
Postencephalitic Parkinsonism: Unique Pathological and Clinical Features—Preliminary Data


 ,
,
Abstract
:1. Introduction
1.1. Although Similar to Idiopathic PD, PEP Shows Significant Differences
1.2. Iron and Neurodegeneration
2. Materials and Methods
Sampling and Processing
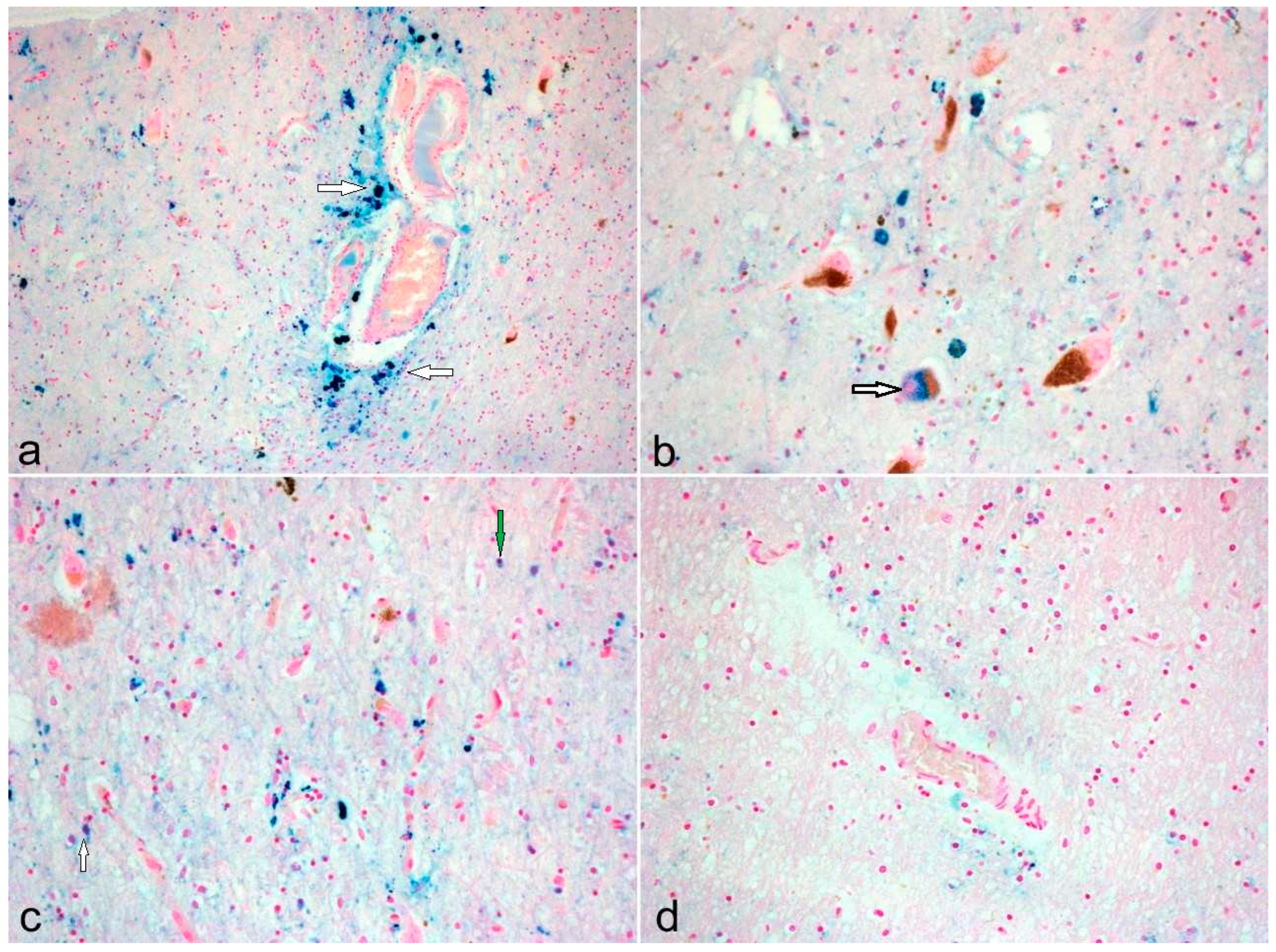
3. Results
Neuropathological Diversity in PD and PEP
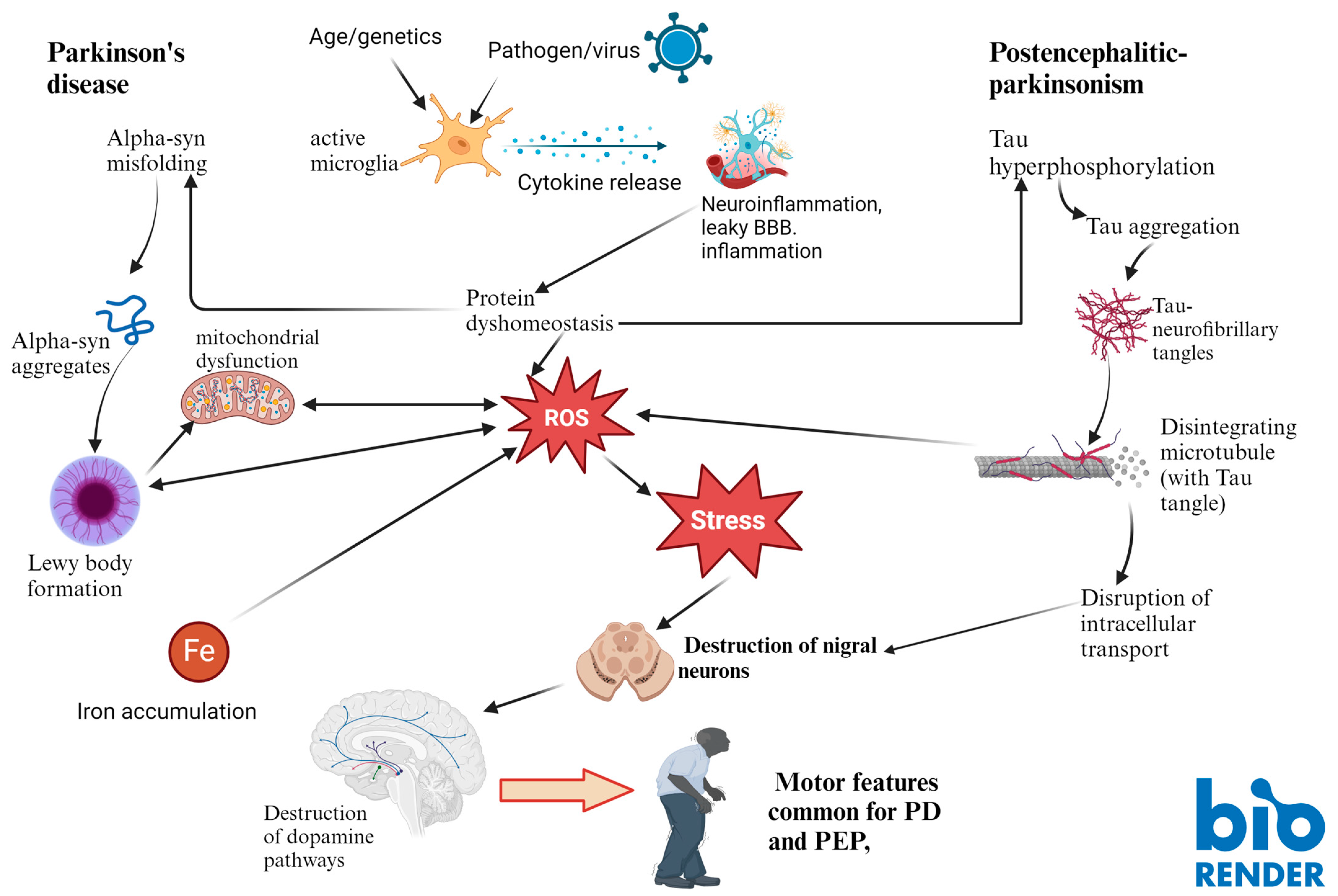
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wüllner, U.; Borghammer, P.; Choe, C.U.; Csoti, I.; Falkenburger, B.; Gasser, T.; Lingor, P.; Riederer, P. The heterogeneity of Parkinson’s disease. J. Neural Transm. 2023, 130, 827–838. [Google Scholar] [CrossRef] [PubMed]
- Riederer, P.; Berg, D.; Casadei, N.; Cheng, F.B.; Classen, J.; Dresel, C.; Jost, W.; Krüger, R.; Müller, T.; Reichmann, H.; et al. α-Synuclein in Parkinson’s disease: Causal or bystander? J. Neural Transm. 2019, 126, 815–840. [Google Scholar] [CrossRef]
- Foley, P.B. Encephalitis lethargica and influenza. I. The role of the influenza virus in the influenza pandemic of 1918/1919. J. Neural Transm. 2009, 116, 143–150. [Google Scholar] [CrossRef]
- Jankovic, J.; Tan, E.K. Parkinson’s disease: Etiopathogenesis and treatment. J. Neurol. Neurosurg. Psychiatry 2020, 91, 795–808. [Google Scholar] [CrossRef] [PubMed]
- Sian-Hulsmann, J.; Riederer, P. Virus-induced brain pathology and the neuroinflammation-inflammation continuum: The neurochemists view. J. Neural Transm. 2024, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Reid, A.H.; Fanning, T.G.; Hultin, J.V.; Taubenberger, J.K. Origin and evolution of the 1918 “Spanish” influenza virus hemagglutinin gene. Proc. Natl. Acad. Sci. USA 1999, 96, 1651–1656. [Google Scholar] [CrossRef]
- Reid, A.H.; Fanning, T.G.; Janczewski, T.A.; Taubenberger, J.K. Characterization of the 1918 “Spanish” influenza virus neuraminidase gene. Proc. Natl. Acad. Sci. USA 2000, 97, 6785–6790. [Google Scholar] [CrossRef] [PubMed]
- Cadar, D.; Jellinger, K.A.; Riederer, P.; Strobel, S.; Monoranu, C.M.; Tappe, D. No Metagenomic Evidence of Causative Viral Pathogens in Postencephalitic Parkinsonism Following Encephalitis Lethargica. Microorganisms 2021, 9, 1716. [Google Scholar] [CrossRef]
- Leta, V.; Urso, D.; Batzu, L.; Lau, Y.H.; Mathew, D.; Boura, I.; Raeder, V.; Falup-Pecurariu, C.; van Wamelen, D.; Chaudhuri, K.R. Viruses, parkinsonism and Parkinson’s disease: The past, present and future. J. Neural Transm. 2022, 129, 1119–1132. [Google Scholar] [CrossRef]
- Casals, J.; Elizan, T.S.; Yahr, M.D. Postencephalitic parkinsonism—A review. J. Neural Transm. 1998, 105, 645–676. [Google Scholar] [CrossRef]
- Donaldson, I.; Marsden, C.D.; Schneider, S.A.; Bhatia, K.P.; Donaldson, I.; Marsden, C.D.; Schneider, S.; Bhatia, K. Postencephalic parkinsonism. In Marsden’s Book of Movement Disorders; Oxford University Press: Oxford, UK, 2012; pp. 125–133. [Google Scholar]
- Buee-Scherrer, V.; Buee, L.; Leveugle, B.; Perl, D.P.; Vermersch, P.; Hof, P.R.; Delacourte, A. Pathological tau proteins in postencephalitic parkinsonism: Comparison with Alzheimer’s disease and other neurodegenerative disorders. Ann. Neurol. 1997, 42, 356–359. [Google Scholar] [CrossRef] [PubMed]
- Caparros-Lefebvre, D.; Cabaret, M.; Godefroy, O.; Steinling, M.; Remy, P.; Samson, Y.; Petit, H. PET study and neuropsychological assessment of a long-lasting post-encephalitic parkinsonism. J. Neural Transm. 1998, 105, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.T.; Allen, I.V.; McQuaid, S.; McConnell, R. An immunohistochemical study of neurofibrillary tangle formation in post-encephalitic Parkinsonism. Clin. Neuropathol. 1996, 15, 22–25. [Google Scholar] [PubMed]
- Wenning, G.K.; Jellinger, K.; Litvan, I. Snpranuclear gaze palsy and eyelid apraxia in postencephalitic parkinsonism. J. Neural Transm. 1997, 104, 845–865. [Google Scholar] [CrossRef]
- Jankovic, J.; Truong, D.D.; Bologna, M. Editorial “Parkinsonism across the spectrum of movement disorders and beyond”. J. Neurol. Sci. 2022, 433, 120013. [Google Scholar] [CrossRef]
- Jellinger, K.A. Neuropathology and pathogenesis of extrapyramidal movement disorders: A critical update-I. Hypokinetic-rigid movement disorders. J. Neural Transm. 2019, 126, 933–995. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A. Absence of alpha-synuclein pathology in postencephalitic parkinsonism. Acta Neuropathol. 2009, 118, 371–379. [Google Scholar] [CrossRef]
- Jang, H.; Boltz, D.A.; Webster, R.G.; Smeyne, R.J. Viral parkinsonism. Biochim. Biophys. Acta 2009, 1792, 714–721. [Google Scholar] [CrossRef]
- Geddes, J.F.; Hughes, A.J.; Lees, A.J.; Daniel, S.E. Pathological overlap in cases of parkinsonism associated with neurofibrillary tangles. A study of recent cases of postencephalitic parkinsonism and comparison with progressive supranuclear palsy and Guamanian parkinsonism-dementia complex. Brain 1993, 116 Pt 1, 281–302. [Google Scholar] [CrossRef]
- Josephs, K.A.; Parisi, J.E.; Dickson, D.W. Alpha-synuclein studies are negative in postencephalic parkinsonism of von Economo. Neurology 2002, 59, 645–646. [Google Scholar] [CrossRef]
- Ishii, T.; Nakamura, Y. Distribution and ultrastructure of Alzheimer’s neurofibrillary tangles in postencephalitic Parkinsonism of Economo type. Acta Neuropathol. 1981, 55, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Akiyama, H.; Kondo, H.; Ikeda, K. Anti-tau-positive glial fibrillary tangles in the brain of postencephalitic parkinsonism of Economo type. Neurosci. Lett. 1993, 162, 176–178. [Google Scholar] [CrossRef] [PubMed]
- Mizukami, K.; Sasaki, M.; Shiraishi, H.; Ikeda, K.; Kosaka, K. A neuropathologic study of long-term, Economo-type postencephalitic parkinsonism with a prolonged clinical course. Psychiatry Clin. Neurosci. 1996, 50, 79–83. [Google Scholar] [CrossRef]
- Hoffman, L.A.; Vilensky, J.A. Encephalitis lethargica: 100 years after the epidemic. Brain 2017, 140, 2246–2251. [Google Scholar] [CrossRef]
- Hof, P.R.; Charpiot, A.; Delacourte, A.; Buee, L.; Purohit, D.; Perl, D.P.; Bouras, C. Distribution of neurofibrillary tangles and senile plaques in the cerebral cortex in postencephalitic parkinsonism. Neurosci. Lett. 1992, 139, 10–14. [Google Scholar] [CrossRef]
- Riederer, P.; Monoranu, C.; Strobel, S.; Iordache, T.; Sian-Hulsmann, J. Iron as the concert master in the pathogenic orchestra playing in sporadic Parkinson’s disease. J. Neural Transm. 2021, 128, 1577–1598. [Google Scholar] [CrossRef]
- Lieu, P.T.; Heiskala, M.; Peterson, P.A.; Yang, Y. The roles of iron in health and disease. Mol. Asp. Med. 2001, 22, 1–87. [Google Scholar] [CrossRef]
- Ward, D.M.; Kaplan, J. Ferroportin-mediated iron transport: Expression and regulation. BBA-Mol. Cell Res. 2012, 1823, 1426–1433. [Google Scholar] [CrossRef] [PubMed]
- Ganz, T.; Nemeth, E. Iron homeostasis in host defence and inflammation. Nat. Rev. Immunol. 2015, 15, 500–510. [Google Scholar] [CrossRef]
- Ganz, T. Hepcidin and iron regulation, 10 years later. Blood 2011, 117, 4425–4433. [Google Scholar] [CrossRef]
- Youdim, M.B.H.; Green, A.R. Iron-Deficiency and Neurotransmitter Synthesis and Function. Proc. Nutr. Soc. 1978, 37, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Long, B.; Koyfman, A. Emergency Medicine Evaluation and Management of Anemia. Emerg. Med. Clin. N. Am. 2018, 36, 609–630. [Google Scholar] [CrossRef] [PubMed]
- Kowdley, K.V. Iron Overload in Patients with Chronic Liver Disease. Gastroenterol. Hepatol. 2016, 12, 695–698. [Google Scholar]
- Yao, F.P.; Cui, X.H.; Zhang, Y.; Bei, Z.C.; Wang, H.Q.; Zhao, D.X.; Wang, H.; Yang, Y.F. Iron regulatory protein 1 promotes ferroptosis by sustaining cellular iron homeostasis in melanoma. Oncol. Lett. 2021, 22, 657. [Google Scholar] [CrossRef] [PubMed]
- Sousa, L.; Oliveira, M.M.; Pessôa, M.T.C.; Barbosa, L.A. Iron overload: Effects on cellular biochemistry. Clin. Chim. Acta 2020, 504, 180–189. [Google Scholar] [CrossRef]
- Ben-Shachar, D.; Youdim, M.B. Selectivity of melaninized nigra-striatal dopamine neurons to degeneration in Parkinson’s disease may depend on iron-melanin interaction. J. Neural Transm. Suppl. 1990, 29, 251–258. [Google Scholar] [CrossRef]
- Jellinger, K.; Kienzl, E.; Rumpelmair, G.; Riederer, P.; Stachelberger, H.; Ben-Shachar, D.; Youdim, M.B. Iron-melanin complex in substantia nigra of parkinsonian brains: An X-ray microanalysis. J. Neurochem. 1992, 59, 1168–1171. [Google Scholar] [CrossRef] [PubMed]
- Sofic, E.; Riederer, P.; Heinsen, H.; Beckmann, H.; Reynolds, G.P.; Hebenstreit, G.; Youdim, M.B.H. Increased Iron (III) and Total Iron Content in Post-Mortem Substantia Nigra of Parkinsonian Brain. J. Neural Transm. 1988, 74, 199–205. [Google Scholar] [CrossRef]
- Dexter, D.T.; Carayon, A.; Javoyagid, F.; Agid, Y.; Wells, F.R.; Daniel, S.E.; Lees, A.J.; Jenner, P.; Marsden, C.D. Alterations in the Levels of Iron, Ferritin and Other Trace-Metals in Parkinsons-Disease and Other Neurodegenerative Diseases Affecting the Basal Ganglia. Brain 1991, 114, 1953–1975. [Google Scholar] [CrossRef]
- Connor, J.R.; Snyder, B.S.; Beard, J.L.; Fine, R.E.; Mufson, E.J. Regional Distribution of Iron and Iron-Regulatory Proteins in the Brain in Aging and Alzheimers-Disease. J. Neurosci. Res. 1992, 31, 327–335. [Google Scholar] [CrossRef]
- De Lury, A.D.; Bisulca, J.A.; Lee, J.S.; Altaf, M.D.; Coyle, P.K.; Duong, T.Q. Magnetic resonance imaging detection of deep gray matter iron deposition in multiple sclerosis: A systematic review. J. Neurol. Sci. 2023, 453, 120816. [Google Scholar] [CrossRef] [PubMed]
- Dusek, P.; Hofer, T.; Alexander, J.; Roos, P.M.; Aaseth, J.O. Cerebral Iron Deposition in Neurodegeneration. Biomolecules 2022, 12, 714. [Google Scholar] [CrossRef] [PubMed]
- Hogarth, P. Neurodegeneration with brain iron accumulation: Diagnosis and management. J. Mov. Disord. 2015, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Levi, S.; Rovida, E. Neuroferritinopathy: From ferritin structure modification to pathogenetic mechanism. Neurobiol. Dis. 2015, 81, 134–143. [Google Scholar] [CrossRef]
- Ward, R.; Zucca, F.A.; Duyn, J.H.; Crichton, R.R.; Zecca, L. The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. 2014, 13, 1045–1060. [Google Scholar] [CrossRef]
- Lee, J.H.; Lee, M.S. Brain Iron Accumulation in Atypical Parkinsonian Syndromes: In Vivo MRI Evidences for Distinctive Patterns. Front. Neurol. 2019, 10, 74. [Google Scholar] [CrossRef]
- Pfefferbaum, A.; Adalsteinsson, E.; Rohlfing, T.; Sullivan, E.V. MRI estimates of brain iron concentration in normal aging: Comparison of field-dependent (FDRI) and phase (SWI) methods. Neuroimage 2009, 47, 493–500. [Google Scholar] [CrossRef]
- Bilgic, B.; Pfefferbaum, A.; Rohlfing, T.; Sullivan, E.V.; Adalsteinsson, E. MRI estimates of brain iron concentration in normal aging using quantitative susceptibility mapping. Neuroimage 2012, 59, 2625–2635. [Google Scholar] [CrossRef]
- Lee, S.; Kovacs, G.G. The Irony of Iron: The Element with Diverse Influence on Neurodegenerative Diseases. Int. J. Mol. Sci. 2024, 25, 4269. [Google Scholar] [CrossRef]
- Ficiara, E.; Stura, I.; Guiot, C. Iron Deposition in Brain: Does Aging Matter? Int. J. Mol. Sci. 2022, 23, 18. [Google Scholar] [CrossRef]
- Zeng, W.Q.; Cai, J.; Zhang, L.; Peng, Q.W. Iron Deposition in Parkinson’s Disease: A Mini-Review. Cell. Mol. Neurobiol. 2024, 44, 26. [Google Scholar] [CrossRef] [PubMed]
- Martin-Bastida, A.; Lao-Kaim, N.P.; Loane, C.; Politis, M.; Roussakis, A.A.; Valle-Guzman, N.; Kefalopoulou, Z.; Paul-Visse, G.; Widner, H.; Xing, Y.; et al. Motor associations of iron accumulation in deep grey matter nuclei in Parkinson’s disease: A cross-sectional study of iron-related magnetic resonance imaging susceptibility. Eur. J. Neurol. 2017, 24, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Mittal, S.O.; Hassan, A.; Sanchez, J.; Robertson, C. Powassan virus causing Post-encephalitic Parkinsonism. Neurology 2017, 88, P1-078. [Google Scholar] [CrossRef]
- Schneider, J.A.; Li, J.L.; Li, Y.; Wilson, R.S.; Kordower, J.H.; Bennett, D.A. Substantia nigra tangles are related to gait impairment in older persons. Ann. Neurol. 2006, 59, 166–173. [Google Scholar] [CrossRef]
- Foguem, C.; Manckoundia, P. Lewy Body Disease: Clinical and Pathological “Overlap Syndrome” between Synucleinopathies (Parkinson Disease) and Tauopathies (Alzheimer Disease). Curr. Neurol. Neurosci. Rep. 2018, 18, 24. [Google Scholar] [CrossRef]
- Das, S.; Zhang, Z.; Ang, L.C. Clinicopathological overlap of neurodegenerative diseases: A comprehensive review. J. Clin. Neurosci. 2020, 78, 30–33. [Google Scholar] [CrossRef]
- Hoglinger, G.U.; Adler, C.H.; Berg, D.; Klein, C.; Outeiro, T.F.; Poewe, W.; Postuma, R.; Stoessl, A.J.; Lang, A.E. A biological classification of Parkinson’s disease: The SynNeurGe research diagnostic criteria. Lancet Neurol. 2024, 23, 191–204. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.S.; Kågedal, K.; Halliday, G.M. Alpha-synuclein biology in Lewy body diseases. Alzheimer’s Res. Ther. 2014, 6, 73. [Google Scholar] [CrossRef]
- Twohig, D.; Nielsen, H.M. α-synuclein in the pathophysiology of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 23. [Google Scholar] [CrossRef]
- Twohig, D.; Rodriguez-Vieitez, E.; Sando, S.B.; Berge, G.; Lauridsen, C.; Moller, I.; Grontvedt, G.R.; Bråthen, G.; Patra, K.; Bu, G.J.; et al. The relevance of cerebrospinal fluid -synuclein levels to sporadic and familial Alzheimer’s disease. Acta Neuropathol. Commun. 2018, 6, 130. [Google Scholar] [CrossRef]
- Zhang, X.; Gao, F.; Wang, D.D.; Li, C.; Fu, Y.; He, W.; Zhang, J.M. Tau Pathology in Parkinson’s Disease. Front. Neurol. 2018, 9, 809. [Google Scholar] [CrossRef]
- Rawat, P.; Sehar, U.; Bisht, J.; Selman, A.; Culberson, J.; Reddy, P.H. Phosphorylated Tau in Alzheimer’s Disease and Other Tauopathies. Int. J. Mol. Sci. 2022, 23, 12841. [Google Scholar] [CrossRef] [PubMed]
- Bai, R.; Guo, J.; Ye, X.Y.; Xie, Y.; Xie, T. Oxidative stress: The core pathogenesis and mechanism of Alzheimer’s disease. Ageing Res. Rev. 2022, 77, 101619. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, G.T.; Chami, B.; Youssef, P.; Witting, P.K. Oxidative stress in Alzheimer’s disease: Primary villain or physiological by-product? Redox Rep. 2013, 18, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Gibb, W.R.; Lees, A.J. The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 1988, 51, 745–752. [Google Scholar] [CrossRef]
- Ishizawa, T.; Mattila, P.; Davies, P.; Wang, D.; Dickson, D.W. Colocalization of tau and alpha-synuclein epitopes in Lewy bodies. J. Neuropathol. Exp. Neurol. 2003, 62, 389–397. [Google Scholar] [CrossRef]
- Riess, O.; Krüger, R. Parkinson’s disease—A multifactorial neurodegenerative disorder. J. Neural Transm. Suppl. 1999, 56, 113–125. [Google Scholar]
- Castellani, R.J.; Siedlak, S.L.; Perry, G.; Smith, M.A. Sequestration of iron by Lewy bodies in Parkinson’s disease. Acta Neuropathol. 2000, 100, 111–114. [Google Scholar] [CrossRef]
- Double, K.L.; Gerlach, M.; Schunemann, V.; Trautwein, A.X.; Zecca, L.; Gallorini, M.; Youdim, M.B.; Riederer, P.; Ben-Shachar, D. Iron-binding characteristics of neuromelanin of the human substantia nigra. Biochem. Pharmacol. 2003, 66, 489–494. [Google Scholar] [CrossRef]
- Zecca, L.; Youdim, M.B.; Riederer, P.; Connor, J.R.; Crichton, R.R. Iron, brain ageing and neurodegenerative disorders. Nat. Rev. Neurosci. 2004, 5, 863–873. [Google Scholar] [CrossRef]
- Riederer, P.; Nagatsu, T.; Youdim, M.B.H.; Wulf, M.; Dijkstra, J.M.; Sian-Huelsmann, J. Lewy bodies, iron, inflammation and neuromelanin: Pathological aspects underlying Parkinson’s disease. J. Neural Transm. 2023, 130, 627–646. [Google Scholar] [CrossRef] [PubMed]
- Dexter, D.T.; Sian, J.; Rose, S.; Hindmarsh, J.G.; Mann, V.M.; Cooper, J.M.; Wells, F.R.; Daniel, S.E.; Lees, A.J.; Schapira, A.H.V.; et al. Indexes of Oxidative Stress and Mitochondrial-Function in Individuals with Incidental Lewy Body Disease. Ann. Neurol. 1994, 35, 38–44. [Google Scholar] [CrossRef]
- Dexter, D.T.; Wells, F.R.; Lees, A.J.; Agid, F.; Agid, Y.; Jenner, P.; Marsden, C.D. Increased nigral iron content and alterations in other metal ions occurring in brain in Parkinson’s disease. J. Neurochem. 1989, 52, 1830–1836. [Google Scholar] [CrossRef]
- Hirsch, E.C.; Brandel, J.P.; Galle, P.; Javoy-Agid, F.; Agid, Y. Iron and aluminum increase in the substantia nigra of patients with Parkinson’s disease: An X-ray microanalysis. J. Neurochem. 1991, 56, 446–451. [Google Scholar] [CrossRef]
- Ma, L.; Gholam Azad, M.; Dharmasivam, M.; Richardson, V.; Quinn, R.J.; Feng, Y.; Pountney, D.L.; Tonissen, K.F.; Mellick, G.D.; Yanatori, I.; et al. Parkinson’s disease: Alterations in iron and redox biology as a key to unlock therapeutic strategies. Redox Biol. 2021, 41, 101896. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, I.; Reimann, K.; Jankuhn, S.; Kirilina, E.; Stieler, J.; Sonntag, M.; Meijer, J.; Weiskopf, N.; Reinert, T.; Arendt, T.; et al. Cell specific quantitative iron mapping on brain slices by immuno-microPIXE in healthy elderly and Parkinson’s disease. Acta Neuropathol. Commun. 2021, 9, 47. [Google Scholar] [CrossRef]
- Lull, M.E.; Block, M.L. Microglial Activation and Chronic Neurodegeneration. Neurotherapeutics 2010, 7, 354–365. [Google Scholar] [CrossRef]
- Ward, R.J.; Dexter, D.T.; Crichton, R.R. Iron, Neuroinflammation and Neurodegeneration. Int. J. Mol. Sci. 2022, 23, 7267. [Google Scholar] [CrossRef] [PubMed]
- Olmedo-Díaz, S.; Estévez-Silva, H.; Orädd, G.; Af Bjerkén, S.; Marcellino, D.; Virel, A. An Altered Blood-Brain Barrier Contributes to Brain Iron Accumulation and Neuroinflammation in the 6-Ohda Rat Model of Parkinson’s Disease. Neuroscience 2017, 362, 141–151. [Google Scholar] [CrossRef]
- Mcgeer, P.L.; Itagaki, S.; Boyes, B.E.; Mcgeer, E.G. Reactive Microglia Are Positive for Hla-Dr in the Substantia Nigra of Parkinsons and Alzheimers-Disease Brains. Neurology 1988, 38, 1285–1291. [Google Scholar] [CrossRef]
- McGeer, P.L.; McGeer, E.G. Glial reactions in Parkinson’s disease. Mov. Disord. 2008, 23, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Dias, V.; Junn, E.; Mouradian, M.M. The Role of Oxidative Stress in Parkinson’s Disease. J. Park. Dis. 2013, 3, 461–491. [Google Scholar] [CrossRef] [PubMed]
- Wardlaw, J.M.; Benveniste, H.; Nedergaard, M.; Zlokovic, B.V.; Mestre, H.; Lee, H.D.; Doubal, F.N.; Brown, R.; Ramirez, J.; MacIntosh, B.J.; et al. Perivascular spaces in the brain: Anatomy, physiology and pathology. Nat. Rev. Neurol. 2020, 16, 137–153. [Google Scholar] [CrossRef] [PubMed]
- Potter, G.M.; Doubal, F.N.; Jackson, C.A.; Chappell, F.M.; Sudlow, C.L.; Dennis, M.S.; Wardlaw, J.M. Enlarged perivascular spaces and cerebral small vessel disease. Int. J. Stroke 2015, 10, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Hernández, M.D.V.; Ballerini, L.; Glatz, A.; Maniega, S.M.; Gow, A.J.; Bastin, M.E.; Starr, J.M.; Deary, I.J.; Wardlaw, J.M. Perivascular spaces in the centrum semiovale at the beginning of the 8th decade of life: Effect on cognition and associations with mineral deposition. Brain Imaging Behav. 2020, 14, 1865–1875. [Google Scholar] [CrossRef]
- Wuerfel, J.; Haertle, M.; Waiczies, H.; Tysiak, E.; Bechmann, I.; Wernecke, K.D.; Zipp, F.; Paul, F. Perivascular spaces—MRI marker of inflammatory activity in the brain? Brain 2008, 131, 2332–2340. [Google Scholar] [CrossRef]
- Shen, T.; Yue, Y.M.; Zhao, S.; Xie, J.J.; Chen, Y.X.; Tian, J.; Lv, W.; Lo, C.Y.Z.; Hsu, Y.C.; Kober, T.; et al. The role of brain perivascular space burden in early-stage Parkinson’s disease. npj Park. Dis. 2021, 7, 12. [Google Scholar] [CrossRef]
- Paul, G.; Elabi, O.F. Microvascular Changes in Parkinson’s Disease-Focus on the Neurovascular Unit. Front. Aging Neurosci. 2022, 14, 853372. [Google Scholar] [CrossRef]
- Khor, S.L.Q.; Ng, K.Y.; Koh, R.Y.; Chye, S.M. Blood-brain Barrier and Neurovascular Unit Dysfunction in Parkinson’s Disease: From Clinical Insights to Pathogenic Mechanisms and Novel Therapeutic Approaches. CNS Neurol. Disord.-Drug Targets 2024, 23, 315–330. [Google Scholar] [CrossRef]
- Kortekaas, R.; Leenders, K.L.; van Oostrom, J.C.H.; Vaalburg, W.; Bart, J.; Willemsen, A.T.M.; Hendrikse, N.H. Blood-brain barrier dysfunction in Parkinsonian midbrain in vivo. Ann. Neurol. 2005, 57, 176–179. [Google Scholar] [CrossRef]
- Gray, M.T.; Woulfe, J.M. Striatal blood-brain barrier permeability in Parkinson’s disease. J. Cereb. Blood Flow Metab. 2015, 35, 747–750. [Google Scholar] [CrossRef] [PubMed]
- Gasca-Salas, C.; Pineda-Pardo, J.A.; Del Alamo, M.; Jimenez, T.; Trompeta, C.; Toltsis, G.; Garcia-Canamaque, L.; Fernandez-Rodriguez, B.; Matarazzo, M.; Plaza de Las Heras, I.; et al. Nigrostriatal blood-brain barrier opening in Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 2024. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.Y.; Wu, C.Y.; Yu, D.; Kim, E.; Wong, M.; Elez, R.; Zebarth, J.; Ouk, M.; Tan, J.; Liao, J.M.; et al. Biofluid markers of blood-brain barrier disruption and neurodegeneration in Lewy body spectrum diseases: A systematic review and meta-analysis. Park. Relat. Disord. 2022, 101, 119–128. [Google Scholar] [CrossRef]
- Hourfar, H.; Aliakbari, F.; Aqdam, S.R.; Nayeri, Z.; Bardania, H.; Otzen, D.E.; Morshedi, D. The impact of alpha-synuclein aggregates on blood-brain barrier integrity in the presence of neurovascular unit cells. Int. J. Biol. Macromol. 2023, 229, 305–320. [Google Scholar] [CrossRef] [PubMed]
- Michalicova, A.; Majerova, P.; Kovac, A. Tau Protein and Its Role in Blood-Brain Barrier Dysfunction. Front. Mol. Neurosci. 2020, 13, 570045. [Google Scholar] [CrossRef]
- Xuan, M.; Guan, X.J.; Gu, Q.Q.; Shen, Z.J.; Yu, X.F.; Qiu, T.T.; Luo, X.; Song, R.R.; Jiaerken, Y.; Xu, X.J.; et al. Different iron deposition patterns in early- and middle-late-onset Parkinson’s disease. Park. Relat. Disord. 2017, 44, 23–27. [Google Scholar] [CrossRef]
- Birkmayer, W.; Mentasti, M. Further experimental studies on the catecholamine metabolism in extrapyramidal diseases (Parkinson and chorea syndromes). Arch Psychiatr. Nervenkr. (1970) 1967, 210, 29–35. [Google Scholar] [CrossRef]
- Quinn, N. Drug-Treatment of Parkinsons-Disease. Brit. Med. J. 1995, 311, 129. [Google Scholar]
- Mercuri, N.B.; Bernardi, G. The ‘magic’ of L-dopa: Why is it the gold standard Parkinson’s disease therapy? Trends Pharmacol. Sci. 2005, 26, 341–344. [Google Scholar] [CrossRef]
- Duvoisin, R.C.; Lobo-Antunes, J.; Yahr, M.D. Response of patients with postencephalitic Parkinsonism to levodopa. J. Neurol. Neurosurg. Psychiatry 1972, 35, 487–495. [Google Scholar] [CrossRef]
- Duda, J.; Potschke, C.; Liss, B. Converging roles of ion channels, calcium, metabolic stress, and activity pattern of Substantia nigra dopaminergic neurons in health and Parkinson’s disease. J. Neurochem. 2016, 139 (Suppl. S1), 156–178. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Dijkstra, J.M.; Nagatsu, T. Parkinson’s Disease: Cells Succumbing to Lifelong Dopamine-Related Oxidative Stress and Other Bioenergetic Challenges. Int. J. Mol. Sci. 2024, 25, 2009. [Google Scholar] [CrossRef]
- Sulzer, D.; Antonini, A.; Leta, V.; Nordvig, A.; Smeyne, R.J.; Goldman, J.E.; Al-Dalahmah, O.; Zecca, L.; Sette, A.; Bubacco, L.; et al. COVID-19 and possible links with Parkinson’s disease and parkinsonism: From bench to bedside. npj Park. Dis. 2020, 6, 18. [Google Scholar] [CrossRef] [PubMed]
- Savica, R.; Rocca, W.A.; Ahlskog, J.E. When Does Parkinson Disease Start? Arch. Neurol. 2010, 67, 798–801. [Google Scholar] [CrossRef]
- Blesa, J.; Trigo-Damas, I.; Dileone, M.; Del Rey, N.L.; Hernandez, L.F.; Obeso, J.A. Compensatory mechanisms in Parkinson’s disease: Circuits adaptations and role in disease modification. Exp. Neurol. 2017, 298, 148–161. [Google Scholar] [CrossRef] [PubMed]
- Turrigiano, G. Homeostatic Synaptic Plasticity: Local and Global Mechanisms for Stabilizing Neuronal Function. Cold Spring Harb. Perspect. Biol. 2012, 4, a005736. [Google Scholar] [CrossRef]
- Clavaguera, F.; Tolnay, M.; Goedert, M. The Prion-Like Behavior of Assembled Tau in Transgenic Mice. Cold Spring Harb. Perspect. Biol. 2017, 7, a024372. [Google Scholar] [CrossRef]
- Jan, A.; Goncalves, N.P.; Vaegter, C.B.; Jensen, P.H.; Ferreira, N. The Prion-Like Spreading of Alpha-Synuclein in Parkinson’s Disease: Update on Models and Hypotheses. Int. J. Mol. Sci. 2021, 22, 8338. [Google Scholar] [CrossRef]
- Zhao, S.; Fan, Z.Q.; Zhang, X.Y.; Li, Z.Y.; Shen, T.; Li, K.C.; Yan, Y.P.; Yuan, Y.F.; Pu, J.L.; Tian, J.; et al. Metformin Attenuates Tau Pathology in Tau-Seeded PS19 Mice. Neurotherapeutics 2023, 20, 452–463. [Google Scholar] [CrossRef]
- Katila, N.; Bhurtel, S.; Shadfar, S.; Srivastav, S.; Neupane, S.; Ojha, U.; Jeong, G.S.; Choi, D.Y. Metformin lowers α-synuclein phosphorylation and upregulates neurotrophic factor in the MPTP mouse model of Parkinson’s disease. Neuropharmacology 2017, 125, 396–407. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Case Number | Extrapyramidal Features | L-Dopa Response | Tendency to Fall |
|---|---|---|---|
| PEP 1 | Bradykinesia, retropulsion, oculogyric crises | + | + |
| PEP 2 | Tremor, hypomimia, gait disturbance | + | + |
| PEP 3 | Gait disturbance | 0 | + |
| PEP 4 | Sialorhea, rigidity, gait disturbance | + | + |
| PEP 5 | Bradikinesia, tremor | 0 | + |
| Case Number | Age at D/E/S | Gender | Braak (LB) | Localization of Iron | α-Syn | Tau |
|---|---|---|---|---|---|---|
| CO 1 | 80 | m | 0 | p | − | − |
| CO 2 | 74 | m | 0 | n, i, p | − | − |
| CO 3 | 65 | m | 0 | i, p | − | − |
| CO 4 | 89 | f | 0 | p | − | − |
| CO 5 | 91 | m | 0 | i | − | − |
| CO 6 | 73 | f | 0 | i, p | − | − |
| CO 7 | 49 | m | 0 | i, p | − | − |
| CO 8 | 60 | f | 0 | i, p | − | − |
| CO 9 | 67 | f | 0 | i | − | − |
| CO 10 | 71 | f | 0 | i | − | − |
| CO 11 | 62 | f | 0 | i | − | − |
| PD 1 | 69 | f | IV | n, i | + | − |
| PD 2 | 58 | m | IV | n, i, p | + | − |
| PD 3 | 85 | m | IV | i | + | − |
| PD 4 | 87 | f | III | i | + | − |
| PD 5 | 70 | f | IV | n, i, p | + | − |
| PD 6 | 72 | m | V | i, p | + | − |
| PD 7 | 83 | m | VI | n, i, p | + | − |
| PD 8 | 69 | f | V | n, i, p | + | − |
| PD 9 | 72 | f | IV | n, i, p | + | − |
| PD 10 | 96 | f | IV | n, i, p | + | − |
| PD 11 | 58 | m | VI | n, i, p | + | − |
| PD 12 | 80 | m | IV | n, i | + | − |
| PD 13 | 63 | m | II | n, i | + | − |
| PD 14 | 77 | m | III | n, i, p | + | − |
| PD 15 | 81 | m | IV | n, i | + | − |
| PD 16 | 87 | m | VI | n, i, p | + | − |
| PD 17 | 58 | w | III | n, i | + | − |
| PD 18 | 72 | m | V | n, p | + | − |
| PEP 1 | 66/26/38 | f | 0 | Ø | − | + |
| PEP 2 | 77/16/60 | f | 0 | Ø | − | + |
| PEP 3 | 66/16/44 | m f | 0 | Ø | − | + |
| PEP 4 | 65/11/32 | f | 0 | p | − | + |
| PEP 5 | 69/17/45 | m | 0 | Ø | − | + |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Strobel, S.; Sian-Hulsmann, J.; Tappe, D.; Jellinger, K.; Riederer, P.; Monoranu, C.-M. Postencephalitic Parkinsonism: Unique Pathological and Clinical Features—Preliminary Data. Cells 2024, 13, 1511. https://doi.org/10.3390/cells13181511
Strobel S, Sian-Hulsmann J, Tappe D, Jellinger K, Riederer P, Monoranu C-M. Postencephalitic Parkinsonism: Unique Pathological and Clinical Features—Preliminary Data. Cells. 2024; 13(18):1511. https://doi.org/10.3390/cells13181511
Chicago/Turabian StyleStrobel, Sabrina, Jeswinder Sian-Hulsmann, Dennis Tappe, Kurt Jellinger, Peter Riederer, and Camelia-Maria Monoranu. 2024. "Postencephalitic Parkinsonism: Unique Pathological and Clinical Features—Preliminary Data" Cells 13, no. 18: 1511. https://doi.org/10.3390/cells13181511
APA StyleStrobel, S., Sian-Hulsmann, J., Tappe, D., Jellinger, K., Riederer, P., & Monoranu, C.-M. (2024). Postencephalitic Parkinsonism: Unique Pathological and Clinical Features—Preliminary Data. Cells, 13(18), 1511. https://doi.org/10.3390/cells13181511