Guzmán-Vega, F.J.; Arold, S.T.; Alkuraya, F.S. MYH1 Is a Candidate Gene for Recurrent Rhabdomyolysis in Humans. Am. J. Med. Genet. A 2021, 185, 2131–2135. [Google Scholar] [CrossRef]
- Eckhardt, J.; Ruiz, A.; Koenig, S.; Frieden, M.; Meier, H.; Schmidt, A.; Treves, S.; Zorzato, F. Quantitative Proteomic Analysis of Skeletal Muscles from Wild-Type and Transgenic Mice Carrying Recessive Ryr1 Mutations Linked to Congenital Myopathies. eLife 2023, 12, e83618. [Google Scholar] [CrossRef]
- Wang, X.; Kong, C.; Liu, P.; Geng, W.; Tang, H. Identification of Potential Biomarkers for Ryanodine Receptor 1 (RYR1) Mutation-Associated Myopathies Using Bioinformatics Approach. Dis. Markers 2022, 2022, 8787782. [Google Scholar] [CrossRef]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative Isoform Regulation in Human Tissue Transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Boulling, A.; Le Maréchal, C.; Trouvé, P.; Raguénès, O.; Chen, J.M.; Férec, C. Functional Analysis of Pancreatitis-Associated Missense Mutations in the Pancreatic Secretory Trypsin Inhibitor (SPINK1) Gene. Eur. J. Hum. Genet. 2007, 15, 936–942. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Miteva, M.A.; Wang, L.; Alexov, E. Analyzing Effects of Naturally Occurring Missense Mutations. Comput. Math. Methods Med. 2012, 2012, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Klos Dehring, D.A.; Vladar, E.K.; Werner, M.E.; Mitchell, J.W.; Hwang, P.; Mitchell, B.J. Deuterosome-Mediated Centriole Biogenesis. Dev. Cell 2013, 27, 103–112. [Google Scholar] [CrossRef]
- Ng, D.C.H.; Ho, U.Y.; Grounds, M.D. Cilia, Centrosomes and Skeletal Muscle. Int. J. Mol. Sci. 2021, 22, 9605. [Google Scholar] [CrossRef]
- Palla, A.R.; Hilgendorf, K.I.; Yang, A.V.; Kerr, J.P.; Hinken, A.C.; Demeter, J.; Kraft, P.; Mooney, N.A.; Yucel, N.; Burns, D.M.; et al. Primary Cilia on Muscle Stem Cells Are Critical to Maintain Regenerative Capacity and Are Lost during Aging. Nat. Commun. 2022, 13, 1439. [Google Scholar] [CrossRef]
- Marg, A.; Haase, H.; Neumann, T.; Kouno, M.; Morano, I. AHNAK1 and AHNAK2 Are Costameric Proteins: AHNAK1 Affects Transverse Skeletal Muscle Fiber Stiffness. Biochem. Biophys. Res. Commun. 2010, 401, 143–148. [Google Scholar] [CrossRef]
- Liu, S.; Li, S.; Zhu, H.; Cheng, S.; Zheng, Q.Y. A Mutation in the Cdh23 Gene Causes Age-Related Hearing Loss in Cdh23nmf308/Nmf308 Mice. Gene 2012, 499, 309–317. [Google Scholar] [CrossRef]
- Dafinger, C.; Liebau, M.C.; Elsayed, S.M.; Hellenbroich, Y.; Boltshauser, E.; Korenke, G.C.; Fabretti, F.; Janecke, A.R.; Ebermann, I.; Nürnberg, G.; et al. Mutations in KIF7 Link Joubert Syndrome with Sonic Hedgehog Signaling and Microtubule Dynamics. J. Clin. Investig. 2011, 121, 2662–2667. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lopergolo, D.; Gallus, G.N.; Pieraccini, G.; Boscaro, F.; Berti, G.; Serni, G.; Volpi, N.; Formichi, P.; Bianchi, S.; Cassandrini, D.; et al. CCDC78: Unveiling the Function of a Novel Gene Associated with Hereditary Myopathy. Cells 2024, 13, 1504. https://doi.org/10.3390/cells13171504
Lopergolo D, Gallus GN, Pieraccini G, Boscaro F, Berti G, Serni G, Volpi N, Formichi P, Bianchi S, Cassandrini D, et al. CCDC78: Unveiling the Function of a Novel Gene Associated with Hereditary Myopathy. Cells. 2024; 13(17):1504. https://doi.org/10.3390/cells13171504
Chicago/Turabian StyleLopergolo, Diego, Gian Nicola Gallus, Giuseppe Pieraccini, Francesca Boscaro, Gianna Berti, Giovanni Serni, Nila Volpi, Patrizia Formichi, Silvia Bianchi, Denise Cassandrini, and et al. 2024. "CCDC78: Unveiling the Function of a Novel Gene Associated with Hereditary Myopathy" Cells 13, no. 17: 1504. https://doi.org/10.3390/cells13171504